
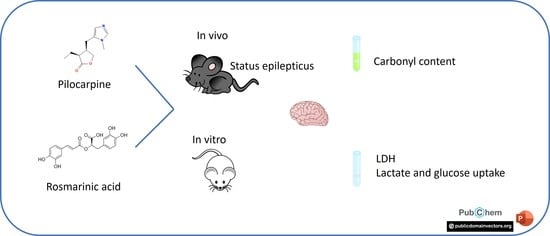
Beneficial Effects of Rosmarinic Acid In Vitro and In Vivo Models of Epileptiform Activity Induced by Pilocarpine

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
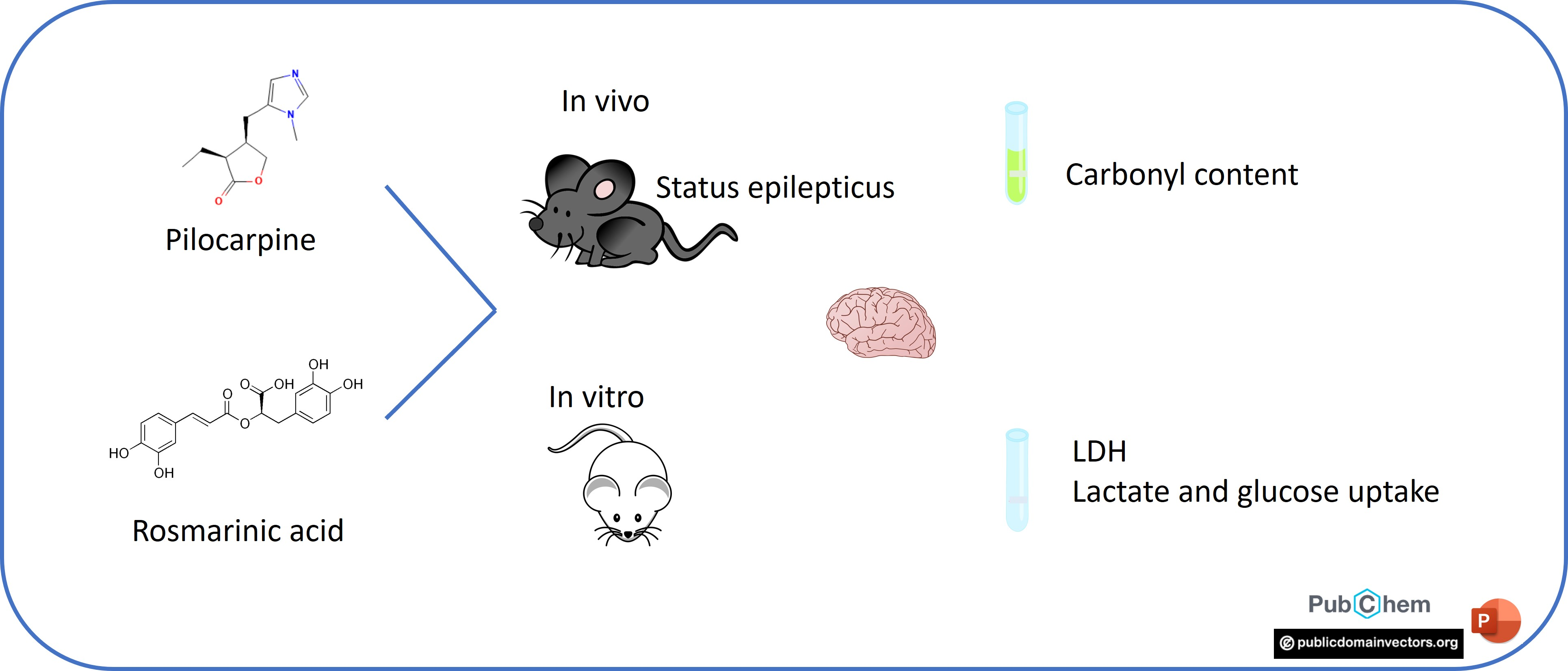
2.1. Animals
2.2. Reagents
2.3. In Vitro Experiments
2.3.1. LDH Release
2.3.2. Lactate Release
2.3.3. Glucose Uptake
2.4. In Vivo Experiments
2.4.1. Neuroscore
2.4.2. Dot Blot Assay
2.4.3. Protein Content
2.5. Statistical Analyses
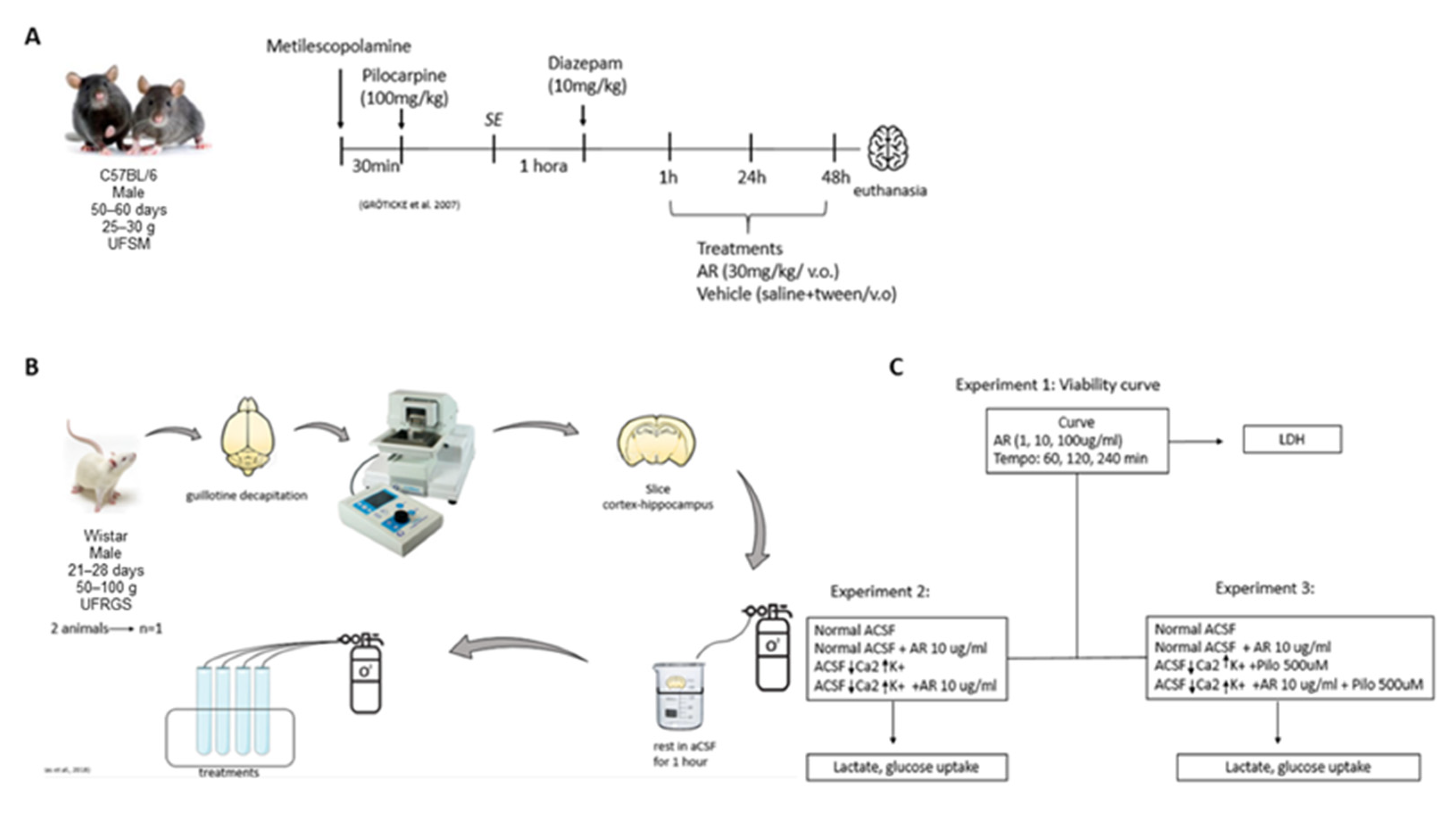
3. Results
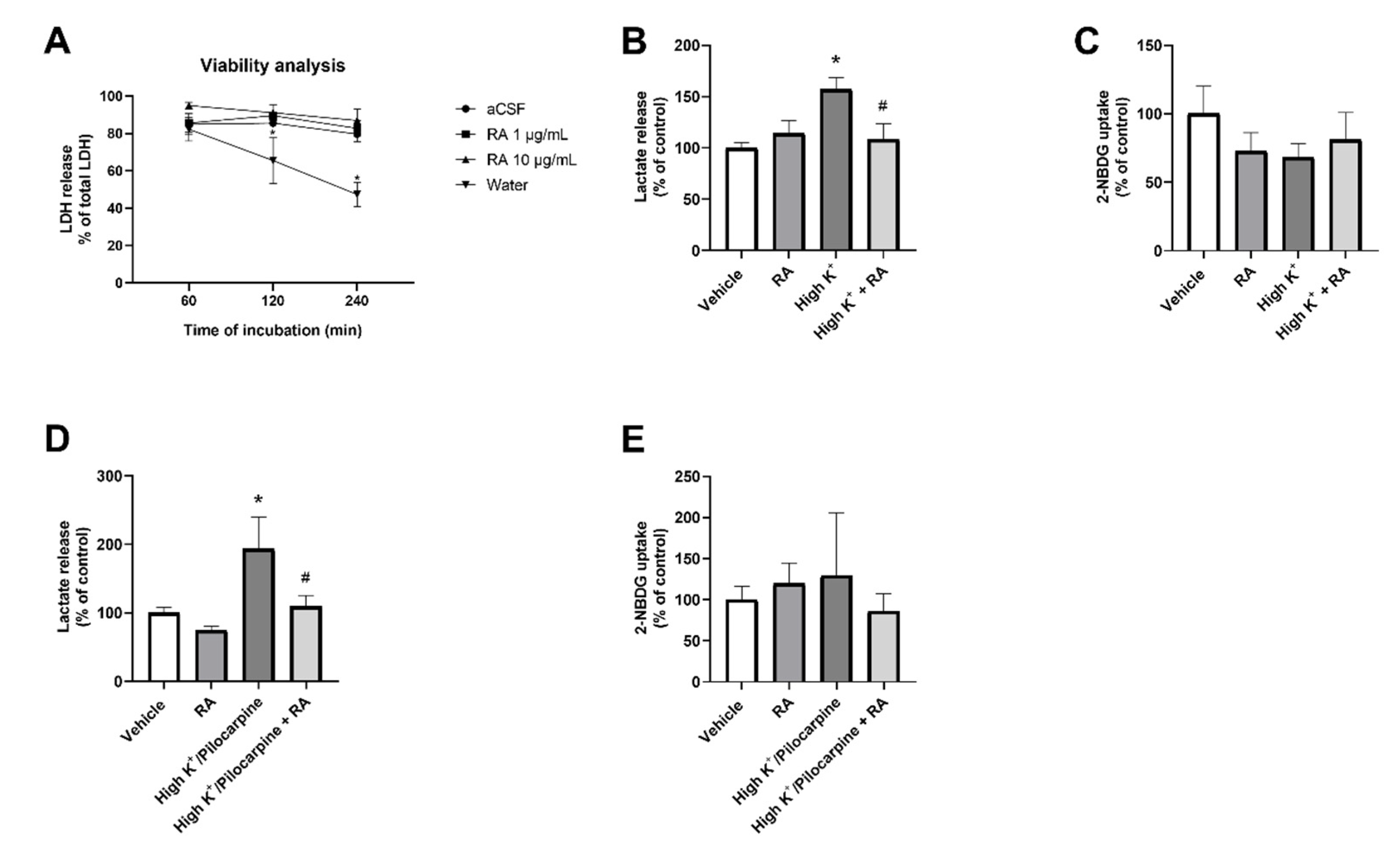
3.1. In Vitro Results
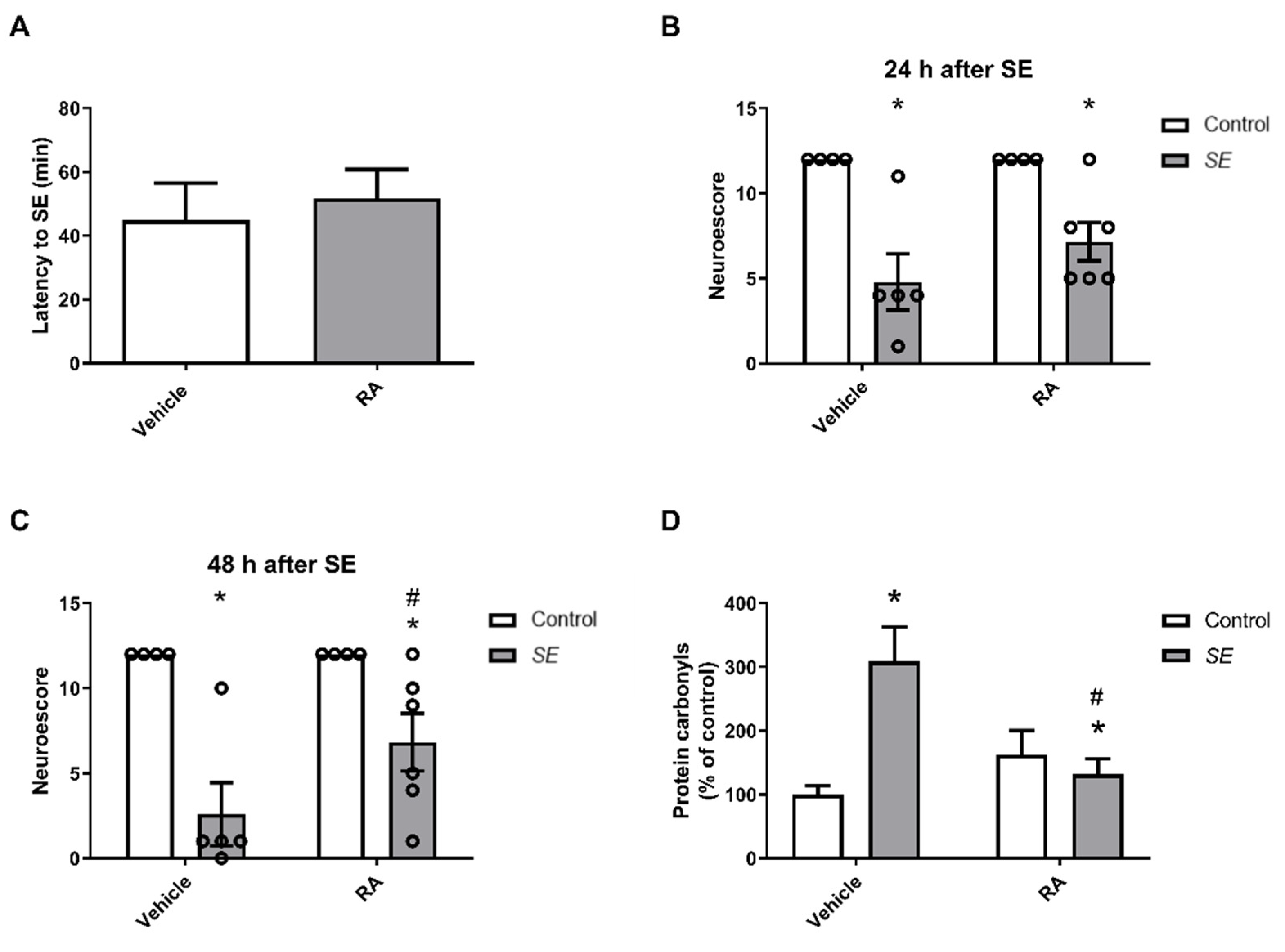
3.2. In Vivo Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S. Redefining epilepsy. Curr. Opin. Neurol. 2015, 28, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Levesque, M.; Biagini, G.; de Curtis, M.; Gnatkovsky, V.; Pitsch, J.; Wang, S.; Avoli, M. The pilocarpine model of mesial temporal lobe epilepsy: Over one decade later, with more rodent species and new investigative approaches. Neurosci. Biobehav. Rev. 2021, 130, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A.; Surgucheva, I.; Sharma, M.; Sharma, R.; Singh, V. Pore-Forming Proteins as Mediators of Novel Epigenetic Mechanism of Epilepsy. Front. Neurol. 2017, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Rowley, S.; Patel, M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic. Biol. Med. 2013, 62, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.D.; Park, Y.S.; Jin, Y.H.; Park, C.S. Production and applications of rosmarinic acid and structurally related compounds. Appl. Microbiol. Biotechnol. 