
Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling

Abstract
:1. Introduction
1.1. Influence of Developmental Stage, Amount and Pattern of Alcohol Use on Neurobehavioral Outcome
1.2. Neurobehavioral Abnormalities in Children with FASD

{kind=link}
{kind=link}
{kind=link}
| Cognitive Function | FASD | References |
|---|---|---|
| General Intelligence | Lower IQ (~70) | [7,23,28] |
| Executive Function | Impaired executive functions such as planning, fluency and working memory | [14,29,30,31,32,33,34] |
| Learning and memory | ||
| Verbal | Impaired initial learning without affecting retention of information already learned due to implicit learning strategies. | [12,35,36,37,38,39,40] |
| Nonverbal | Impaired nonverbal learning and memory but impairment of retention of information is inconsistent. | [29,36,39,41,42,43,44] |
| Motor function | Deficits in motor abilities and visual-motor tasks. | [11,35,42,45,46,47,48,49,50,51] |
| Attention and hyperactivity | Deficits in attention and exhibit hyperactivity. | [10,48,52,53,54,55,56,57,58,59,60] |
1.3. Neurobehavioral Abnormalities in Animal Models of FASD
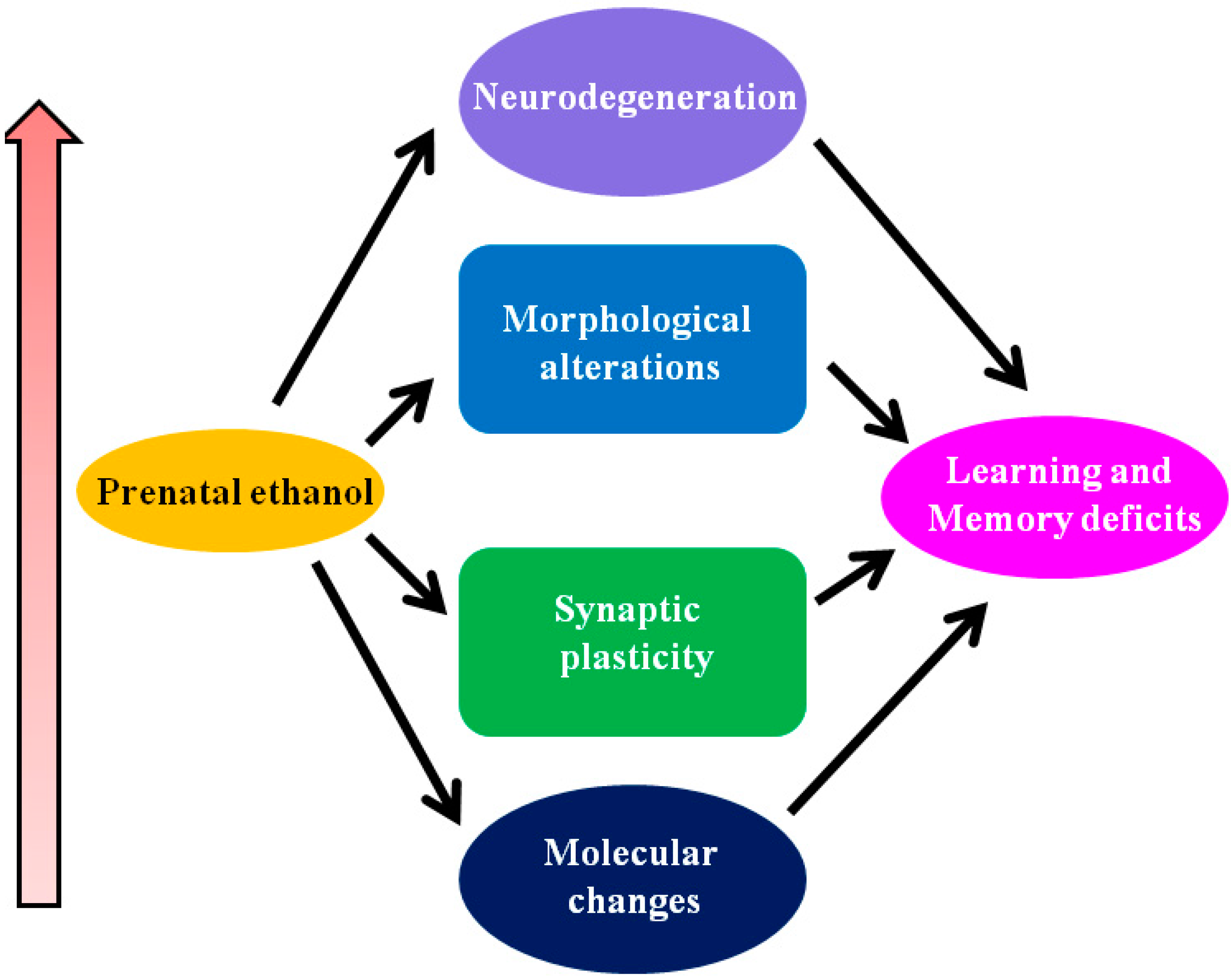
1.3.1. Potential Mechanisms Responsible for FASD
1.3.2. Role of the Endocannabinoid System during Brain Development and in Ethanol-Induced Brain Abnormalities
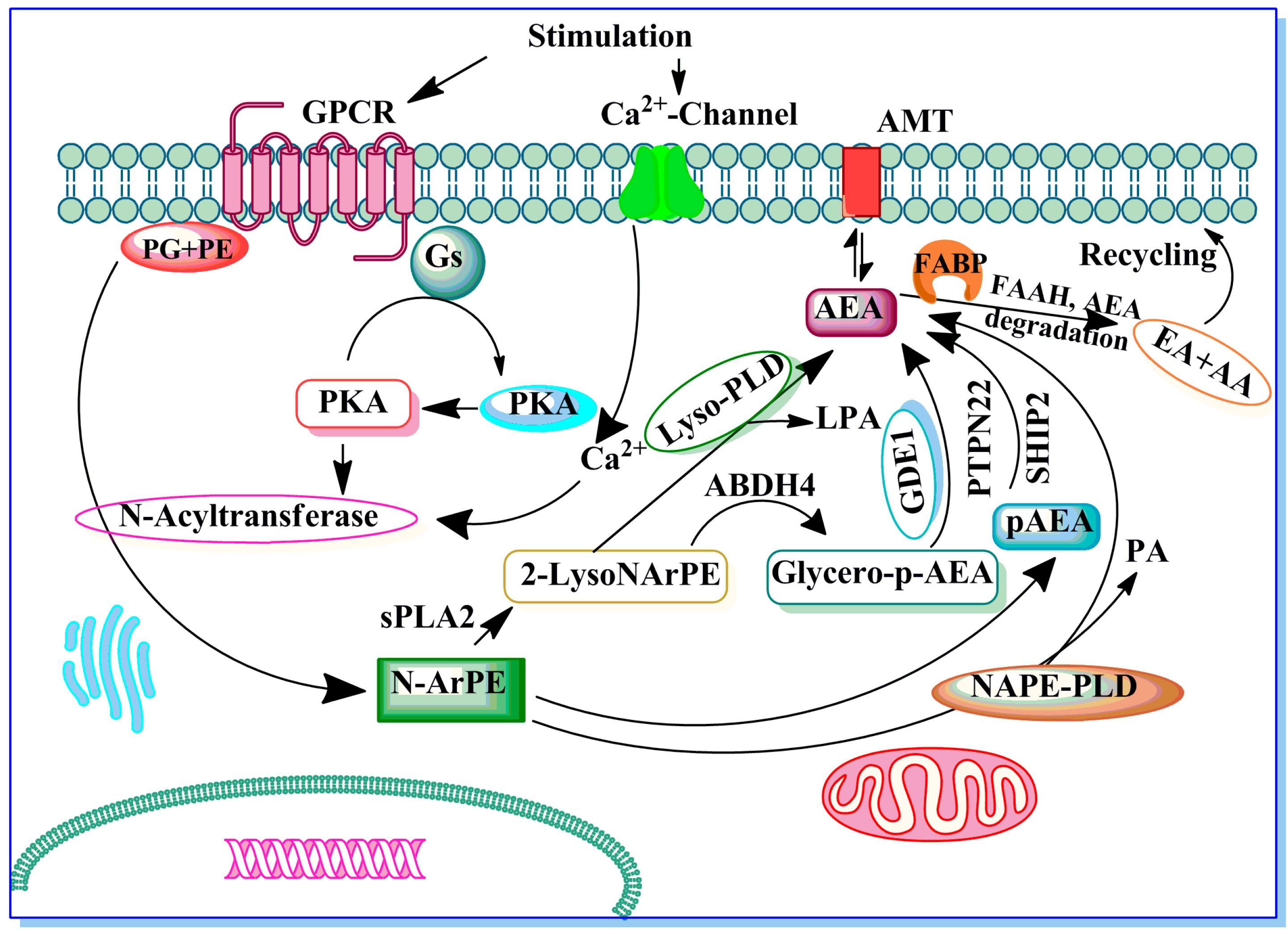
Potential Role of Endocannabinoids and Their Metabolic Enzymes

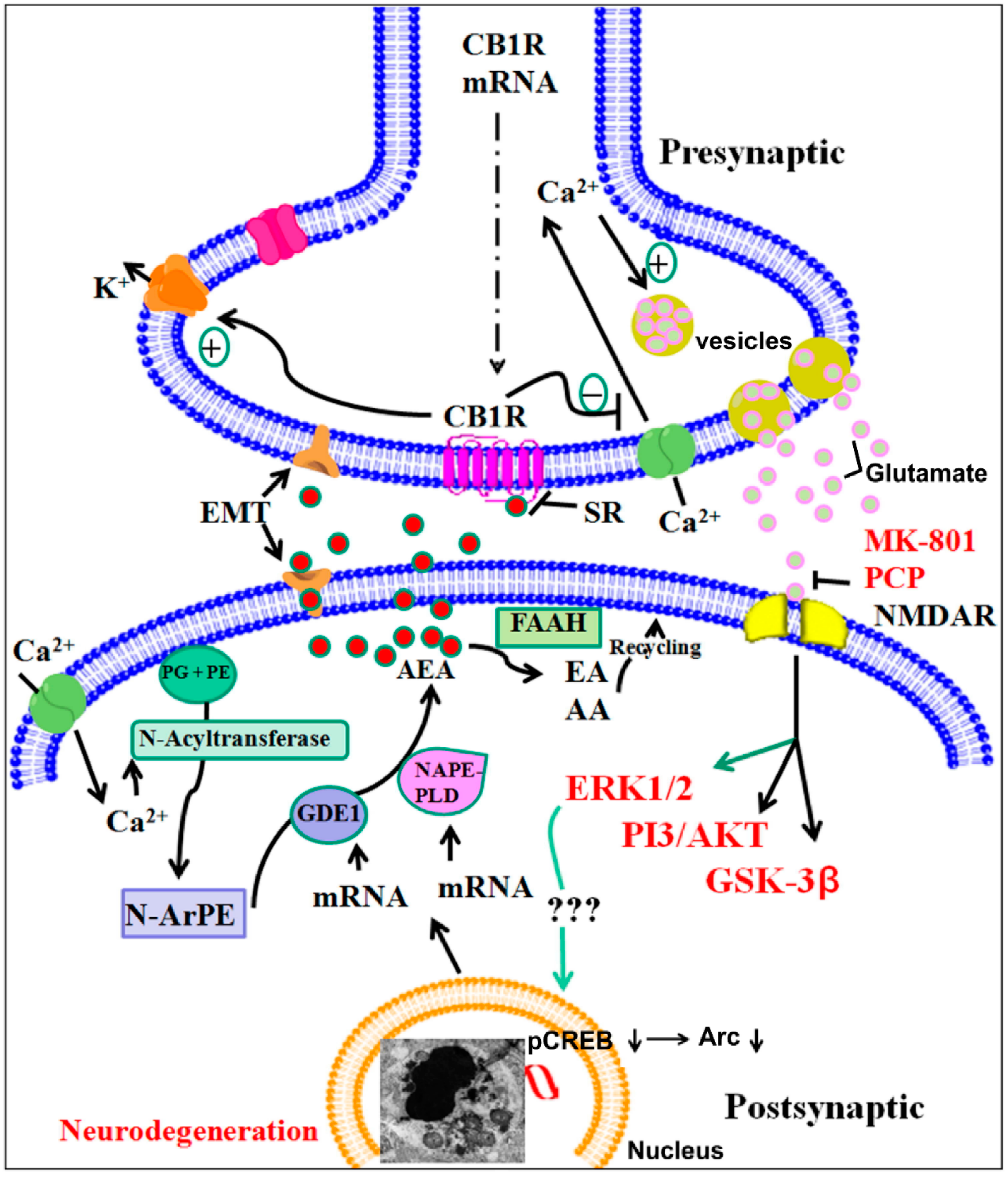
A Major Role of Cannabinoid Receptors and Their Signaling

| Cannabinoid Exposure | References | |
|---|---|---|
| Learning and memory | Learning deficits in the Morris water maze. | [305] |
| Impairment in the inhibitory avoidance test and impaired olfactory short-term memory in the social discrimination task. | [306] | |
| Poorer performance in homing behavior and impaired active avoidance performance. | [307] | |
| Disruption of memory retention in the passive avoidance task. | [308] | |
| Reduced novel object recognition. | [309,310] | |
| Impaired working memory on Y maze. | [311] | |
| Reduced social interaction. | [309,310] |
2. Conclusions

Acknowledgments
Conflict of Interest
Abbreviations
References
- Jones, K.L.; Smith, D.W.; Ulleland, C.N.; Streissguth, P. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet 1973, 1, 1267–1271. [Google Scholar] [CrossRef]
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 2, 999–1001. [Google Scholar] [CrossRef]
- Alati, R.; al Mamun, A.; Williams, G.M.; O’Callaghan, M.; Najman, J.M.; Bor, W. In utero alcohol exposure and prediction of alcohol disorders in early adulthood: A birth cohort study. Arch. Gen. Psychiatry 2006, 63, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Autti-Ramo, I.; Fagerlund, A.; Ervalahti, N.; Loimu, L.; Korkman, M.; Hoyme, H.E. Fetal alcohol spectrum disorders in Finland: Clinical delineation of 77 older children and adolescents. Am. J. Med. Genet. Part A 2006, 140, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ceccanti, M.; Alessandra Spagnolo, P.; Tarani, L.; Luisa Attilia, M.; Chessa, L.; Mancinelli, R.; Stegagno, M.; Francesco Sasso, G.; Romeo, M.; Jones, K.L.; et al. Clinical delineation of fetal alcohol spectrum disorders (FASD) in Italian children: Comparison and contrast with other racial/ethnic groups and implications for diagnosis and prevention. Neurosci. Biobehav. Rev. 2007, 31, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Spohr, H.L.; Willms, J.; Steinhausen, H.C. Fetal alcohol spectrum disorders in young adulthood. J. Pediatr. 2007, 150, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Aase, J.M.; Clarren, S.K.; Randels, S.P.; LaDue, R.A.; Smith, D.F. Fetal alcohol syndrome in adolescents and adults. JAMA 1991, 265, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Delaney-Black, V.; Nordstrom, B. Fetal alcohol spectrum disorder. JAMA 2003, 290, 2996–2999. [Google Scholar] [CrossRef] [PubMed]
- Burd, L.; Klug, M.G.; Martsolf, J.T.; Kerbeshian, J. Fetal alcohol syndrome: Neuropsychiatric phenomics. Neurotoxicol. Teratol. 2003, 25, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Crocker, N.; Nguyen, T.T. Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychol. Rev. 2011, 21, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Riley, E.P. A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol. Clin. Exp. Res. 1998, 22, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Riley, E.P.; Gramling, L.; Delis, D.C.; Jones, K.L. Neuropsychological comparison of alcohol-exposed children with or without physical features of fetal alcohol syndrome. Neuropsychology 1998, 12, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Delis, D.C.; Mattson, S.N. Normative data for 4-year-old children on the California Verbal Learning Test-Children’s Version. Clin. Neuropsychol. 1999, 13, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Goodman, A.M.; Caine, C.; Delis, D.C.; Riley, E.P. Executive functioning in children with heavy prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 1999, 23, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Sood, B.; Delaney-Black, V.; Covington, C.; Nordstrom-Klee, B.; Ager, J.; Templin, T.; Janisse, J.; Martier, S.; Sokol, R.J. Prenatal alcohol exposure and childhood behavior at age 6 to 7 years: I. dose-response effect. Pediatrics 2001, 108, E34. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Bookstein, F.L.; Sampson, P.D.; Barr, H.M. Neurobehavioral effects of prenatal alcohol: Part III. PLS analyses of neuropsychologic tests. Neurotoxicol. Teratol. 1989, 11, 493–507. [Google Scholar] [CrossRef]
- Bailey, B.N.; Delaney-Black, V.; Covington, C.Y.; Ager, J.; Janisse, J.; Hannigan, J.H.; Sokol, R.J. Prenatal exposure to binge drinking and cognitive and behavioral outcomes at age 7 years. Am. J. Obstet. Gynecol. 2004, 191, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; Goodlett, C.R.; West, J.R. Blood alcohol concentration and severity of microencephaly in neonatal rats depend on the pattern of alcohol administration. Alcohol 1988, 5, 209–214. [Google Scholar] [CrossRef]
- Guerri, C.; Bazinet, A.; Riley, E.P. Foetal Alcohol Spectrum Disorders and alterations in brain and behaviour. Alcohol Alcohol. 2009, 44, 108–114. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Gossage, J.P.; Kalberg, W.O.; Robinson, L.K.; Buckley, D.; Manning, M.; Hoyme, H.E. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev. Disabil. Res. Rev. 2009, 15, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Floyd, R.L.; Weber, M.K.; O’Connor, M.; Riley, E.P.; Johnson, K.A.; Cohen, D.E. Fetal Alcohol Syndrome: Guidelines for Referral and Diagnosis; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2004. [Google Scholar]
- Dalen, K.; Bruaroy, S.; Wentzel-Larsen, T.; Laegreid, L.M. Cognitive functioning in children prenatally exposed to alcohol and psychotropic drugs. Neuropediatrics 2009, 40, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Riley, E.P.; Gramling, L.; Delis, D.C.; Jones, K.L. Heavy prenatal alcohol exposure with or without physical features of fetal alcohol syndrome leads to IQ deficits. J. Pediatr. 1997, 131, 718–721. [Google Scholar] [CrossRef]
- Alati, R.; Clavarino, A.; Najman, J.M.; O’Callaghan, M.; Bor, W.; Mamun, A.A.; Williams, G.M. The developmental origin of adolescent alcohol use: Findings from the Mater University Study of Pregnancy and its outcomes. Drug Alcohol Depend. 2008, 98, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fried, P.A.; Watkinson, B. 12- and 24-month neurobehavioural follow-up of children prenatally exposed to marihuana, cigarettes and alcohol. Neurotoxicol. Teratol. 1988, 10, 305–313. [Google Scholar] [CrossRef]
