
Immunomodulators as Therapeutic Agents in Mitigating the Progression of Parkinson’s Disease

{kind=link}
Abstract
:1. Parkinson’s Disease
2. The Interaction with Aging
3. Role of Neuroinflammation
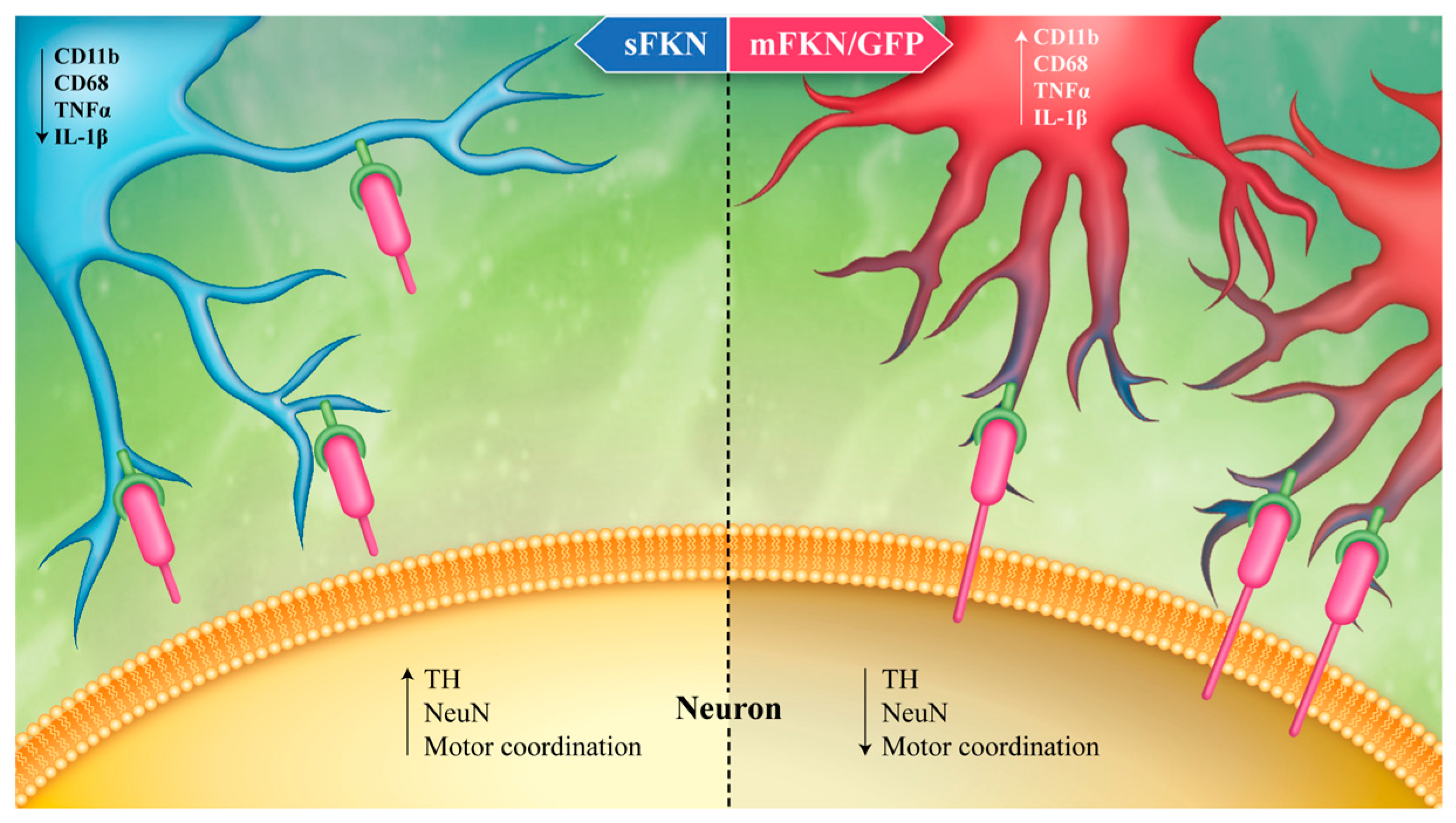
4. Fractalkine as an Anti-Inflammatory Treatment
5. Therapeutic Potential of Astaxanthin
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kowal, S.L.; Dall, T.M.; Chakrabarti, R.; Storm, M.V.; Jain, A. The current and projected economic burden of Parkinson’s disease in the United States. Mov. Disord. 2013, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.S.; Zhang, J. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, A.N.; Stöckl, M.T.; Claessens, M.M.; Subramaniam, V. α-Synuclein oligomers distinctively permeabilize complex model membranes. FEBS J. 2014, 281, 2838–2850. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Hurtig, H.I.; Trojanowski, J.Q.; Galvin, J.; Ewbank, D.; Schmidt, M.L.; Lee, V.M.; Clark, C.M.; Glosser, G.; Stern, M.B.; Gollomp, S.M.; et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 2000, 54, 1916–1921. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kim, Y.M.; Lee, G.; Junn, E.; Iwatsubo, T.; Mouradian, M.M. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 2004, 279, 4625–4631. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [PubMed]
- Bharath, S.; Hsu, M.; Kaur, D.; Rajagopalan, S.; Andersen, J.K. Glutathione, iron and Parkinson’s disease. Biochem. Pharmacol. 2002, 64, 1037–1048. [Google Scholar] [CrossRef]
- Venkateshappa, C.; Harish, G.; Mythri, R.B.; Mahadevan, A.; Bharath, M.M.; Shankar, S.K. Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: Implications for Parkinson’s disease. Neurochem. Res. 2012, 37, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Gross, C.T. Microglia in development: Linking brain wiring to brain environment. Neuron Glia Biol. 2011, 7, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, H.A.; Johnson, R.W. Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation. Exp. Neurol. 2012, 233, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Ruiz, C.R.; Lebson, L.; Selenica, M.-L.B.L.; Rizer, J.; Hunt, J.B.; Rojiani, R.; Reid, P.; Kammath, S.; Nash, K.; et al. Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol. Aging 2013, 34, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Akiyama, H.; McGeer, E.G. Rate of cell death in parkinsonism indicates active neuropathological process. Ann. Neurol. 1988, 24, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Trender-Gerhard, I.; Turkheimer, F.; Quinn, N.P.; Bhatia, K.P.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov. Disord. 2006, 21, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M.; Tanner, C.M.; Oakes, D.; Bhudhikanok, G.S.; Gupta, A.; Langston, J.W. Head injury and Parkinson’s disease risk in twins. Ann. Neurol. 