2015, 99, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Rahbardar, M.G.; Hosseinzadeh, H. Effects of rosmarinic acid on nervous system disorders: An updated review. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 1779–1795. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, K.; Saul, N.; Chakrabarti, S.; Sturzenbaum, S.R.; Menzel, R.; Steinberg, C.E. Hormetins, antioxidants and prooxidants: Defining quercetin-, caffeic acid- and rosmarinic acid-mediated life extension in C. elegans. Biogerontology 2011, 12, 329–347. [Google Scholar] [CrossRef]
- Gamaro, G.D.; Suyenaga, E.; Borsoi, M.; Lermen, J.; Pereira, P.; Ardenghi, P. Effect of rosmarinic and caffeic acids on inflammatory and nociception process in rats. ISRN Pharmacol. 2011, 2011, 451682. [Google Scholar] [CrossRef]
- Jalali, A.; Firouzabadi, N.; Zarshenas, M.M. Pharmacogenetic-based management of depression: Role of traditional Persian medicine. Phytother. Res. 2021, 35, 5031–5052. [Google Scholar] [CrossRef]
- Khamse, S.; Sadr, S.S.; Roghani, M.; Hasanzadeh, G.; Mohammadian, M. Rosmarinic acid exerts a neuroprotective effect in the kainate rat model of temporal lobe epilepsy: Underlying mechanisms. Pharm. Biol. 2015, 53, 1818–1825. [Google Scholar] [CrossRef]
