
The Neuroprotective Effects and Therapeutic Potential of the Chalcone Cardamonin for Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods for Literature Review
3. Neurodegenerative Diseases
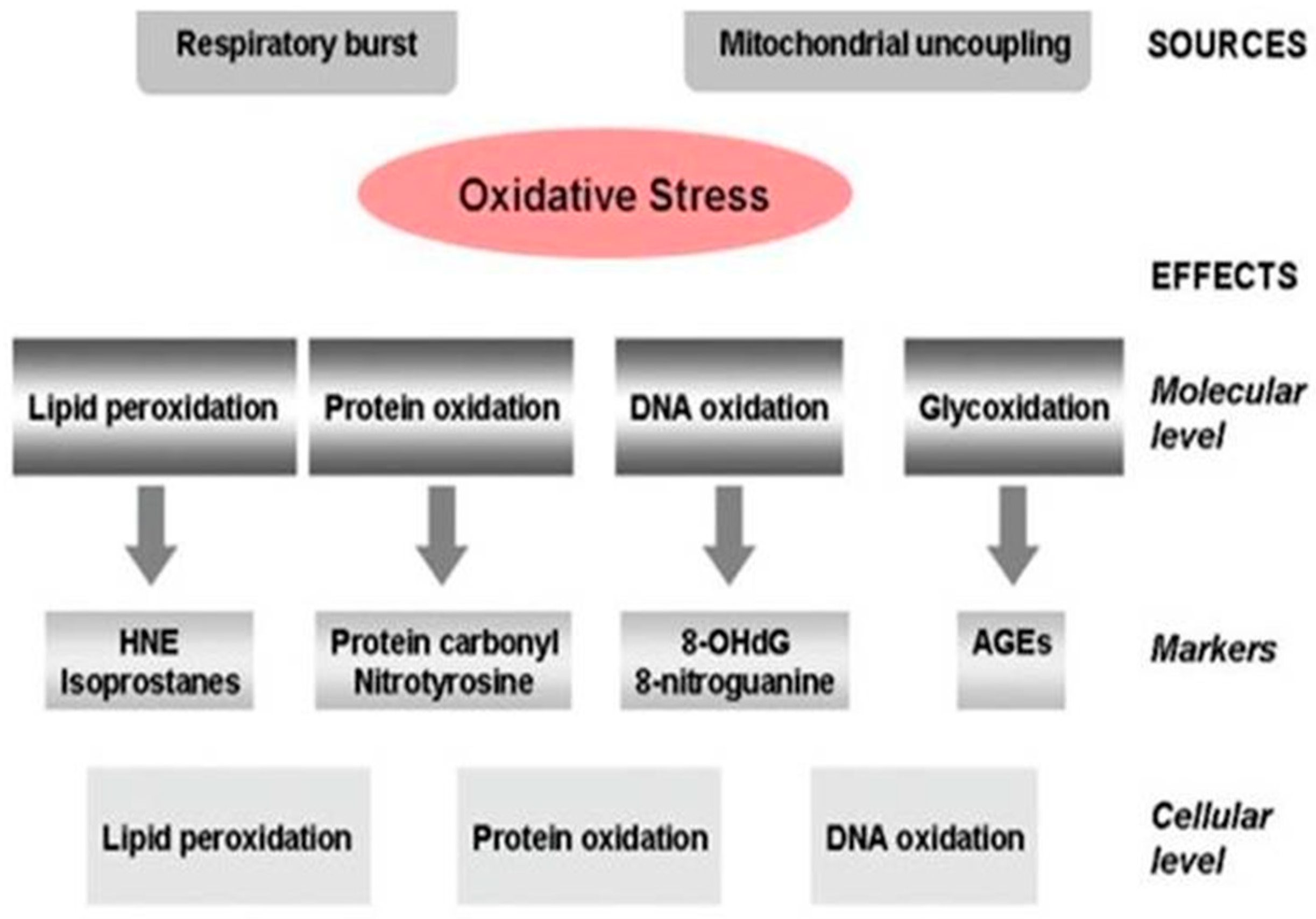
Alzheimer’s Disease Pathogenesis, Risk Factors, and Oxidative Stress
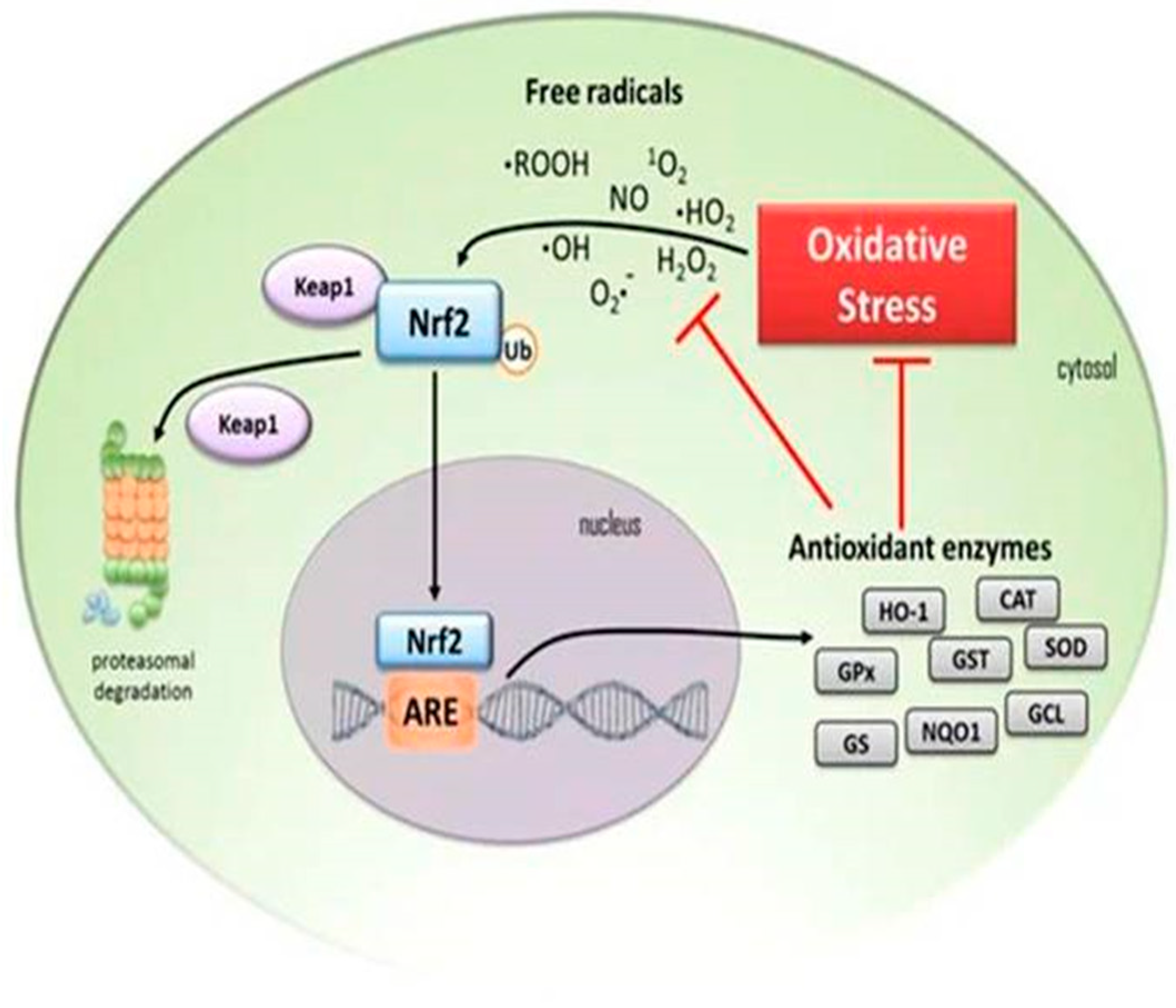
4. Down-Regulation of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Antioxidant Activity in the Elderly
5. Biomarkers for Diagnosis and Treatment of Alzheimer’s Disease
5.1. The Role of Neuroinflammation in Alzheimer’s Disease
5.2. Microglia Role in Neuroinflammation
5.3. Intracellular Signaling Pathways Activated in Neuroinflammation
5.3.1. MAPK Signaling in Neuroinflammation
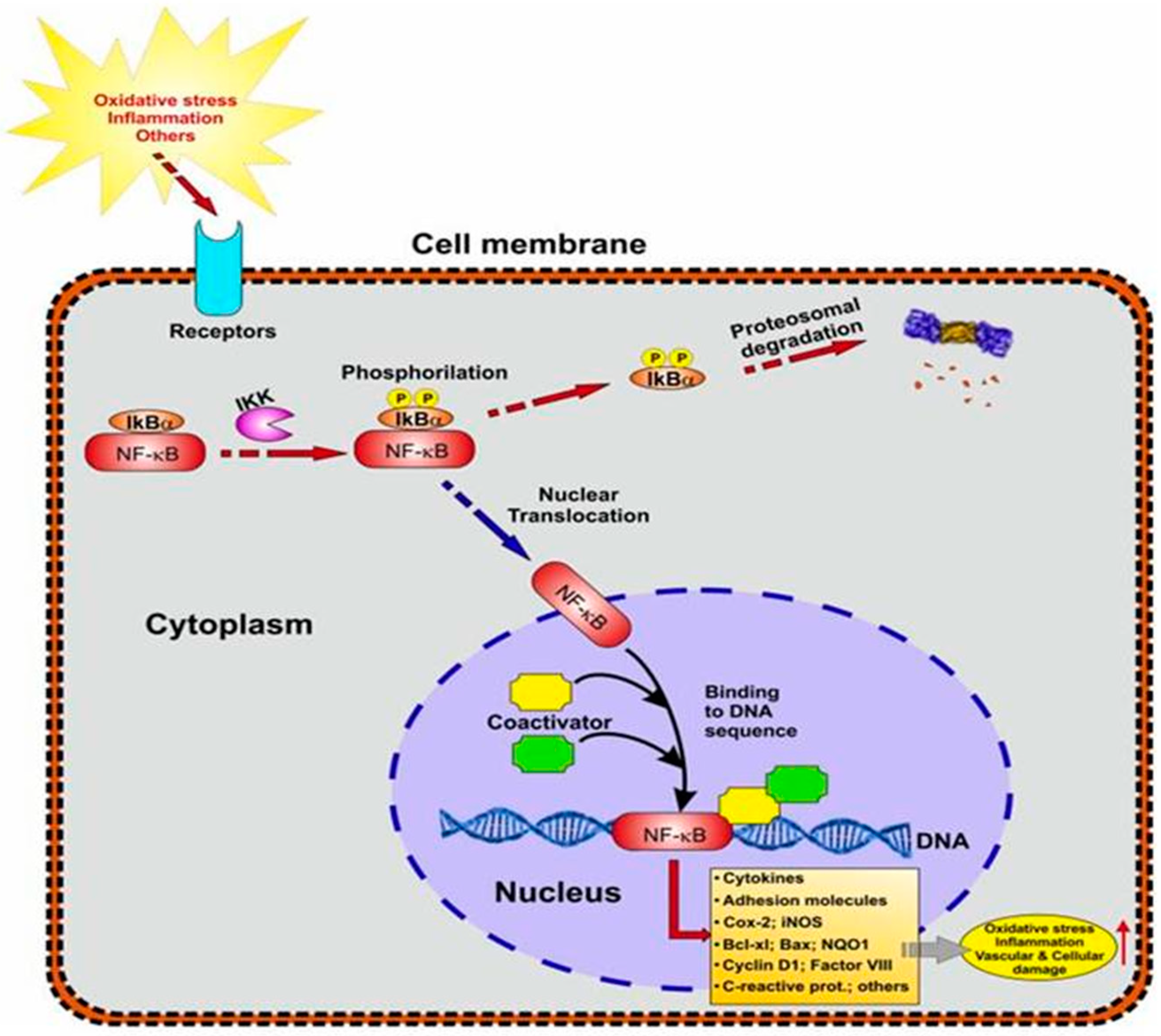
5.3.2. NF-kB Signaling in Neuroinflammation
5.4. Cytokines and Neuroinflammation
5.5. The Association between NF-kB and Nrf2
6. Current FDA-Approved AD Medications
7. The Use of Natural Compounds to Prevent Alzheimer’s Disease
7.1. Cardamonin
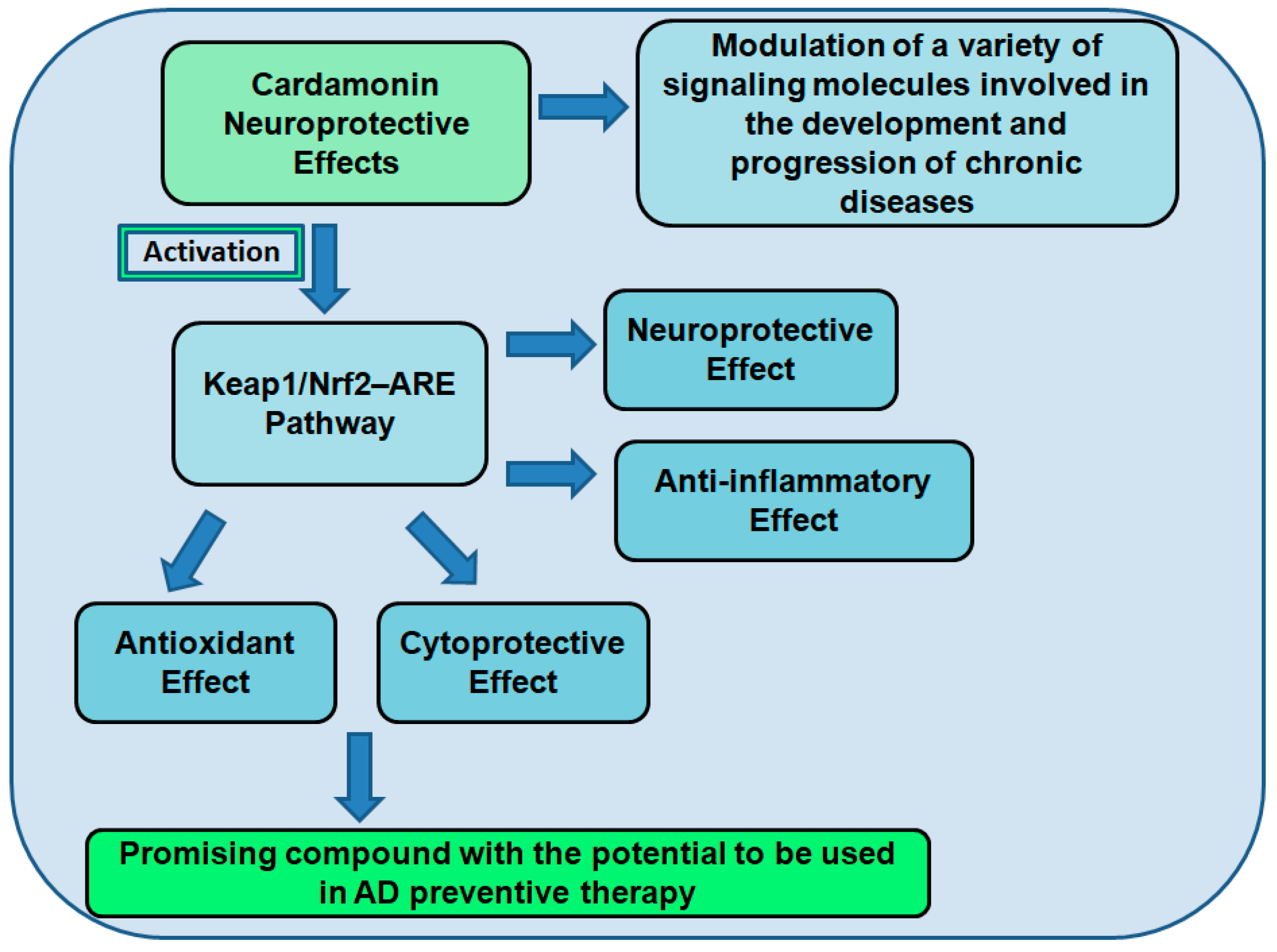
7.2. Cardamonin as a Neuroprotective Compound
7.3. The Role of Cardamonin as an Antioxidant and Nrf2 Activator
Neuroprotective Effects of Nrf2 Activation
7.4. Cardamonin and Neuroinflammation
7.5. Cardamonin and Autophagy
7.6. Cardamonin and Microglia
7.7. Cardamonin and MicroRNA
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garofalo, M.; Pandini, C.; Bordoni, M.; Pansarasa, O.; Rey, F.; Costa, A.; Minafra, B.; Diamanti, L.; Zucca, S.; Carelli, S.; et al. Alzheimer’s, Parkinson’s Disease and Amyotrophic Lateral Sclerosis Gene Expression Patterns Divergence Reveals Different Grade of RNA Metabolism Involvement. Int. J. Mol. Sci. 2020, 21, 9500. [Google Scholar] [CrossRef]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Park. Dis. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.-J.; Zhu, X.-X.; Yang, X.; Jin, B.; Lu, J.-J.; Ding, B.; Ding, Z.-S.; Chen, S.-H. Cardamonin inhibits angiotensin II-induced vascular smooth muscle cell proliferation and migration by downregulating p38 MAPK, Akt, and ERK phosphorylation. J. Nat. Med. 2014, 68, 623–629. [Google Scholar] [CrossRef] [PubMed]