- Streissguth, A.P.; Barr, H.M.; Sampson, P.D. Moderate prenatal alcohol exposure: Effects on child IQ and learning problems at age 7 1/2 years. Alcohol. Clin. Exp. Res. 1990, 14, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Willford, J.A.; Leech, S.L.; Day, N.L. Moderate prenatal alcohol exposure and cognitive status of children at age 10. Alcoholism 2006, 30, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Chasnoff, I.J.; Wells, A.M.; Telford, E.; Schmidt, C.; Messer, G. Neurodevelopmental functioning in children with FAS, pFAS, and ARND. J. Dev. Behav. Pediatr. 2010, 31, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Aragon, A.S.; Kalberg, W.O.; Buckley, D.; Barela-Scott, L.M.; Tabachnick, B.G.; May, P.A. Neuropsychological study of FASD in a sample of American Indian children: Processing simple versus complex information. Alcohol. Clin. Exp. Res. 2008, 32, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Mihic, A.M.; Nikkel, S.M.; Stade, B.C.; Rasmussen, C.; Munoz, D.P.; Reynolds, J.N. Executive function deficits in children with fetal alcohol spectrum disorders (FASD) measured using the Cambridge Neuropsychological Tests Automated Battery (CANTAB). J. Child Psychol. Psychiatry 2009, 50, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Kodituwakku, P.W.; Adnams, C.M.; Hay, A.; Kitching, A.E.; Burger, E.; Kalberg, W.O.; Viljoen, D.L.; May, P.A. Letter and category fluency in children with fetal alcohol syndrome from a community in South Africa. J. Stud. Alcohol. 2006, 67, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Kodituwakku, P.W.; Handmaker, N.S.; Cutler, S.K.; Weathersby, E.K.; Handmaker, S.D. Specific impairments in self-regulation in children exposed to alcohol prenatally. Alcohol. Clin. Exp. Res. 1995, 19, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, A.M.; Mattson, S.N.; Lang, A.R.; Delis, D.C.; Riley, E.P. Verbal and nonverbal fluency in children with heavy prenatal alcohol exposure. J. Stud. Alcohol. 2001, 62, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Vaurio, L.; Riley, E.P.; Mattson, S.N. Differences in executive functioning in children with heavy prenatal alcohol exposure or attention-deficit/hyperactivity disorder. J. Int. Neuropsychol. Soc. 2008, 14, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Riley, E.P.; Delis, D.C.; Stern, C.; Jones, K.L. Verbal learning and memory in children with fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1996, 20, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Roebuck, T.M. Acquisition and retention of verbal and nonverbal information in children with heavy prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2002, 26, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Roebuck-Spencer, T.M.; Mattson, S.N. Implicit strategy affects learning in children with heavy prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2004, 28, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Willford, J.A.; Richardson, G.A.; Leech, S.L.; Day, N.L. Verbal and visuospatial learning and memory function in children with moderate prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2004, 28, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Kaemingk, K.L.; Mulvaney, S.; Halverson, P.T. Learning following prenatal alcohol exposure: Performance on verbal and visual multitrial tasks. Arch. Clin. Neuropsychol. 2003, 18, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, K.A.; Sheard, E.D.; Nash, K.; Rovet, J. Effects of prenatal alcohol exposure on hippocampal volume, verbal learning, and verbal and spatial recall in late childhood. J. Int. Neuropsychol. Soc. 2008, 14, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.A.; Kodituwakku, P.; Sutherland, R.J.; Savage, D.D. Children with Fetal Alcohol Syndrome are impaired at place learning but not cued-navigation in a virtual Morris water task. Behav. Brain Res. 2003, 143, 85–94. [Google Scholar] [CrossRef]
- Kaemingk, K.L.; Halverson, P.T. Spatial memory following prenatal alcohol exposure: More than a material specific memory deficit. Child Neuropsychol. 2000, 6, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, C.; Horne, K.; Witol, A. Neurobehavioral functioning in children with fetal alcohol spectrum disorder. Child Neuropsychol. 2006, 12, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Uecker, A.; Nadel, L. Spatial but not object memory impairments in children with fetal alcohol syndrome. Am. J. Ment. Retard. 1998, 103, 12–18. [Google Scholar] [CrossRef]
- Aronson, M.; Hagberg, B. Neuropsychological disorders in children exposed to alcohol during pregnancy: A follow-up study of 24 children to alcoholic mothers in Goteborg, Sweden. Alcohol. Clin. Exp. Res. 1998, 22, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Chiodo, L.M.; Janisse, J.; Delaney-Black, V.; Sokol, R.J.; Hannigan, J.H. A metric of maternal prenatal risk drinking predicts neurobehavioral outcomes in preschool children. Alcohol. Clin. Exp. Res. 2009, 33, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Conry, J. Neuropsychological deficits in fetal alcohol syndrome and fetal alcohol effects. Alcohol. Clin. Exp. Res. 1990, 14, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Janzen, L.A.; Nanson, J.L.; Block, G.W. Neuropsychological evaluation of preschoolers with fetal alcohol syndrome. Neurotoxicol. Teratol. 1995, 17, 273–279. [Google Scholar] [CrossRef]
- Simmons, R.W.; Thomas, J.D.; Levy, S.S.; Riley, E.P. Motor response programming and movement time in children with heavy prenatal alcohol exposure. Alcohol 2010, 44, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.W.; Wass, T.; Thomas, J.D.; Riley, E.P. Fractionated simple and choice reaction time in children with prenatal exposure to alcohol. Alcohol. Clin. Exp. Res. 2002, 26, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Wass, T.S.; Simmons, R.W.; Thomas, J.D.; Riley, E.P. Timing accuracy and variability in children with prenatal exposure to alcohol. Alcohol. Clin. Exp. Res. 2002, 26, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Roesch, S.C.; Fagerlund, A.; Autti-Ramo, I.; Jones, K.L.; May, P.A.; Adnams, C.M.; Konovalova, V.; Riley, E.P. Collaborative Initiative on Fetal Alcohol Spectrum Disorders (CIFASD). Toward a neurobehavioral profile of fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2010, 34, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Aragon, A.S.; Coriale, G.; Fiorentino, D.; Kalberg, W.O.; Buckley, D.; Gossage, J.P.; Ceccanti, M.; Mitchell, E.R.; May, P.A. Neuropsychological characteristics of Italian children with fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2008, 32, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.T.; Coles, C.D.; Smith, I.E.; Platzman, K.A.; Silverstein, J.; Erickson, S.; Falek, A. Effects of prenatal alcohol exposure at school age. II. Attention and behavior. Neurotoxicol. Teratol. 1991, 13, 369–376. [Google Scholar] [CrossRef]
- Burden, M.J.; Jacobson, S.W.; Sokol, R.J.; Jacobson, J.L. Effects of prenatal alcohol exposure on attention and working memory at 7.5 years of age. Alcohol. Clin. Exp. Res. 2005, 29, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Burden, M.J.; Mitchell, D.B. Implicit memory development in school-aged children with attention deficit hyperactivity disorder (ADHD): Conceptual priming deficit? Dev. Neuropsychol. 2005, 28, 779–807. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.W.; Jacobson, J.L.; Sokol, R.J. Effects of fetal alcohol exposure on infant reaction time. Alcohol. Clin. Exp. Res. 1994, 18, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.W.; Jacobson, J.L.; Sokol, R.J.; Martier, S.S.; Ager, J.W. Prenatal alcohol exposure and infant information processing ability. Child Dev. 1993, 64, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.; Rovet, J.; Greenbaum, R.; Fantus, E.; Nulman, I.; Koren, G. Identifying the behavioural phenotype in Fetal Alcohol Spectrum Disorder: Sensitivity, specificity and screening potential. Arch. N.A. Ment. Health 2006, 9, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Sampson, P.D.; Olson, H.C.; Bookstein, F.L.; Barr, H.M.; Scott, M.; Feldman, J.; Mirsky, A.F. Maternal drinking during pregnancy: Attention and short-term memory in 14-year-old offspring—A longitudinal prospective study. Alcohol. Clin. Exp. Res. 1994, 18, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.K.; Lynch, M.E.; Platzman, K.A.; Smith, G.H.; Coles, C.D. Prenatal alcohol exposure and ability, academic achievement, and school functioning in adolescence: A longitudinal follow-up. J. Pediatr. Psychol. 2006, 31, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Coles, C.D.; Platzman, K.A.; Lynch, M.E.; Freides, D. Auditory and visual sustained attention in adolescents prenatally exposed to alcohol. Alcohol. Clin. Exp. Res. 2002, 26, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Calarco, K.E.; Lang, A.R. Focused and shifting attention in children with heavy prenatal alcohol exposure. Neuropsychology 2006, 20, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Fryer, S.L.; McGee, C.L.; Matt, G.E.; Riley, E.P.; Mattson, S.N. Evaluation of psychopathological conditions in children with heavy prenatal alcohol exposure. Pediatrics 2007, 119, e733–e741. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L. Frontal-subcortical circuits and human behavior. Arch. Neurol. 1993, 50, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Fryer, S.L.; Tapert, S.F.; Mattson, S.N.; Paulus, M.P.; Spadoni, A.D.; Riley, E.P. Prenatal alcohol exposure affects frontal-striatal BOLD response during inhibitory control. Alcohol. Clin. Exp. Res. 2007, 31, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Riley, E.P.; Sowell, E.R.; Jernigan, T.L.; Sobel, D.F.; Jones, K.L. A decrease in the size of the basal ganglia in children with fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1996, 20, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- McGee, C.L.; Fryer, S.L.; Bjorkquist, O.A.; Mattson, S.N.; Riley, E.P. Deficits in social problem solving in adolescents with prenatal exposure to alcohol. Am. J. Drug Alcohol. Abuse 2008, 34, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, A.M.; Mattson, S.N.; Riley, E.P. Moral maturity and delinquency after prenatal alcohol exposure. J. Stud. Alcohol. 2005, 66, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Timler, G.R.; Olswang, L.B. Variable structure/variable performance: Parent and teacher perspectives on a school-age child with FAS. J. Posit. Behav. Interv. 2001, 3, 48–56. [Google Scholar] [CrossRef]