- Grigoletto, J.; Oliveira, C.V.; Grauncke, A.C.; Souza, T.L.; Souto, N.S.; Freitas, M.L.; Furian, A.F.; Santos, A.R.; Oliveira, M.S. Rosmarinic acid is anticonvulsant against seizures induced by pentylenetetrazol and pilocarpine in mice. Epilepsy Behav. 2016, 62, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Coelho, V.R.; Vieira, C.G.; de Souza, L.P.; Moyses, F.; Basso, C.; Papke, D.K.; Pires, T.R.; Siqueira, I.R.; Picada, J.N.; Pereira, P. Antiepileptogenic, antioxidant and genotoxic evaluation of rosmarinic acid and its metabolite caffeic acid in mice. Life Sci. 2015, 122, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Berdichevsky, Y.; Saponjian, Y.; Park, K.I.; Roach, B.; Pouliot, W.; Lu, K.; Swiercz, W.; Dudek, F.E.; Staley, K.J. Staged anticonvulsant screening for chronic epilepsy. Ann. Clin. Transl. Neurol. 2016, 3, 908–923. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.L.; Oliveira, C.V.; Mello, F.K.; Funck, V.R.; Fighera, M.R.; Royes, L.F.F.; Furian, A.F.; Larrick, J.W.; Oliveira, M.S. Na(+), K(+)-ATPase Activating Antibody Displays in vitro and in vivo Beneficial Effects in the Pilocarpine Model of Epilepsy. Neuroscience 2018, 377, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Kim, H.B.; Choi, G.Y.; Lee, S.; Lee, S.O.; Kim, S.; Park, J.H. Acute rosmarinic acid treatment enhances long-term potentiation, BDNF and GluR-2 protein expression, and cell survival rate against scopolamine challenge in rat organotypic hippocampal slice cultures. Biochem. Biophys. Res. Commun. 2016, 475, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.S.; Yaari, Y. Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J. Neurophysiol. 