- El-Naga, R.N. Pre-treatment with cardamonin protects against cisplatin-induced nephrotoxicity in rats: Impact on NOX-1, inflammation and apoptosis. Toxicol. Appl. Pharmacol. 2014, 274, 87–95. [Google Scholar] [CrossRef]
- Lee, M.Y.; Seo, C.S.; Lee, J.A.; Shin, I.S.; Kim, S.J.; Ha, H.; Shin, H.K. Alpinia katsumadai H(AYATA) seed extract inhibit LPS-induced inflammation by induction of heme oxygenase-1 in RAW264.7 cells. Inflammation 2012, 35, 746–757. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [Green Version]
- WHO. Dementia; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 101–108. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjians, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Dhikav, V.; Anand, K.S. Hippocampus in health and disease: An overview. Ann. Indian Acad. Neurol. 2012, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Vasic, V.; Barth, K.; Schmidt, M.H. Neurodegeneration and Neuro-Regeneration-Alzheimer’s Disease and Stem Cell Therapy. Int. J. Mol. Sci. 2019, 20, 4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabłocka, A. Alzheimer’s disease as neurodegenerative disorder. Postepy Hig. Med. Dosw. (Online) 2006, 60, 209–216. [Google Scholar] [PubMed]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R. Risk factors for Alzheimer’s disease. Folia Neuropathologica. 2019, 57, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Killin, L.O.J.; Starr, J.M.; Shiue, I.J.; Russ, T.C. Environmental risk factors for dementia: A systematic review. BMC Geriatr. 2016, 16, 175. [Google Scholar] [CrossRef]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-J.; Kang, K.S.; Choi, K.-C.; Ko, H. Cardamonin induces autophagy and an antiproliferative effect through JNK activation in human colorectal carcinoma HCT116 cells. Bioorganic Med. Chem. Lett. 2015, 25, 2559–2564. [Google Scholar] [CrossRef]