- Timler, G.R.; Olswang, L.B.; Coggins, T.E. “Do I know what I need to do?” A social communication intervention for children with complex clinical profiles. Lang. Speech Hear. Serv. Sch. 2005, 36, 73–85. [Google Scholar] [CrossRef]
- Newmann, F.; Wehlage, G.; Lamborn, S. The significance and sources of student engagement. In Student Engagement and Achievement in American Secondary Schools; Newman, F., Ed.; Teachers College Press: New York, NY, USA, 1992; pp. 11–39. [Google Scholar]
- Floyd, R.L.; Weber, M.K.; Denny, C.; O’Connor, M.J. Prevention of fetal alcohol spectrum disorders. Dev. Disabil. Res. Rev. 2009, 15, 193–199. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J.; Paley, B. Psychiatric conditions associated with prenatal alcohol exposure. Dev. Disabil. Res. Rev. 2009, 15, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Paley, B.; O’Connor, M.J. Intervention for individuals with fetal alcohol spectrum disorders: Treatment approaches and case management. Dev. Disabil. Res. Rev. 2009, 15, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Abdollah, S.; Catlin, M.C.; Brien, J.F. Ethanol neuro-behavioural teratogenesis in the guinea pig: Behavioural dysfunction and hippocampal morphologic change. Can. J. Physiol. Pharmacol. 1993, 71, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Butters, N.S.; Gibson, M.A.; Reynolds, J.N.; Brien, J.F. Effects of chronic prenatal ethanol exposure on hippocampal glutamate release in the postnatal guinea pig. Alcohol 2000, 21, 1–9. [Google Scholar] [CrossRef]
- Christie, B.R.; Swann, S.E.; Fox, C.J.; Froc, D.; Lieblich, S.E.; Redila, V.; Webber, A. Voluntary exercise rescues deficits in spatial memory and long-term potentiation in prenatal ethanol-exposed male rats. Eur. J. Neurosci. 2005, 21, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, C.D.; Streissguth, A.P.; Riley, E.P. Prenatal alcohol exposure: Comparability of effects in humans and animal models. Neurotoxicol. Teratol. 1990, 12, 231–237. [Google Scholar] [CrossRef]
- Iqbal, U.; Rikhy, S.; Dringenberg, H.C.; Brien, J.F.; Reynolds, J.N. Spatial learning deficits induced by chronic prenatal ethanol exposure can be overcome by non-spatial pre-training. Neurotoxicol. Teratol. 2006, 28, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.B.; Goodlett, C.R. Selective and enduring deficits in spatial learning after limited neonatal binge alcohol exposure in male rats. Alcohol. Clin. Exp. Res. 2002, 26, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Reyes, E.; Wolfe, J.; Savage, D.D. The effects of prenatal alcohol exposure on radial arm maze performance in adult rats. Physiol. Behav. 1989, 46, 45–48. [Google Scholar] [CrossRef]
- Richardson, D.P.; Byrnes, M.L.; Brien, J.F.; Reynolds, J.N.; Dringenberg, H.C. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur. J. Neurosci. 2002, 16, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.D.; Becher, M.; de la Torre, A.J.; Sutherland, R.J. Dose-dependent effects of prenatal ethanol exposure on synaptic plasticity and learning in mature offspring. Alcohol. Clin. Exp. Res. 2002, 26, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.J.; McDonald, R.J.; Savage, D.D. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus 1997, 7, 232–238. [Google Scholar] [CrossRef]
- Tan, S.E.; Berman, R.F.; Abel, E.L.; Zajac, C.S. Prenatal alcohol exposure alters hippocampal slice electrophysiology. Alcohol 1990, 7, 507–511. [Google Scholar] [CrossRef]
- Cebral, E.; Lasserre, A.; Rettori, V.; de Gimeno, M.A. Impaired mouse fertilization by low chronic alcohol treatment. Alcohol Alcohol. 1997, 32, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Cebral, E.; Lasserre, A.; Rettori, V.; de Gimeno, M.A. Alterations in preimplantation in vivo development after preconceptional chronic moderate alcohol consumption in female mice. Alcohol Alcohol. 2000, 35, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Coll, T.A.; Tito, L.P.; Sobarzo, C.M.; Cebral, E. Embryo developmental disruption during organogenesis produced by CF-1 murine periconceptional alcohol consumption. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2011, 92, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, R.J.; Hammond, P.; O’Leary-Moore, S.K.; Ament, J.J.; Pecevich, S.J.; Jiang, Y.; Budin, F.; Parnell, S.E.; Suttie, M.; Godin, E.A.; et al. Ethanol-induced face-brain dysmorphology patterns are correlative and exposure-stage dependent. PLOS ONE 2012, 7, e43067. [Google Scholar] [CrossRef] [PubMed]
- Sulik, K.K. Craniofacial defects from genetic and teratogen-induced deficiencies in presomite embryos. Birth Defects Orig. Artic. Ser. 1984, 20, 79–98. [Google Scholar] [PubMed]
- Sulik, K.K.; Johnston, M.C. Sequence of developmental alterations following acute ethanol exposure in mice: Craniofacial features of the fetal alcohol syndrome. Am. J. Anat. 1983, 166, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Sulik, K.K.; Johnston, M.C.; Webb, M.A. Fetal alcohol syndrome: Embryogenesis in a mouse model. Science 1981, 214, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, M.M.; Smith, S.M. Stage-dependent effects of ethanol on cranial neural crest cell development: Partial basis for the phenotypic variations observed in fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1995, 19, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Guerri, C.; Pascual, M.; Renau-Piqueras, J. Glia and fetal alcohol syndrome. Neurotoxicology 2001, 22, 593–599. [Google Scholar] [CrossRef]
- Rubert, G.; Minana, R.; Pascual, M.; Guerri, C. Ethanol exposure during embryogenesis decreases the radial glial progenitorpool and affects the generation of neurons and astrocytes. J. Neurosci. Res. 2006, 84, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Fred, B.L.; Sampson, P.D.; Connor, P.D.; Streissguth, A.P. Midline Corpus Callosum is a Neuroanatomical Focus of Fetal Alcohol Damage. New Anat. Rec. 2002, 269, 162–174. [Google Scholar]
- Guerri, C. Neuroanatomical and neurophysiological mechanisms involved in central nervous system dysfunctions induced by prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 1998, 22, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Mazer, C.; Muneyyirci, J.; Taheny, K.; Raio, N.; Borella, A.; Whitaker-Azmitia, P. Serotonin depletion during synaptogenesis leads to decreased synaptic density and learning deficits in the adult rat: A possible model of neurodevelopmental disorders with cognitive deficits. Brain Res. 1997, 760, 68–73. [Google Scholar] [CrossRef]
- Thomas, J.D.; Goodlett, C.R.; West, J.R. Alcohol-induced Purkinje cell loss depends on developmental timing of alcohol exposure and correlates with motor performance. Brain Res. Dev. Brain Res. 1998, 105, 159–166. [Google Scholar] [CrossRef]
- Hamre, K.M.; West, J.R. The effects of the timing of ethanol exposure during the brain growth spurt on the number of cerebellar Purkinje and granule cell nuclear profiles. Alcohol. Clin. Exp. Res. 1993, 17, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; West, J.R. Alcohol-induced neuronal loss in developing rats: Increased brain damage with binge exposure. Alcohol. Clin. Exp. Res. 1990, 14, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; West, J.R. Permanent neuronal deficits in rats exposed to alcohol during the brain growth spurt. Teratology 1991, 44, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Livy, D.J.; Miller, E.K.; Maier, S.E.; West, J.R. Fetal alcohol exposure and temporal vulnerability: Effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol. Teratol. 2003, 25, 447–458. [Google Scholar] [CrossRef]