2006, 60, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Sharma, N.; Nehru, B. Characterization of the lipopolysaccharide induced model of Parkinson’s disease: Role of oxidative stress and neuroinflammation. Neurochem. Int. 2015, 87, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Glanzer, J.G.; Kadiu, I.; Ricardo-Dukelow, M.; Chaudhuri, A.; Ciborowski, P.; Cerny, R.; Gelman, B.; Thomas, M.P.; Mosley, R.L.; et al. Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J. Neurochem. 2008, 104, 1504–1525. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Theodore, S.; Standaert, D.G. Fcγ receptors are required for NF-κB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson’s disease. Mol. Neurodegener. 2010, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Standaert, D.G.; Harms, A.S. The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflamm. 2012, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Polinski, N.K.; Gombash, S.E.; Manfredsson, F.P.; Lipton, J.W.; Kemp, C.J.; Cole-Strauss, A.; Kanaan, N.M.; Steece-Collier, K.; Kuhn, N.C.; Wohlgenant, S.L.; et al. Recombinant adenoassociated virus 2/5-mediated gene transfer is reduced in the aged rat midbrain. Neurobiol. Aging 2015, 36, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.R.; Moran, P.; Finneran, D.J.; Hudson, C.; Robinson, J.; Morgan, D.; Bickford, P.C. Fractalkine over expression suppresses α-synuclein-mediated neurodegeneration. Mol. Ther. 2015, 23, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Nash, K.R.; Grimmig, B.A.; Ranjit, S.; Small, B.; Bickford, P.C.; Gemma, C. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 2012, 32, 14592–14601. [Google Scholar] [CrossRef] [PubMed]
- Pabon, M.M.; Jernberg, J.N.; Morganti, J.; Contreras, J.; Hudson, C.E.; Klein, R.L.; Bickford, P.C. A spirulina-enhanced diet provides neuroprotection in an α-synuclein model of Parkinson’s disease. PLoS ONE 2012, 7, e45256. [Google Scholar] [CrossRef] [PubMed]
- Pabon, M.M.; Bachstetter, A.D.; Hudson, C.E.; Gemma, C.; Bickford, P.C. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflamm. 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Rees, K.; Stowe, R.; Patel, S.; Ives, N.; Breen, K.; Clarke, C.E.; Ben-Shlomo, Y. Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: Evidence from observational studies. Cochrane Database Syst. Rev. 2011, 11, CD008454. [Google Scholar] [PubMed]
- Du, Y.; Ma, Z.; Lin, S.; Dodel, R.C.; Gao, F.; Bales, K.R.; Triarhou, L.C.; Chernet, E.; Perry, K.W.; Nelson, D.L.; et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 14669–14674. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Yune, T.Y.; Kim, S.J.; Kim, Y.C.; Oh, Y.J.; Markelonis, G.J.; Oh, T.H. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J. Neurochem. 2004, 91, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A potential new target for inflammatory diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Thome, A.D.; Standaert, D.G.; Harms, A.S. Fractalkine signaling regulates the inflammatory response in an α-synuclein model of Parkinson disease. PLoS ONE 2015, 10, e0140566. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.R.; Lee, D.C.; Hunt, J.B.; Morganti, J.M.; Selenica, M.-L.L.; Moran, P.; Reid, P.; Brownlow, M.; Yang, C.G.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Xu, G.; Jay, T.R.; Bhatta, S.; Kim, K.-W.W.; Jung, S.; Landreth, G.E.; Ransohoff, R.M.; Lamb, B.T. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J. Neurosci. 2014, 34, 12538–12546. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Morganti, J.M.; Jernberg, J.; Schlunk, A.; Mitchell, S.H.; Brewster, K.W.; Hudson, C.E.; Cole, M.J.; Harrison, J.K.; Bickford, P.C.; et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 2011, 32, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Wynne, A.M.; Henry, C.J.; Huang, Y.; Cleland, A.; Godbout, J.P. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 2010, 24, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium 2010, 47, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.; Guzman, J.; Sanchez-Padilla, J.; Goldberg, J. What causes the death of dopaminergic neurons in Parkinson’s Disease? Prog. Brain Res. 2010, 183, 59–77. [Google Scholar] [PubMed]
- Kim, Y.H.; Koh, H.-K.K.; Kim, D.-S.S. Down-regulation of IL-6 production by astaxanthin via ERK-, MSK-, and NF-κB-mediated signals in activated microglia. Int. Immunopharmacol. 2010, 10, 1560–1572. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shibata, T.; Hisaka, S.; Osawa, T. Astaxanthin inhibits reactive oxygen species-mediated cellular toxicity in dopaminergic SH-SY5Y cells via mitochondria-targeted protective mechanism. Brain Res. 2009, 1254, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.H.; Kim, C.-S.S.; Lee, Y.J. Astaxanthin protects against MPTP/MPP+-induced mitochondrial dysfunction and ROS production in vivo and in vitro. Food Chem. Toxicol. 2011, 49, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Kidd, P. Astaxanthin, a cell membrane nutrient with diverse clinical benefits and anti aging potential. Am. Med. Rev. 2011, 16, 355–364. [Google Scholar]
- Park, J.; Mathison, B.; Hayek, M.; Zhang, J.; Reinhart, G.; Chew, B. Astaxanthin modulates age-associated mitochondrial dysfunction in healthy dogs. J. Anim. Sci. 2012, 91, 268275. [Google Scholar] [CrossRef] [PubMed]
- Otton, R.; Marin, D.P.; Bolin, A.P.; Santos, R.C.; Polotow, T.G.; Sampaio, S.C.; de Barros, M.P. Astaxanthin ameliorates the redox imbalance in lymphocytes of experimental diabetic rats. Chem. Biol. Interact. 2010, 186, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Augusti, P.R.; Quatrin, A.; Somacal, S.; Conterato, G.M.; Sobieski, R.; Ruviaro, A.R.; Maurer, L.H.; Duarte, M.M.; Roehrs, M.; Emanuelli, T. Astaxanthin prevents changes in the activities of thioredoxin reductase and paraoxonase in hypercholesterolemic rabbits. J. Clin. Biochem. Nutr. 2012, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Liu, H.; Chen, X.; Shi, H.; Fan, Y.; Hou, D.; Zhang, X. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated phase II enzymes through activation of PI3K/Akt. Mol. Vis. 2013, 19, 1656–1666. [Google Scholar] [PubMed]