1997, 77, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Oby, E.; Batra, A.; Uva, L.; De Curtis, M.; Hernandez, N.; Van Boxel-Dezaire, A.; Najm, I.; Janigro, D. In vivo and in vitro effects of pilocarpine: Relevance to ictogenesis. Epilepsia 2007, 48, 1934–1946. [Google Scholar] [CrossRef]
- Groticke, I.; Hoffmann, K.; Loscher, W. Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Exp. Neurol. 2007, 207, 329–349. [Google Scholar] [CrossRef]
- Raghupathi, R.; Fernandez, S.C.; Murai, H.; Trusko, S.P.; Scott, R.W.; Nishioka, W.K.; McIntosh, T.K. BCL-2 overexpression attenuates cortical cell loss after traumatic brain injury in transgenic mice. J. Cereb. Blood Flow Metab. 1998, 18, 1259–1269. [Google Scholar] [CrossRef]
- Souza, T.L.; Grauncke, A.C.B.; Ribeiro, L.R.; Mello, F.K.; Oliveira, S.M.; Brant, F.; Machado, F.S.; Oliveira, M.S. Cerebral Malaria Causes Enduring Behavioral and Molecular Changes in Mice Brain without Causing Gross Histopathological Damage. Neuroscience 2018, 369, 66–75. [Google Scholar] [CrossRef]
- Wehr, N.B.; Levine, R.L. Quantitation of protein carbonylation by dot blot. Anal. Biochem. 2012, 423, 241–245. [Google Scholar] [CrossRef]
- Ghasemzadeh Rahbardar, M.; Hosseinzadeh, H. Therapeutic effects of rosemary (Rosmarinus officinalis L.) and its active constituents on nervous system disorders. Iran. J. Basic Med. Sci. 2020, 23, 1100–1112. [Google Scholar] [CrossRef]