- Tran, T.D.; Kelly, S.J. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicol. Teratol. 2003, 25, 519–528. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Peterson, S.D.; Lundahl, K.R.; Pearlman, A.D. Binge-like alcohol exposure of neonatal rats via intragastric intubation induces both Purkinje cell loss and cortical astrogliosis. Alcohol. Clin. Exp. Res. 1997, 21, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Kitabayashi, R.; Funatsu, M.; Izumi, M.; Yuede, C.; Hartman, R.E.; Wozniak, D.F.; Zorumski, C.F. A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-d-aspartate receptor function and ethanol sensitivity. Neuroscience 2005, 136, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Noel, M.; Norris, E.H.; Strickland, S. Tissue plasminogen activator is required for the development of fetal alcohol syndrome in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 5069–5074. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Basavarajappa, B.S. Pre-administration of G9a/GLP inhibitor during synaptogenesis prevents postnatal ethanol-induced LTP deficits and neurobehavioral abnormalities in adult mice. Exp. Neurol. 2014, 261, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagaraja, N.N.; Umapathy, N.S.; Pace, B.S.; Basavarajappa, B.S. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int. J. Neuropsychopharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Anandamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic and memory deficits. J. Neuosci. 2013, 33, 6350–6366. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.A.; Peterson, J.; Basavaraj, B.S.; Saito, M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcohol. Clin. Exp. Res. 2011, 35, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. Effect of pre- or postnatal exposure to ethanol on the total number of neurons in the principal sensory nucleus of the trigeminal nerve: Cell proliferation and neuronal death. Alcohol. Clin. Exp. Res. 1995, 19, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Noctor, S.C.; Flint, A.C.; Weissman, T.A.; Dammerman, R.S.; Kriegstein, A.R. Neurons derived from radial glial cells establish radial units in neocortex. Nature 2001, 409, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.; Pitarch, J.; Renau-Piqueras, J.; Guerri, C. Ethanol exposure affects glial fibrillary acidic protein gene expression and transcription during rat brain development. J. Neurochem. 1997, 69, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.; Sancho-Tello, M.; Minana, R.; Climent, E.; Renau-Piqueras, J.; Guerri, C. Glial fibrillary acidic protein expression in rat brain and in radial glia culture is delayed by prenatal ethanol exposure. J. Neurochem. 1996, 67, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, M.M.; Tessmer, L.L.; Smith, S.M. Ethanol-induced neural crest apoptosis is coincident with their endogenous death, but is mechanistically distinct. Alcohol. Clin. Exp. Res. 1998, 22, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Liesi, P. Ethanol-exposed central neurons fail to migrate and undergo apoptosis. J. Neurosci. Res. 1997, 48, 439–448. [Google Scholar] [CrossRef]
- Camarillo, C.; Miranda, R.C. Ethanol exposure during neurogenesis induces persistent effects on neural maturation: Evidence from an ex vivo model of fetal cerebral cortical neuroepithelial progenitor maturation. Gene Exp. 2008, 14, 159–171. [Google Scholar]
- Prock, T.L.; Miranda, R.C. Embryonic cerebral cortical progenitors are resistant to apoptosis, but increase expression of suicide receptor DISC-complex genes and suppress autophagy following ethanol exposure. Alcohol. Clin. Exp. Res. 2007, 31, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Hörster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Guerri, C.; Climent, E.; Pascual, M. Ethanol exposure emhances apoptosis during brain development and affects brain-derived neurotrophic factor and its TrkB receptors. Alcohol Alcohol. 2001, 36, 437. [Google Scholar]
- Sadrian, B.; Subbanna, S.; Wilson, D.A.; Basavarajappa, B.S.; Saito, M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience 2012, 206, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Shivakumar, M.; Umapathy, N.S.; Saito, M.; Mohan, P.S.; Kumar, A.; Nixon, R.A.; Verin, A.D.; Psychoyos, D.; Basavarajappa, B.S. G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol. Dis. 2013, 54, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Horn, K.H.; Zhou, F.C. Alcohol teratogenesis: Mechanisms of damage and strategies for intervention. Exp. Biol. Med. 2005, 230, 394–406. [Google Scholar]
- Guerri, C. Mechanisms involved in central nervous system dysfunctions induced by prenatal ethanol exposure. Neurotox. Res. 2002, 4, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Psychoyos, D.; Shan, X.; Basavarajappa, B.S. Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol (MAGL) does not cause neurodegeneration in neonatal mice. J. Neurochem. 2015, 134, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Deltour, L.; Ang, H.L.; Duester, G. Ethanol inhibition of retinoic acid synthesis as a potential mechanism for fetal alcohol syndrome. FASEB J. 1996, 10, 1050–1057. [Google Scholar] [PubMed]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wasney, G.A.; Dong, A.; Barsyte, D.; Kozieradzki, I.; Senisterra, G.; Chau, I.; et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J. Med. Chem. 2009, 52, 7950–7953. [Google Scholar] [CrossRef] [PubMed]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B.S. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A and DNA methylation. J. Neurochem. 2015, 132, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Rifas, L.; Towler, D.A.; Avioli, L.V. Gestational exposure to ethanol suppresses msx2 expression in developing mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 7549–7554. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Umapathy, N.S.; Psychoyos, D.; Basavarajappa, B.S. Ethanol induced acetylation of histone at G9a Exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 2014, 258C, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.C. MicroRNAs and fetal brain development: Implications for ethanol teratology during the second trimester period of neurogenesis. Front. Genet. 2012, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Sathyan, P.; Golden, H.B.; Miranda, R.C. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: Evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 2007, 27, 8546–8557. [Google Scholar] [CrossRef] [PubMed]
- Miller, M. Early exposure to ethanol affects the proliferation of neuronal precursors. In Brain Development Normal Processes and Effects of Alcohol and Nicotine; Miller, M., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 182–198. [Google Scholar]
- Mooney, S.M.; Miller, M.W.; Henderson, G.I. Intracellular events in ethanol-induced neuronal death. In Brain Development Normal Processes and Effects of Alcohol and Nicotine; Miller, M., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 267–278. [Google Scholar]
- Siegenthaler, J.A.; Miller, M.W. Mechanisms of ethanol-induced alterations in neuronal migration. In Brain Development Normal Processes and Effects of Alcohol and Nicotine; Miller, M.W., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 216–229. [Google Scholar]
- Cui, S.J.; Tewari, M.; Schneider, T.; Rubin, R. Ethanol promotes cell death by inhibition of the insulin-like growth factor I receptor. Alcohol. Clin. Exp. Res. 1997, 21, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Minana, R.; Climent, E.; Barettino, D.; Segui, J.M.; Renau-Piqueras, J.; Guerri, C. Alcohol exposure alters the expression pattern of neural cell adhesion molecules during brain development. J. Neurochem. 2000, 75, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Wilkemeyer, M.F.; Mittal, B.; Perides, G.; Charness, M.E. Alcohol inhibits cell-cell adhesion mediated by human L1. J. Cell Biol. 1996, 133, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Wilkemeyer, M.F.; Menkari, C.E.; Charness, M.E. Novel antagonists of alcohol inhibition of l1-mediated cell adhesion: Multiple mechanisms of action. Mol. Pharmacol. 2002, 62, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Wilkemeyer, M.F.; Sebastian, A.B.; Smith, S.A.; Charness, M.E. Antagonists of alcohol inhibition of cell adhesion. Proc. Natl. Acad. Sci. USA 2000, 97, 3690–3695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.X.; Rubin, R.; Rooney, T.A. Ethanol induces apoptosis in cerebellar granule neurons by inhibiting insulin-like growth factor 1 signaling. J. Neurochem. 1998, 71, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Guerri, C.; Montoliu, C.; Renau-Piqueras, J. Involvement of free radical mechanism in the toxic effects of alcohol: Implications for fetal alcohol syndrome. Adv. Exp. Med. Biol. 1994, 366, 291–305. [Google Scholar] [PubMed]