- Wang, H.-Q.Q.; Sun, X.-B.B.; Xu, Y.-X.X.; Zhao, H.; Zhu, Q.-Y.Y.; Zhu, C.-Q.Q. Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against beta-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res. 2010, 1360, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Sun, X.; Shao, X.; Cheng, C.; Feng, J.; Sun, W.; Gu, D.; Liu, W.; Xu, F.; Duan, Y. Macrophage-microglia networks drive M1 microglia polarization after mycobacterium infection. Inflammation 2015, 38, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Minogue, A.M.; Jones, R.S.; Kelly, R.J.; McDonald, C.L.; Connor, T.J.; Lynch, M.A. Age-associated dysregulation of microglial activation is coupled with enhanced blood-brain barrier permeability and pathology in APP/PS1 mice. Neurobiol. Aging 2014, 35, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-K.K.; Park, Y.-S.S.; Choi, D.-K.K.; Chang, H.-I.I. Effects of astaxanthin on the production of NO and the expression of COX-2 and iNOS in LPS-stimulated BV2 microglial cells. J. Microbiol. Biotechnol. 2008, 18, 1990–1996. [Google Scholar] [PubMed]
- Sanchez-Padilla, J.; Guzman, J.N.; Ilijic, E.; Kondapalli, J.; Galtieri, D.J.; Yang, B.; Schieber, S.; Oertel, W.; Wokosin, D.; Schumacker, P.T.; et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci. 2014, 17, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.; Miller, R.J.; Schumacker, P.T.; Surmeier, D.J. Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J.; Schumacker, P.T. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience 2011, 198, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson’s disease (STEADY-PD). Mov. Disord. 2013, 28, 1823–1831. [Google Scholar]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Huang, B.; Zhang, X.; Zhu, Y.; Chen, X. Astaxanthin protects against MPP(+)-induced oxidative stress in PC12 cells via the HO-1/NOX2 axis. BMC Neurosci. 2012, 13, 156. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grimmig, B.; Morganti, J.; Nash, K.; Bickford, P.C. Immunomodulators as Therapeutic Agents in Mitigating the Progression of Parkinson’s Disease. Brain Sci. 2016, 6, 41. https://doi.org/10.3390/brainsci6040041
Grimmig B, Morganti J, Nash K, Bickford PC. Immunomodulators as Therapeutic Agents in Mitigating the Progression of Parkinson’s Disease. Brain Sciences. 2016; 6(4):41. https://doi.org/10.3390/brainsci6040041
Chicago/Turabian StyleGrimmig, Bethany, Josh Morganti, Kevin Nash, and Paula C Bickford. 2016. "Immunomodulators as Therapeutic Agents in Mitigating the Progression of Parkinson’s Disease" Brain Sciences 6, no. 4: 41. https://doi.org/10.3390/brainsci6040041
APA StyleGrimmig, B., Morganti, J., Nash, K., & Bickford, P. C. (2016). Immunomodulators as Therapeutic Agents in Mitigating the Progression of Parkinson’s Disease. Brain Sciences, 6(4), 41. https://doi.org/10.3390/brainsci6040041