- Naderali, E.; Nikbakht, F.; Ofogh, S.N.; Rasoolijazi, H. The role of rosemary extract in degeneration of hippocampal neurons induced by kainic acid in the rat: A behavioral and histochemical approach. J. Integr. Neurosci. 2018, 17, 19–25. [Google Scholar] [CrossRef]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends. Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef]
- Walker, M.C. Reactive oxygen species in status epilepticus. Epilepsia Open 2023, 00, 1–7. [Google Scholar] [CrossRef]
- Gorter, J.A.; van Vliet, E.A.; Aronica, E.; Breit, T.; Rauwerda, H.; Lopes da Silva, F.H.; Wadman, W.J. Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J. Neurosci. 2006, 26, 11083–11110. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Liu, K.; Wake, H.; Teshigawara, K.; Yoshino, T.; Takahashi, H.; Mori, S.; Nishibori, M. Therapeutic effects of anti-HMGB1 monoclonal antibody on pilocarpine-induced status epilepticus in mice. Sci Rep 2017, 7, 1179. [Google Scholar] [CrossRef] [PubMed]
- Vizuete, A.F.K.; Hansen, F.; Negri, E.; Leite, M.C.; de Oliveira, D.L.; Goncalves, C.A. Effects of dexamethasone on the Li-pilocarpine model of epilepsy: Protection against hippocampal inflammation and astrogliosis. J. Neuroinflamm. 2018, 15, 68. [Google Scholar] [CrossRef]
- Dyomina, A.V.; Zubareva, O.E.; Smolensky, I.V.; Vasilev, D.S.; Zakharova, M.V.; Kovalenko, A.A.; Schwarz, A.P.; Ischenko, A.M.; Zaitsev, A.V. Anakinra Reduces Epileptogenesis, Provides Neuroprotection, and Attenuates Behavioral Impairments in Rats in the Lithium-Pilocarpine Model of Epilepsy. Pharmaceuticals 2020, 13, 340. [Google Scholar] [CrossRef]