- Olney, J.W.; Ishimaru, M.J.; Bittigau, P.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing brain. Apoptosis 2000, 5, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Muglia, L.J.; Jermakowicz, W.J.; D’Sa, C.; Roth, K.A. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol. Dis. 2002, 9, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Wozniak, D.F.; Farber, N.B.; Jevtovic-Todorovic, V.; Bittigau, P.; Ikonomidou, C. The enigma of fetal alcohol neurotoxicity. Ann. Med. 2002, 34, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Young, C.; Wozniak, D.F.; Jevtovic-Todorovic, V.; Ikonomidou, C. Do pediatric drugs cause developing neurons to commit suicide? Trends Pharmacol. Sci. 2004, 25, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Chakraborty, G.; Hegde, M.; Ohsie, J.; Paik, S.M.; Vadasz, C. Involvement of ceramide in ethanol-induced apoptotic neurodegeneration in the neonatal mouse brain. J. Neurochem. 2010, 115, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Young, C.; Roth, K.A.; Klocke, B.J.; West, T.; Holtzman, D.M.; Labruyere, J.; Qin, Y.Q.; Dikranian, K.; Olney, J.W. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol. Dis. 2005, 20, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Guerri, C.; Rubert, G.; Pascual, M. Glial targets of developmental exposure to ethanol. In Brain Development; Miller, M.W., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 295–312. [Google Scholar]
- Mechoulam, R.; Parker, L.A. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, A.C.; Regehr, W.G. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 2001, 29, 717–727. [Google Scholar] [CrossRef]
- Ohno-Shosaku, T.; Maejima, T.; Kano, M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminal. Neuron 2001, 29, 729–738. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001, 410, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Alger, B.E. Retrograde signaling in the regulation of synaptic transmission: Focus on endocannabinoids. Prog. Neurobiol. 2002, 68, 247–286. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Arancio, O. Synaptic plasticity: Emerging role for endocannabinoid system. In Synaptic Plasticity New Research; Kaiser, T.F., Peters, F.J., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2008; pp. 77–112. [Google Scholar]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Cadas, H.; di Tomaso, E.; Piomelli, D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J. Neurosci. 1997, 17, 1226–1242. [Google Scholar] [PubMed]
- Mechoulam, R.; Fride, E.; di Marzo, V. Endocannabinoids. Eur. J. Pharmacol. 1998, 359, 1–18. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Hungund, B.L. Chronic ethanol increases the cannabinoid receptor agonist, anandamide and its precursor N-arachidonyl phosphatidyl ethanolamine in SK-N-SH Cells. J. Neurochem. 1999, 72, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S.; Saito, M.; Cooper, T.B.; Hungund, B.L. Stimulation of cannabinoid receptor agonist 2-arachidonylglycerol by chronic ethanol and its modulation by specific neuromodulators in cerebellar granule neurons. Biochem. Biophys. Acta 2000, 1535, 78–86. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Saito, M.; Cooper, T.B.; Hungund, B.L. Chronic ethanol inhibits the anandamide transport and increases extracellular anandamide levels in cerebellar granule neurons. Eur. J. Pharmacol. 2003, 466, 73–83. [Google Scholar] [CrossRef]
- Giuffrida, A.; Parsons, L.H.; Kerr, T.M.; Rodriguez de Fonseca, F.; Navarro, M.; Piomelli, D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 1999, 2, 358–363. [Google Scholar] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Hanus, L.; Abu-Lafi, S.; Fride, E.; Breuer, A.; Vogel, Z.; Shalev, D.E.; Kustanovich, I.; Mechoulam, R. 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. 2001, 98, 3662–3665. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.C.; Sauer, J.M.; Knierman, M.D.; Becker, G.W.; Berna, M.J.; Bao, J.; Nomikos, G.G.; Carter, P.; Bymaster, F.P.; Leese, A.B.; et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J. Pharmacol. Exp. Ther. 2002, 301, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Bisogno, T.; Trevisani, M.; Al-Hayani, A.; de Petrocellis, L.; Fezza, F.; Tognetto, M.; Petros, T.J.; Krey, J.F.; Chu, C.J.; et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8400–8405. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. Neuropharmacology of the endocannabinoid signaling system-Molecular mechanisms, biological actions and synaptic plasticity. Curr. Neuropharmacol. 2007, 5, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Morishita, J.; Tsuboi, K.; Tonai, T.; Ueda, N. Molecular characterization of a phospholipase D generating anandamide and its congeners. J. Biol. Chem. 2004, 279, 5298–5305. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Kurahashi, Y.; Yamamoto, S.; Tokunaga, T. Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J. Biol. Chem. 1995, 270, 23823–23827. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.M.; Cravatt, B.F. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 2008, 283, 9341–9349. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Harvey-White, J.; Huang, B.X.; Kim, H.Y.; Luquet, S.; Palmiter, R.D.; Krystal, G.; Rai, R.; Mahadevan, A.; et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 2008, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [CrossRef] [PubMed]
- Morishita, J.; Okamoto, Y.; Tsuboi, K.; Ueno, M.; Sakamoto, H.; Maekawa, N.; Ueda, N. Regional distribution and age-dependent expression of N-acylphosphatidylethanolamine-hydrolyzing phospholipase D in rat brain. J. Neurochem. 2005, 94, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Nyilas, R.; Dudok, B.; Urban, G.M.; Mackie, K.; Watanabe, M.; Cravatt, B.F.; Freund, T.F.; Katona, I. Enzymatic machinery for endocannabinoid biosynthesis associated with calcium stores in glutamatergic axon terminals. J. Neurosci. 2008, 28, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Okamoto, Y.; Ikematsu, N.; Inoue, M.; Shimizu, Y.; Uyama, T. Enzymatic formation of N-acylethanolamines from N-acylethanolamine plasmalogen through N-acylphosphatidylethanolamine-hydrolyzing phospholipase D-dependent and -independent pathways. Biochim. Biophys. Acta 2011, 1811, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.; Saghatelian, A.; Simon, G.M.; Cravatt, B.F. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry (Mosc.) 2006, 45, 4720–4726. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.M.; Cravatt, B.F. Characterization of mice lacking candidate N-acyl ethanolamine biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo. Mol. Biosyst. 2010, 6, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; Piomelli, D. Carrier-mediated transport and enzymatic hydrolysis of the endogenous cannabinoid 2-arachidonylglycerol. Neuroreport 2000, 11, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; MacCarrone, M.; de Petrocellis, L.; Jarrahian, A.; Finazzi-Agro, A.; Hillard, C.; di Marzo, V. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur. J. Biochem. 2001, 268, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Hillard, C.J.; Edgemond, W.S.; Jarrahian, A.; Campbell, W.B. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997, 69, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; Stella, N.; Calignano, A.; Lin, S.Y.; Makriyannis, A.; Piomelli, D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 1997, 277, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Hillard, C.J.; Jarrahian, A. The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem. Phys. Lipids 2000, 108, 123–134. [Google Scholar] [CrossRef]
- Maccarrone, M.; van der Stelt, M.; Rossi, A.; Veldink, G.A.; Vliegenthart, J.F.; Agro, A.F. Anandamide hydrolysis by human cells in culture and brain. J. Biol. Chem. 1998, 273, 32332–32339. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, A.; Beltramo, M.; Piomelli, D. Mechanisms of endocannabinoid inactivation: Biochemistry and pharmacology. J. Pharmacol. Exp. Ther. 2001, 298, 7–14. [Google Scholar] [PubMed]