- Kim, E.J.; Kim, H.J.; Park, M.K.; Kang, G.J.; Byun, H.J.; Lee, H.; Lee, C.H. Cardamonin Suppresses TGF-β1-Induced Epithelial Mesenchymal Transition via Restoring Protein Phosphatase 2A Expression. Biomol. Ther. 2015, 23, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, Y.; Picciano, M.; La Francois, J.; Duff, K. Fibrillar beta-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer’s disease. Neuroscience 2001, 104, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free. Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [Green Version]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Humpel, C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011, 29, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Llerena, C.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.; Vafeiadou, K.; Williams, R.J.; Vauzour, D. Neuroinflammation: Modulation by flavonoids and mechanisms of action. Mol. Asp. Med. 2012, 33, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K. Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimers Dis. 2010, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- McGeer, E.G.; McGeer, P.L. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J Alzheimers Dis. 2010, 19, 355–361. [Google Scholar] [CrossRef]
- Brown, G.C.; Bal-Price, A. Inflammatory Neurodegeneration Mediated by Nitric Oxide, Glutamate, and Mitochondria. Mol. Neurobiol. 2003, 27, 325–355. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, H.S.; Baron, J.; Hagl, S.; Eckert, G.P.; Fiebich, B.L. Rice bran derivatives alleviate microglia activation: Possible involvement of MAPK pathway. J. Neuroinflammation 2016, 13, 148. [Google Scholar] [CrossRef] [Green Version]
- Albert-Gascó, H.; Ros-Bernal, F.; Castillo-Gómez, E.; Olucha-Bordonau, F.E. MAP/ERK Signaling in Developing Cognitive and Emotional Function and Its Effect on Pathological and Neurodegenerative Processes. Int. J. Mol. Sci. 2020, 21, 4471. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, T.G. Role of Nuclear Factor Kappa B (NF-κB) Signalling in Neurodegenerative Diseases: An Mechanistic Approach. Curr. Neuropharmacol. 2020, 18, 918–935. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms, and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Sharma, V.; Thakur, V.; Singh, S.N.; Guleria, R. Tumor Necrosis Factor, and Alzheimer’s Disease: A Cause and Consequence Relationship. Klin. Psikofarmakol. Bülteni Bull. Clin. Psychopharmacol. 2012, 22, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry. 