- Terrone, G.; Balosso, S.; Pauletti, A.; Ravizza, T.; Vezzani, A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology 2020, 167, 107742. [Google Scholar] [CrossRef]
- Dulla, C.G.; Janigro, D.; Jiruska, P.; Raimondo, J.V.; Ikeda, A.; Lin, C.K.; Goodkin, H.P.; Galanopoulou, A.S.; Bernard, C.; de Curtis, M. How do we use in vitro models to understand epileptiform and ictal activity? A report of the TASK1-WG4 group of the ILAE/AES Joint Translational Task Force. Epilepsia Open 2018, 3, 460–473. [Google Scholar] [CrossRef]
- Castillo, M.; Smith, J.K.; Kwock, L. Proton MR spectroscopy in patients with acute temporal lobe seizures. AJNR Am. J. Neuroradiol. 2001, 22, 152–157. [Google Scholar]
- Mosienko, V.; Teschemacher, A.G.; Kasparov, S. Is L-lactate a novel signaling molecule in the brain? J. Cereb. Blood Flow Metab. 2015, 35, 1069–1075. [Google Scholar] [CrossRef]
- Boling, W.W.; Lancaster, M.; Kraszpulski, M.; Palade, A.; Marano, G.; Puce, A. Fluorodeoxyglucose-positron emission tomographic imaging for the diagnosis of mesial temporal lobe epilepsy. Neurosurgery 2008, 63, 1130–1138; discussion 1138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, Y.; Hu, H.; Wang, J.; Liu, Z.; Gao, F. FDG-PET and NeuN-GFAP immunohistochemistry of hippocampus at different phases of the pilocarpine model of temporal lobe epilepsy. Int. J. Med. Sci. 2015, 12, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Tsytsarev, V.; Maslov, K.I.; Yao, J.; Parameswar, A.R.; Demchenko, A.V.; Wang, L.V. In vivo imaging of epileptic activity using 2-NBDG, a fluorescent deoxyglucose analog. J. Neurosci. Methods 2012, 203, 136–140. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuberger, B.; Mello, F.K.; Mallmann, M.P.; da Costa Sobral, K.G.; Fighera, M.R.; Royes, L.F.F.; Furian, A.F.; Sampaio, T.B.; Oliveira, M.S. Beneficial Effects of Rosmarinic Acid In Vitro and In Vivo Models of Epileptiform Activity Induced by Pilocarpine. Brain Sci. 2023, 13, 289. https://doi.org/10.3390/brainsci13020289
Neuberger B, Mello FK, Mallmann MP, da Costa Sobral KG, Fighera MR, Royes LFF, Furian AF, Sampaio TB, Oliveira MS. Beneficial Effects of Rosmarinic Acid In Vitro and In Vivo Models of Epileptiform Activity Induced by Pilocarpine. Brain Sciences. 2023; 13(2):289. https://doi.org/10.3390/brainsci13020289
Chicago/Turabian StyleNeuberger, Bruna, Fernanda Kulinski Mello, Michele Pereira Mallmann, Karine Gabriela da Costa Sobral, Michele Rechia Fighera, Luiz Fernando Freire Royes, Ana Flávia Furian, Tuane Bazanella Sampaio, and Mauro Schneider Oliveira. 2023. "Beneficial Effects of Rosmarinic Acid In Vitro and In Vivo Models of Epileptiform Activity Induced by Pilocarpine" Brain Sciences 13, no. 2: 289. https://doi.org/10.3390/brainsci13020289
APA StyleNeuberger, B., Mello, F. K., Mallmann, M. P., da Costa Sobral, K. G., Fighera, M. R., Royes, L. F. F., Furian, A. F., Sampaio, T. B., & Oliveira, M. S. (2023). Beneficial Effects of Rosmarinic Acid In Vitro and In Vivo Models of Epileptiform Activity Induced by Pilocarpine. Brain Sciences, 13(2), 289. https://doi.org/10.3390/brainsci13020289