- Deutsch, D.G.; Glaser, S.T.; Howell, J.M.; Kunz, J.S.; Puffenbarger, R.A.; Hillard, C.J.; Abumrad, N. The cellular uptake of anandamide is coupled to its breakdown by fatty-acid amide hydrolase. J. Biol. Chem. 2001, 276, 6967–6973. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.T.; Abumrad, N.A.; Fatade, F.; Kaczocha, M.; Studholme, K.M.; Deutsch, D.G. Evidence against the presence of an anandamide transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 4269–4274. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Johnson, D.S.; Mileni, M.; Beidler, D.; Long, J.Z.; McKinney, M.K.; Weerapana, E.; Sadagopan, N.; Liimatta, M.; Smith, S.E.; et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009, 16, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valino, F.; Duranti, A.; Tontini, A.; Giustino, A.; Tattoli, M.; Palmery, M.; Cuomo, V.; et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.T.; Ralph, B.P.; Kaczocha, M.; Sun, J.; Balius, T.E.; Rizzo, R.C.; Haj-Dahmane, S.; Ojima, I.; Deutsch, D.G. Targeting fatty acid binding protein (FABP) anandamide transporters—A novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS ONE 2012, 7, e50968. [Google Scholar] [CrossRef] [PubMed]
- Kaczocha, M.; Glaser, S.T.; Deutsch, D.G. Identification of intracellular carriers for the endocannabinoid anandamide. Proc. Natl. Acad. Sci. USA 2009, 106, 6375–6380. [Google Scholar] [CrossRef] [PubMed]
- Kaczocha, M.; Rebecchi, M.J.; Ralph, B.P.; Teng, Y.H.; Berger, W.T.; Galbavy, W.; Elmes, M.W.; Glaser, S.T.; Wang, L.; Rizzo, R.C.; et al. Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS ONE 2014, 9, e94200. [Google Scholar] [CrossRef] [PubMed]
- Kaczocha, M.; Vivieca, S.; Sun, J.; Glaser, S.T.; Deutsch, D.G. Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J. Biol. Chem. 2012, 287, 3415–3424. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S.H.; Rossetti, R.G.; Yagen, B.; Zurier, R.B. Oxidative metabolism of anandamide. Prostaglandins Other Lipid Mediat. 2000, 61, 29–41. [Google Scholar] [CrossRef]
- Kozak, K.R.; Crews, B.C.; Morrow, J.D.; Wang, L.H.; Ma, Y.H.; Weinander, R.; Jakobsson, P.J.; Marnett, L.J. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem. 2002, 277, 44877–44885. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A.; Craib, S.J.; Stevenson, L.A.; Pertwee, R.G.; Henderson, A.; Toole, J.; Ellington, H.C. Pharmacological characterization of the anandamide cyclooxygenase metabolite: Prostaglandin E2 ethanolamide. J. Pharmacol. Exp. Ther. 2002, 301, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Morozov, Y.M.; Ben-Ari, Y.; Freund, T.F. The spatial and temporal pattern of fatty acid amide hydrolase expression in rat hippocampus during postnatal development. Eur. J. Neurosci. 2004, 20, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Yoshinaga, N.; Kondo, S.; Waku, K.; Ishima, Y. Generation of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand, in picrotoxinin-administered rat brain. Biochem. Biophys. Res. Commun. 2000, 271, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. Critical Enzymes Involved in Endocannabinoid Metabolism. Protein Peptide Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.Y.; Strassle, B.W.; Lu, P.; et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Day, T.A.; Rakhshan, F.; Deutsch, D.G.; Barker, E.L. Role of fatty acid amide hydrolase in the transport of the endogenous cannabinoid anandamide. Mol. Pharmacol. 2001, 59, 1369–1375. [Google Scholar] [PubMed]
- Konrad, R.J.; Major, C.D.; Wolf, B.A. Diacylglycerol hydrolysis to arachidonic acid is necessary for insulin secretion from isolated pancreatic islets: Sequential actions of diacylglycerol and monoacylglycerol lipases. Biochemistry (Mosc.) 1994, 33, 13284–13294. [Google Scholar] [CrossRef]
- Harkany, T.; Guzman, M.; Galve-Roperh, I.; Berghuis, P.; Devi, L.A.; Mackie, K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol. Sci. 2007, 28, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.; di Marzo, V. Endocannabinoid synthesis and degradation, and their regulation in the framework of energy balance. J. Endocrinol. Investig. 2006, 29, 15–26. [Google Scholar]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Van der Stelt, M.; Fox, S.H.; Hill, M.; Crossman, A.R.; Petrosino, S.; di Marzo, V.; Brotchie, J.M. A role for endocannabinoids in the generation of parkinsonism and levodopa-induced dyskinesia in MPTP-lesioned non-human primate models of Parkinson’s disease. FASEB J. 2005, 19, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Berrendero, F.; Sepe, N.; Ramos, J.A.; di Marzo, V.; Fernandez-Ruiz, J.J. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse 1999, 33, 181–191. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Berrendero, F.; Hernandez, M.L.; Ramos, J.A. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef]
- Paria, B.C.; Song, H.; Wang, X.; Schmid, P.C.; Krebsbach, R.J.; Schmid, H.H.; Bonner, T.I.; Zimmer, A.; Dey, S.K. Dysregulated cannabinoid signaling disrupts uterine receptivity for embryo implantation. J. Biol. Chem. 2001, 276, 20523–20528. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M. Cannabinoids: Potential anticancer agents. Nat. Rev. Cancer 2003, 3, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Sanchez, C.; Galve-Roperh, I. Cannabinoids and cell fate. Pharmacol. Ther. 2002, 95, 175–184. [Google Scholar] [CrossRef]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urban, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabas, K.; Ballester Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc. Natl. Acad. Sci. USA 2008, 105, 8760–8765. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Dobszay, M.B.; Wang, X.; Spano, S.; Ledda, F.; Sousa, K.M.; Schulte, G.; Ernfors, P.; Mackie, K.; Paratcha, G.; et al. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 19115–19120. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Dobszay, M.B.; Ibanez, R.M.; Ernfors, P.; Harkany, T. Turning the heterogeneous into homogeneous: Studies on selectively isolated GABAergic interneuron subsets. Int. J. Dev. Neurosci. 2004, 22, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Thayer, S.A. Cannabinoids inhibit the formation of new synapses between hippocampal neurons in culture. J. Neurosci. 2001, 21, RC146. [Google Scholar] [PubMed]
- Galve-Roperh, I.; Aguado, T.; Rueda, D.; Velasco, G.; Guzman, M. Endocannabinoids: A new family of lipid mediators involved in the regulation of neural cell development. Curr. Pharm. Des. 2006, 12, 2319–2325. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.D.; He, J.C.; Eungdamrong, N.J.; Gomes, I.; Ali, W.; Nguyen, T.; Bivona, T.G.; Philips, M.R.; Devi, L.A.; Iyengar, R. Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through Gαo/I-triggered proteasomal degradation of Rap1GAPII. J. Biol. Chem. 2005, 280, 11413–11421. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.J.; Walsh, F.S.; Doherty, P. The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth response. J. Cell Biol. 2003, 160, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Johnson, M.R.; Melvin, L.S.; Milne, G.M. Nonclassical cannabinoid analgetics inhibit adenylate cyclase: Development of a cannabinoid receptor model. Mol. Pharmacol. 1988, 33, 297–302. [Google Scholar] [PubMed]
- Devane, W.A.; Dysarz, F.A.I.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar] [PubMed]
- Jarai, Z.; Wagner, J.A.; Varga, K.; Lake, K.D.; Compton, D.R.; Martin, B.R.; Zimmer, A.M.; Bonner, T.I.; Buckley, N.E.; Mezey, E.; et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 14136–14141. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Varga, K.; Jarai, Z.; Kunos, G. Mesenteric vasodilation mediated by endothelial anandamide receptors. Hypertension 1999, 33, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Breivogel, C.S.; Tao, Q.; Bridgen, D.T.; Razdan, R.K.; Zimmer, A.M.; Zimmer, A.; Martin, B.R. Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: Evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J. Neurochem. 2000, 75, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Breivogel, C.S.; Griffin, G.; di Marzo, V.; Martin, B.R. Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol. Pharmacol. 2001, 60, 155–163. [Google Scholar] [PubMed]
- Basavarajappa, B.S. Endocannabinoid signaling and alcohol addiction. In New Research on Alcoholism; Baye, D.R., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2007; pp. 1–55. [Google Scholar]