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Liao, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tweedie, D.; Ferguson, R.A.; Fishman, K.; Frankola, K.A.; Van Praag, H.; Holloway, H.W.; Luo, W.; Li, Y.; Caracciolo, L.; Russo, I.; et al. Tumor necrosis factor-α synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflammation 2012, 9, 106. [Google Scholar] [CrossRef]
- Kornman, K.S. Interleukin 1 genetics, inflammatory mechanisms, and nutrigenetic opportunities to modulate diseases of aging. Am. J. Clin. Nutr. 2006, 83, 475S–483S. [Google Scholar] [CrossRef] [Green Version]
- Honma, T.; Hatta, K.; Hitomi, Y.; Kambayashi, Y.; Hibino, Y.; Konoshita, T.; Nakamura, H. Increased systemic inflammatory interleukin-1ß and interleukin-6 during agitation as predictors of Alzheimer’s disease. Int. J. Geriatr. Psychiatry. 2013, 28, 233–241. [Google Scholar] [CrossRef]
- Rogers, J.T.; Leiter, L.M.; McPhee, J.; Cahill, C.M.; Zhan, S.-S.; Potter, H.; Nilsson, L.N.G. Translation of the Alzheimer Amyloid Precursor Protein mRNA Is Up-regulated by Interleukin-1 through 5′-Untranslated Region Sequences. J. Biol. Chem. 1999, 274, 6421–6431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serretti, A.; Olgiati, P.; De Ronchi, D. Genetics of Alzheimer’s disease. A rapidly evolving field. J. Alzheimers Dis. 2007, 12, 73–92. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tones, Y.; Tong, Y.; Song, W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.-P.; Qu, Z.-Y.; Duan, S.-R.; Wei, S.-Q.; Wen, S.-R.; Bi, S. IL-6-174 G/C and -572 C/G Polymorphisms and Risk of Alzheimer’s Disease. PLOS ONE 2012, 7, e37858. [Google Scholar] [CrossRef]
- Talalay, P.; Dinkova-Kostova, A.T.; Holtzclaw, W. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv. Enzym. Regul. 2003, 43, 121–134. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Guo, X.; Huang, Q.; Kong, Y.; Lu, X. Association between interleukin-6 -174G/C polymorphism and the risk of Alzheimer’s disease: A meta-analysis. Int. J. Neurosci. 2013, 123, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Szczepanik, A.M.; Funes, S.; Petko, W.; Ringheim, G.E. IL-4, IL-10, and IL-13 modulate A beta(1--42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J. Neuroimmunol. 2001, 113, 49–62. [Google Scholar] [CrossRef]