- Basavarajappa, B.S.; Yalamanchili, R.; Cooper, T.B.; Hungund, B.L. Endocannabinoid system. In Handbook of Neurochemistry and Molecular Neurobiology; Lajtha, A., Sylvester, E.V., Eds.; Springer: New York, NY, USA, 2008; pp. 343–384. [Google Scholar]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Cost, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 16, 8057–8066. [Google Scholar]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Facci, L.; dal Toso, R.; Romanello, S.; Buriani, A.; Skaper, S.D.; Leon, A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. USA 1995, 92, 3376–3380. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann. N. Y. Acad. Sci. 2006, 1074, 514–536. [Google Scholar] [CrossRef] [PubMed]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: From mice to human subjects. PLoS ONE 2008, 3, e1640. [Google Scholar] [CrossRef] [PubMed]
- Berrendero, F.; Garcia-Gil, L.; Hernandez, M.L.; Romero, J.; Cebeira, M.; de Miguel, R.; Ramos, J.A.; Fernández-Ruiz, J.J. Localization of mRNA expression and activation of signal transduction mechanisms for cannabinoid receptor in rat brain during fetal development. Development 1998, 125, 3179–3188. [Google Scholar] [PubMed]
- Buckley, N.E.; Hansson, S.; Harta, G.; Mezey, E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience 1998, 82, 1131–1149. [Google Scholar] [CrossRef]
- Romero, J.; Garcia-Palomero, E.; Berrendero, F.; Garcia-Gil, L.; Hernandez, M.L.; Ramos, J.A.; Fernández-Ruiz, J.J. Atypical location of cannabinoid receptors in white matter areas during rat brain development. Synapse 1997, 26, 317–323. [Google Scholar] [CrossRef]
- Insel, T.R. The Development of Brain and Behavior. In Psychopharmacology: The Four Generation of Progress; Bloom, F.E., Kupfer, D.J., Eds.; Raven Press: New York, NY, USA, 1995; pp. 683–694. [Google Scholar]
- Glass, M.; Dragunow, M.; Faull, R.L. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef]
- Mailleux, P.; Vanderhaeghen, J.J. Distribution of neuronal cannabinoid receptor in the adult rat brain: A comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience 1992, 48, 655–668. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; McIntosh, H.H.; Houston, D.B.; Howlett, A.C. The CB(1) cannabinoid receptor juxtamembrane C-terminal peptide confers activation to specific G proteins in brain. Mol. Pharmacol. 2000, 57, 162–170. [Google Scholar] [PubMed]
- Childers, S.R.; Sexton, T.; Roy, M.B. Effects of anandamide on cannabinoid receptors in rat brain membranes. Biochem. Pharmacol. 1994, 47, 711–715. [Google Scholar] [CrossRef]
- Pinto, J.C.; Potie, F.; Rice, K.C.; Boring, D.; Johnson, M.R.; Evans, D.M.; Wilken, G.H.; Cantrell, C.H.; Howlett, A.C. Cannabinoid receptor binding and agonist activity of amides and esters of arachidonic acid. Mol. Pharmacol. 1994, 46, 516–522. [Google Scholar] [PubMed]
- Howlett, A.C.; Mukhopadhyay, S. Cellular signal transduction by anandamide and 2-arachidonoylglycerol. Chem. Phys. Lipids 2000, 108, 53–70. [Google Scholar] [CrossRef]
- Caulfield, M.P.; Brown, D.A. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br. J. Pharmacol. 1992, 106, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K.; Hille, B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3825–3829. [Google Scholar] [CrossRef] [PubMed]
- Nogueron, M.I.; Porgilsson, B.; Schneider, W.E.; Stucky, C.L.; Hillard, C.J. Cannabinoid receptor agonists inhibit depolarization-induced calcium influx in cerebellar granule neurons. J. Neurochem. 2001, 79, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Ikeda, S.R.; Lewis, D.L. Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol. Pharmacol. 1996, 49, 707–714. [Google Scholar] [PubMed]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Zhuang, S.Y.; Kirby, M.T.; Hampson, R.E.; Deadwyler, S.A. Cannabinoid receptors differentially modulate potassium A and D currents in hippocampal neurons in culture. J. Pharmacol. Exp. Ther. 1999, 291, 893–902. [Google Scholar] [PubMed]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef] [PubMed]
- Wartmann, M.; Campbell, D.; Subramanian, A.; Burstein, S.H.; Davis, R.J. The MAP kinase signal transduction pathway is activated by the endogenous cannabinoid anandamide. FEBS Lett. 1995, 359, 133–136. [Google Scholar] [CrossRef]
- Bouaboula, M.; Poinot-Chazel, C.; Bourrie, B.; Canat, X.; Calandra, B.; Rinaldi-Carmona, M.; le Fur, G.; Casellas, P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem. J. 1995, 312, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Derkinderen, P.; Toutant, M.; Burgaya, F.; le Bert, M.; Siciliano, J.C.; de Franciscis, V.; Gelman, M.; Girault, J.A. Regulation of a neuronal form of focal adhesion kinase by anandamide. Science 1996, 273, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Derkinderen, P.; Toutant, M.; Kadare, G.; Ledent, C.; Parmentier, M.; Girault, J.A. Dual role of Fyn in the regulation of FAK+6,7 by cannabinoids in hippocampus. J. Biol. Chem. 2001, 276, 38289–38296. [Google Scholar] [PubMed]
- Ferna’ndez-Ruiz, J.J.; Bonnin, A.; Cebeira, M.; Ramos, J.A. Ontogenic and adults changes in the activity of hypothalamic and extrahypothalamic dopaminergic neurons after perinatal cannabinoid exposure. In Strategies for Studying Brain Disorders; Palomo, T., Archer, T., Eds.; Farrand Press: England, UK, 1994; pp. 357–390. [Google Scholar]
- Ferna’ndez-Ruiz, J.J.; Rodriguez, F.; Navarro, M.; Ramos, J.A. Maternal cannabinoid exposure and brain development: Changes in the ontogeny of dopaminergic neurons. In Neurobiology and Neurophysiology of Cannabinoids: Biochemistry and Physiology of Substance Abuse; Bartke AaM, L.L., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 119–164. [Google Scholar]
- Ferna’ndez-Ruiz, J.J.; Romero, J.; Garcı’a-Gil, L.; Garcı’a-Palomero, E.; Ramos, J.A. Dopaminergic neurons as neurochemical substrates of neurobehavioral effects of marihuana: Developmental and adult studies. In Dopamine Disease States; Beninger, R.J., Archer, T., Palomo, T., Eds.; CYM Press: Madrid, Spain, 1996; pp. 359–387. [Google Scholar]
- Fernandez-Ruiz, J.J.; Berrendero, F.; Hernandez, M.L.; Romero, J.; Ramos, J.A. Role of endocannabinoids in brain development. Life Sci. 1999, 65, 725–736. [Google Scholar] [CrossRef]
- Dalterio, S.L. Perinatal or adult exposure to cannabinoids alters male reproductive functions in mice. Pharmacol. Biochem. Behav. 1980, 12, 143–153. [Google Scholar] [CrossRef]
- Navarro, M.; Rodriguez de Fonseca, F.; Hernandez, M.L.; Ramos, J.A.; Fernandez-Ruiz, J.J. Motor behavior and nigrostriatal dopaminergic activity in adult rats perinatally exposed to cannabinoids. Pharmacol. Biochem. Behav. 1994, 47, 47–58. [Google Scholar] [CrossRef]
- Dalterio, S.L. Cannabinoid exposure: Effects on development. Neurobehav. Toxicol. Teratol. 1986, 8, 345–352. [Google Scholar] [PubMed]
- Mokler, D.J.; Robinson, S.E.; Johnson, J.H.; Hong, J.S.; Rosecrans, J.A. Neonatal administration of delta-9-tetrahydrocannabinol (THC) alters the neurochemical response to stress in the adult Fischer-344 rat. Neurotoxicol. Teratol. 1987, 9, 321–327. [Google Scholar] [CrossRef]
- Vela, G.; Fuentes, J.A.; Bonnin, A.; Fernandez-Ruiz, J.; Ruiz-Gayo, M. Perinatal exposure to Δ9-tetrahydrocannabinol (Δ9-THC) leads to changes in opioid-related behavioral patterns in rats. Brain Res. 1995, 680, 142–147. [Google Scholar] [CrossRef]
- Navarro, M.; de Miguel, R.; Rodriguez de Fonseca, F.; Ramos, J.A.; Fernandez-Ruiz, J.J. Perinatal cannabinoid exposure modifies the sociosexual approach behavior and the mesolimbic dopaminergic activity of adult male rats. Behav. Brain Res. 1996, 75, 91–98. [Google Scholar] [CrossRef]
- Vela, G.; Martin, S.; Garcia-Gil, L.; Crespo, J.A.; Ruiz-Gayo, M.; Fernandez-Ruiz, J.J.; García-Lecumberri, C.; Pélaprat, D.; Fuentes, J.A.; Ramos, J.A.; et al. Maternal exposure to Δ9-tetrahydrocannabinol facilitates morphine self-administration behavior and changes regional binding to central mu opioid receptors in adult offspring female rats. Brain Res. 1998, 807, 101–109. [Google Scholar] [CrossRef]
- Dalterio, S.; Bartke, A. Perinatal exposure to cannabinoids alters male reproductive function in mice. Science 1979, 205, 1420–1422. [Google Scholar] [CrossRef] [PubMed]
- Shivachar, A.C.; Martin, B.R.; Ellis, E.F. Anandamide- and Δ9-tetrahydrocannabinol-evoked arachidonic acid mobilization and blockade by SR141716A [N-(Piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride]. Biochem. Pharmacol. 1996, 51, 669–676. [Google Scholar] [CrossRef]