- Kim, J.; Onstead, L.; Randle, S.; Price, R.; Smithson, L.; Zwizinski, C.; Dickson, D.W.; Golde, T.; McGowan, E. Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 2007, 27, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, A.J.; Yamamoto, M.; De Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Kishi, T.; Iwata, N. Memantine Monotherapy for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0123289. [Google Scholar] [CrossRef] [Green Version]
- Farlow, M.; Anand, R.; Messina, J., Jr.; Hartman, R.; Veach, J. A 52-Week Study of the Efficacy of Rivastigmine in Patients with Mild to Moderately Severe Alzheimer’s Disease. Eur. Neurol. 2000, 44, 236–241. [Google Scholar] [CrossRef]
- Rountree, S.D.; Atri, A.; Lopez, O.L.; Doody, R.S. Effectiveness of antidementia drugs in delaying Alzheimer’s disease progression. Alzheimer Dement. 2013, 9, 338–345. [Google Scholar] [CrossRef]
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef]
- Prasathkumar, M.; Anisha, S.; Dhrisya, C.; Becky, R.; Sadhasivam, S. Therapeutic and pharmacological efficacy of selective Indian medicinal plants—A review. Phytomedicine Plus 2021, 1, 100029. [Google Scholar] [CrossRef]
- Daimary, U.D.; Parama, D.; Rana, V.; Banik, K.; Kumar, A.; Harsha, C.; Kunnumakkara, A.B. Emerging roles of cardamonin, a multitargeted nutraceutical in the prevention and treatment of chronic diseases. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, L.; Chen, J.; Li, Q.; Huo, L.; Wang, Y.; Wang, H.; Du, J. Pharmacological Modulation of Nrf2/HO-1 Signaling Pathway as a Therapeutic Target of Parkinson’s Disease. Front. Pharmacol. 2021, 12, 757161. [Google Scholar] [CrossRef]
- Nawaz, J.; Rasul, A.; Shah, M.A.; Hussain, G.; Riaz, A.; Sarfraz, I.; Zafar, S.; Adnan, M.; Khan, A.H.; Selamoglu, Z. Cardamonin: A new player to fight cancer via multiple cancer signaling pathways. Life Sci. 2020, 250, 117591. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.M.; Valente, I.M.; Rodrigues, J.A. An Overview on Cardamonin. J. Med. Food 2014, 17, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatziieremia, S.; Gray, A.I.; Ferro, V.A.; Paul, A.; Plevin, R. The effects of cardamonin on lipopolysaccharide-induced inflammatory protein production and MAP kinase and NFkappaB signalling pathways in monocytes/macrophages. Br. J. Pharmacol. 2006, 149, 188–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Jung, H.; Giang, P.; Jin, X.; Lee, S.; Son, P.; Lee, D.; Hong, Y.S.; Lee, K.; Lee, J.J. Blockade of Nuclear Factor- B Signaling Pathway and Anti-Inflammatory Activity of Cardamomin, a Chalcone Analog from Alpinia conchigera. J. Pharmacol. Exp. Ther. 2006, 316, 271–278. [Google Scholar] [CrossRef]
- Peng, Y.-J.; Lu, J.-W.; Lee, C.-H.; Lee, H.-S.; Chu, Y.-H.; Ho, Y.-J.; Liu, F.-C.; Huang, C.-J.; Wu, C.-C.; Wang, C.-C. Cardamonin Attenuates Inflammation and Oxidative Stress in Interleukin-1β-Stimulated Osteoarthritis Chondrocyte through the Nrf2 Pathway. Antioxidants 2021, 10, 862. [Google Scholar] [CrossRef]
- Singh, S.; Nagalakshmi, D.; Sharma, K.; Ravichandiran, V. Natural antioxidants for neuroinflammatory disorders and possible involvement of Nrf2 pathway: A review. Heliyon 2021, 7, e06216. [Google Scholar] [CrossRef]
- An, J.; Chen, B.; Kang, X.; Zhang, R.; Guo, Y.; Zhao, J.; Yang, H. Neuroprotective effects of natural compounds on LPS-induced inflammatory responses in microglia. Am. J. Transl. Res. 2020, 12, 2353–2378. [Google Scholar] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer’s disease. Cell Adh. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Hou, Y.; Yao, J.; Fang, J. Activation of Nrf2-driven antioxidant enzymes by cardamonin confers neuroprotection of PC12 cells against oxidative damage. Food Funct. 2017, 8, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Francisqueti-Ferron, F.V.; Ferron, A.J.T.; Garcia, J.L.; Silva, C.C.V.D.A.; Costa, M.R.; Gregolin, C.S.; Moreto, F.; Ferreira, A.L.A.; Minatel, I.O.; Correa, C.R. Basic Concepts on the Role of Nuclear Factor Erythroid-Derived 2-Like 2 (Nrf2) in Age-Related Diseases. Int. J. Mol. Sci. 2019, 20, 3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Yang, T.; Leak, R.K.; Chen, J.; Zhang, F. Preventive and Protective Roles of Dietary Nrf2 Activators Against Central Nervous System Diseases. CNS Neurol. Disord. Drug Targets 2017, 16, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Fakhri, S.; Pesce, M.; Patruno, A.; Moradi, S.Z.; Iranpanah, A.; Farzaei, M.H.; Sobarzo-Sánchez, E. Attenuation of Nrf2/Keap1/ARE in Alzheimer’s Disease by Plant Secondary Metabolites: A Mechanistic Review. Molecules 2020, 25, 4926. [Google Scholar] [CrossRef]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical Antioxidants as Novel Neuroprotective Agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [Green Version]
- Chow, Y.L.; Lee, K.H.; Vidyadaran, S.; Lajis, N.H.; Akhtar, M.N.; Israf, D.A.; Syahida, A. Cardamonin from Alpinia rafflesiana inhibits inflammatory responses in IFN-γ/LPS-stimulated BV2 microglia via NF-κB signalling pathway. Int. Immunopharmacol. 2012, 12, 657–665. [Google Scholar] [CrossRef]
- Arroyo, D.S.; Gaviglio, E.A.; Peralta Ramos, J.M.; Bussi, C.; Rodriguez-Galan, M.C.; Iribarren, P. Autophagy in inflammation, infection, neurodegeneration, and cancer. Int. Immunopharmacol. 2014, 18, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Law, B.Y.K.; Mok, S.W.F.; Wu, A.G.; Lam, C.W.K.; Yu, M.X.Y.; Wong, V.K.W. New Potential Pharmacological Functions of Chinese Herbal Medicines via Regulation of Autophagy. Molecules 2016, 21, 359. [Google Scholar] [CrossRef] [PubMed]
- Frake, R.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, A.; Terro, F.; Janet, T.; Bilan, A.R.; Paccalin, M.; Page, G. Involvement of interleukin-1β in the autophagic process of microglia: Relevance to Alzheimer’s disease. J. Neuroinflammation 2013, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Stachowiak, A.; Al Mamun, A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rahman, M.S.; Rahman, M.D.H.; Rasheduzzaman, M.; Mamun-Or-Rashid, A.; Uddin, M.J.; Rahman, M.R.; Hwang, H.; Pang, M.G.; Rhim, H. Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease. Biomedicines 2020, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Niu, P.; Heng, X.; Chen, L.; Zhu, Y.; Zhou, J. Autophagy induced by cardamonin is associated with mTORC1 inhibition in SKOV3 cells. Pharmacol. Rep. 2018, 70, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, G.; Gao, Y.; Zhan, X.; Qin, N.; Fu, S.; Li, R.; Niu, M.; Wang, J.; Liu, Y.; et al. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm. Sin. B 2019, 9, 734–744. [Google Scholar] [CrossRef]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Drew, J.; Berney, W.; Lei, W. Neuroprotective Natural Products for Alzheimer’s Disease. Cells 2021, 10, 1309. [Google Scholar] [CrossRef]
- Liu, M.; Shan, G.; Jiang, H.; Zeng, L.; Zhao, K.; Li, Y.; Ashraf, G.; Li, Z.; Liu, R. Identification of miRNA and Their Regulatory Effects Induced by Total Flavonoids From Dracocephalum moldavica in the Treatment of Vascular Dementia. Front. Pharmacol. 2021, 12, 796628. [Google Scholar] [CrossRef]
- Benameur, T.; Soleti, R.; Porro, C. The Potential Neuroprotective Role of Free and Encapsulated Quercetin Mediated by miRNA against Neurological Diseases. Nutrients 2021, 13, 1318. [Google Scholar] [CrossRef]
- James, S.; Aparna, J.S.; Paul, A.M.; Lankadasari, M.B.; Mohammed, S.; Binu, V.S.; Santhoshkumar, T.R.; Reshmi, G.; Harikumar, K.B. Cardamonin inhibits colonic neoplasia through modulation of MicroRNA expression. Sci. Rep. 2017, 7, 13945. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barber, K.; Mendonca, P.; Soliman, K.F.A. The Neuroprotective Effects and Therapeutic Potential of the Chalcone Cardamonin for Alzheimer’s Disease. Brain Sci. 2023, 13, 145. https://doi.org/10.3390/brainsci13010145
Barber K, Mendonca P, Soliman KFA. The Neuroprotective Effects and Therapeutic Potential of the Chalcone Cardamonin for Alzheimer’s Disease. Brain Sciences. 2023; 13(1):145. https://doi.org/10.3390/brainsci13010145
Chicago/Turabian StyleBarber, Kimberly, Patricia Mendonca, and Karam F. A. Soliman. 2023. "The Neuroprotective Effects and Therapeutic Potential of the Chalcone Cardamonin for Alzheimer’s Disease" Brain Sciences 13, no. 1: 145. https://doi.org/10.3390/brainsci13010145
APA StyleBarber, K., Mendonca, P., & Soliman, K. F. A. (2023). The Neuroprotective Effects and Therapeutic Potential of the Chalcone Cardamonin for Alzheimer’s Disease. Brain Sciences, 13(1), 145. https://doi.org/10.3390/brainsci13010145