- Sanchez, C.; Galve-Roperh, I.; Rueda, D.; Guzman, M. Involvement of sphingomyelin hydrolysis and the mitogen-activated protein kinase cascade in the Δ9-tetrahydrocannabinol-induced stimulation of glucose metabolism in primary astrocytes. Mol. Pharmacol. 1998, 54, 834–843. [Google Scholar] [PubMed]
- Sanchez, C.; Velasco, G.; Guzman, M. Δ9-tetrahydrocannabinol stimulates glucose utilization in C6 glioma cells. Brain Res. 1997, 767, 64–71. [Google Scholar] [CrossRef]
- Bouaboula, M.; Bourrie, B.; Rinaldi-Carmona, M.; Shire, D.; le Fur, G.; Casellas, P. Stimulation of cannabinoid receptor CB1 induces krox-24 expression in human astrocytoma cells. J. Biol. Chem. 1995, 270, 13973–13980. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Krutz, B.; Sifringer, M.; Stefovska, V.; Bittigau, P.; Pragst, F.; Marsicano, G.; Lutz, B.; Ikonomidou, C. Cannabinoids enhance susceptibility of immature brain to ethanol neurotoxicity. Ann. Neurol. 2008, 64, 42–52. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; Gomez del Pulgar, T.; Izquierdo, M.; Guzman, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [PubMed]
- Stefanis, N.C.; Delespaul, P.; Henquet, C.; Bakoula, C.; Stefanis, C.N.; van Os, J. Early adolescent cannabis exposure and positive and negative dimensions of psychosis. Addiction 2004, 99, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Jew, C.P.; Lu, H.C. Lasting impacts of prenatal cannabis exposure and the role of endogenous cannabinoids in the developing brain. Future Neurol. 2011, 6, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Twitchell, W.; Brown, S.; Mackie, K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997, 78, 43–50. [Google Scholar] [PubMed]
- Hampson, R.E.; Miller, F.; Palchik, G.; Deadwyler, S.A. Cannabinoid receptor activation modifies NMDA receptor mediated release of intracellular calcium: Implications for endocannabinoid control of hippocampal neural plasticity. Neuropharmacology 2011, 60, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S.; Ninan, I.; Arancio, O. Acute Ethanol Suppresses Glutamatergic Neurotransmission through Endocannabinoids in Hippocampal Neurons. J. Neurochem. 2008, 107, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vockler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Gerdeman, G.; Lovinger, D.M. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J. Neurophysiol. 2001, 85, 468–471. [Google Scholar] [PubMed]
- Huang, C.C.; Lo, S.W.; Hsu, K.S. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J. Physiol. 2001, 532, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Chevaleyre, V.; Castillo, P.E. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron 2004, 43, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.M. Cellular and molecular mechanisms underlying learning and memory impairments produced by cannabinoids. Learn. Mem. 2000, 7, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid Signaling in the Brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Mameli, M.; Zamudio, P.A.; Carta, M.; Valenzuela, C.F. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J. Neurosci. 2005, 25, 8027–8036. [Google Scholar] [CrossRef] [PubMed]
- Sadrian, B.; Lopez-Guzman, M.; Wilson, D.A.; Saito, M. Distinct neurobehavioral dysfunction based on the timing of developmental binge-like alcohol exposure. Neuroscience 2014, 280C, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Vaglenova, J.; Pandiella, N.; Wijayawardhane, N.; Vaithianathan, T.; Birru, S.; Breese, C.; Suppiramaniam, V.; Randal, C. Aniracetam reversed learning and memory deficits following prenatal ethanol exposure by modulating functions of synaptic AMPA receptors. Neuropsychopharmacology 2008, 33, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Margret, C.P.; Li, C.X.; Chappell, T.D.; Elberger, A.J.; Matta, S.G.; Waters, R.S. Prenatal alcohol exposure delays the development of the cortical barrel field in neonatal rats. Exp. Brain Res. 2006, 172, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Medina, A.E.; Krahe, T.E.; Ramoa, A.S. Early alcohol exposure induces persistent alteration of cortical columnar organization and reduced orientation selectivity in the visual cortex. J. Neurophysiol. 2005, 93, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Powrozek, T.A.; Zhou, F.C. Effects of prenatal alcohol exposure on the development of the vibrissal somatosensory cortical barrel network. Brain Res. Dev. Brain Res. 2005, 155, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Sari, Y.; Powrozek, T.A. Fetal alcohol exposure reduces serotonin innervation and compromises development of the forebrain along the serotonergic pathway. Alcohol. Clin. Exp. Res. 2005, 29, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Bohme, G.A.; Laville, M.; Ledent, C.; Parmentier, M.; Imperato, A. Enhanced long-term potentiation in mice lacking cannabinoid CB1 receptors. Neuroscience 2000, 95, 5–7. [Google Scholar] [CrossRef]
- Reibaud, M.; Obinu, M.C.; Ledent, C.; Parmentier, M.; Bohme, G.A.; Imperato, A. Enhancement of memory in cannabinoid CB1 receptor knock-out mice. Eur. J. Pharmacol. 1999, 379, R1–R2. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Gomez, M.; Hernandez, M.; de Miguel, R.; Ramos, J.A. Cannabinoids and gene expression during brain development. Neurotox. Res. 2004, 6, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dow-Edwards, D.; Anderson, V.; Minkoff, H.; Hurd, Y.L. Discrete opioid gene expression impairment in the human fetal brain associated with maternal marijuana use. Pharm. J. 2006, 6, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M. Cannabis use in pregnancy and early life and its consequences: Animal models. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Trezza, V.; Ratano, P.; Palmery, M.; Cuomo, V. Developmental consequences of perinatal cannabis exposure: Behavioral and neuroendocrine effects in adult rodents. Psychopharmacology (Berl.) 2011, 214, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Kawash, G.F.; Yeung, D.L.; Berg, S.D. Effects of administration of cannabis resin during pregnancy on emotionality and learning in rats’ offspring. Percept. Mot. Skills 1980, 50, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Trezza, V.; Cassano, T.; Gaetani, S.; Morgese, M.G.; Ubaldi, M.; Soverchia, L.; Antonelli, T.; Ferraro, L.; Massi, M.; et al. Perinatal exposure to Δ-9-tetrahydrocannabinol causes enduring cognitive deficits associated with alteration of cortical gene expression and neurotransmission in rats. Addict. Biol. 2007, 12, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, T.; Tomasini, M.C.; Tattoli, M.; Cassano, T.; Tanganelli, S.; Finetti, S.; Mazzoni, E.; Trabace, L.; Steardo, L.; Cuomo, V.; et al. Prenatal exposure to the CB1 receptor agonist WIN 55,212-2 causes learning disruption associated with impaired cortical NMDA receptor function and emotional reactivity changes in rat offspring. Cereb. Cortex. 2005, 15, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Mereu, G.; Fa, M.; Ferraro, L.; Cagiano, R.; Antonelli, T.; Tattoli, M.; Ghiglieri, V.; Tanganelli, S.; Gessa, G.L.; Cuomo, V. Prenatal exposure to a cannabinoid agonist produces memory deficits linked to dysfunction in hippocampal long-term potentiation and glutamate release. Proc. Natl. Acad. Sci. USA 2003, 100, 4915–4920. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, M.; McGregor, I.S.; Mallet, P.E. Repeated cannabinoid exposure during perinatal, adolescent or early adult ages produces similar longlasting deficits in object recognition and reduced social interaction in rats. J. Psychopharmacol. 2006, 20, 611–621. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, M.; Singh, M.E.; McGregor, I.S.; Mallet, P.E. Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J. Psychopharmacol. 2004, 18, 502–508. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, M.; Mallet, P.E. Impaired learning in adulthood following neonatal Δ9-THC exposure. Behav. Pharmacol. 2005, 16, 455–461. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basavarajappa, B.S. Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain Sci. 2015, 5, 456-493. https://doi.org/10.3390/brainsci5040456
Basavarajappa BS. Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain Sciences. 2015; 5(4):456-493. https://doi.org/10.3390/brainsci5040456
Chicago/Turabian StyleBasavarajappa, Balapal S. 2015. "Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling" Brain Sciences 5, no. 4: 456-493. https://doi.org/10.3390/brainsci5040456
APA StyleBasavarajappa, B. S. (2015). Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain Sciences, 5(4), 456-493. https://doi.org/10.3390/brainsci5040456