Parkinson’s Disease and Metal Storage Disorders: A Systematic Review


Abstract
:1. Introduction
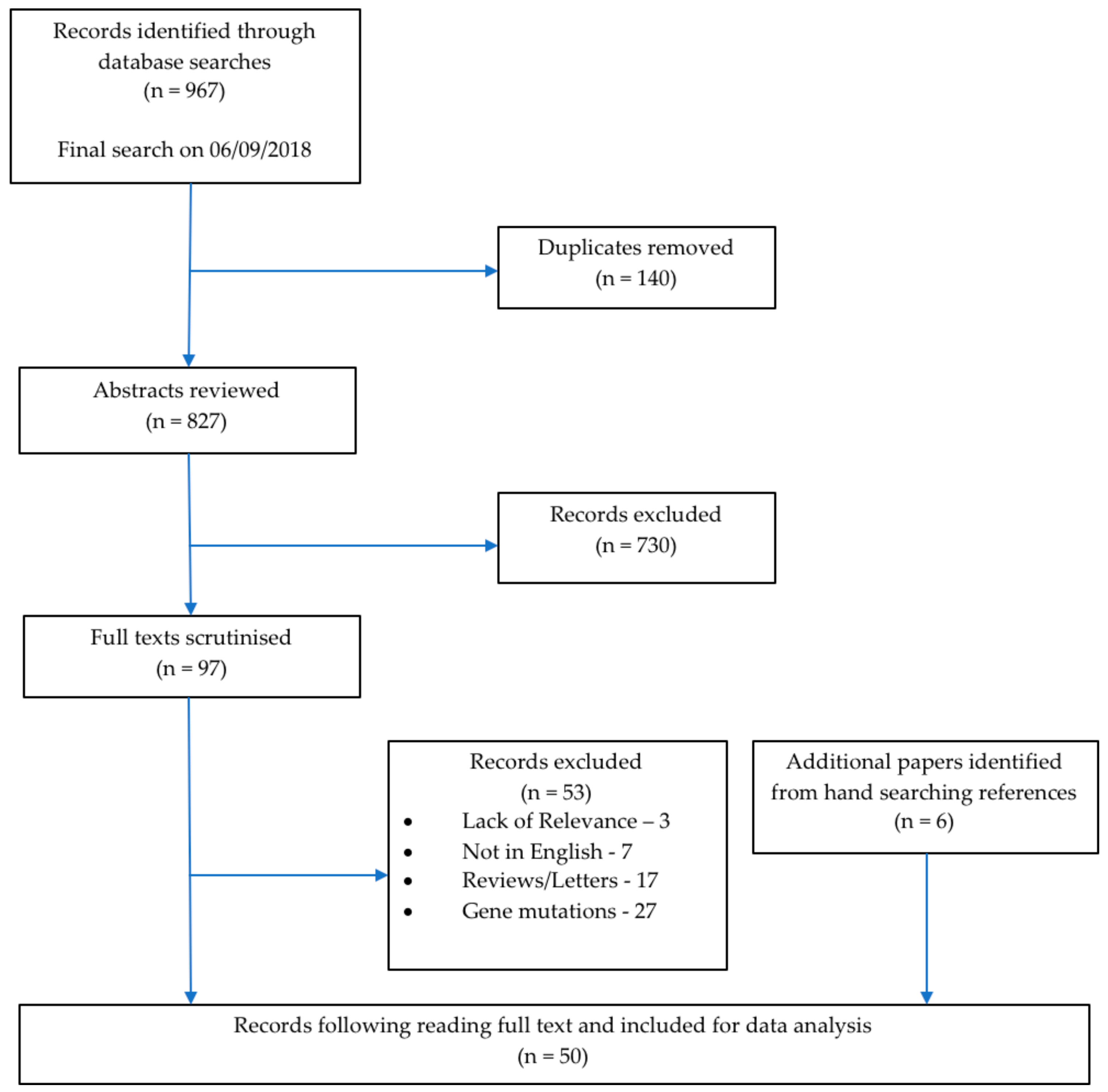
2. Materials and Methods
2.1. Search Terms
2.2. Inclusion Criteria
2.3. Exclusion Criteria
2.4. Selection Process
2.5. Data Extraction
3. Results
4. Discussion
5. Conclusions
- There is evidence of Parkinsonism coexisting with metal storage disorders in particular neurodegenerative brain iron accumulation disorders.
- Patients with these metal storage disorders have an earlier age of onset of Parkinsonism than sporadic PD patients, which suggests additional underlying pathological processes are taking place. The ratio of males to females seen in many of these also differs significantly to the sporadic PD population, which further indicates a differing pathogenesis.
- Future research must be conducted at a higher level than individual case reports to better assess the relationship between metal storage disorders and Parkinsonism. Cohort studies or case control studies using large cohorts will lead to a reliable dataset. At the same time, research in sporadic PD patients will identify whether any of the pathological mutations or processes are involved in the disorders discussed in relation to the development of Parkinsonism.
- Smoking status and ethnicity should be documented in all future studies of Parkinsonism since Caucasian ethnicity is a large risk factor in sporadic PD while cigarette smoking appears to be protective. Recording these demographics will allow for the investigation of their presence in patients with metal storage disorders.
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
| Paper | Type of Paper | Condition | Male/Female | Average Age at Onset of Parkinsonism (years) | Ethnicity | Smoking Status | Typical Parkinsonism | Atypical Parkinsonism | Parkinsonism Features |
|---|---|---|---|---|---|---|---|---|---|
| Alberca, R. et al., 1987. [20] | Case report | PKAN | 1M/1F | 27 | NA | NA | ✓ | ✓ | Female siblings: Typical features. Male sibling: associated with dystonia. Fast progression. |
| Batla, A. et al., 015. [48] | Case report | Neuroferritinopathy | 1F | 79 | NA | NA | ✓ | Associated with dystonia. | |
| Behrens, M.I. et al., 2010. [49] | Case Series | Kufor-Rakeb Syndrome | 4M/1F | NA | Chilean | NA | ✓ | Parkinsonian features in all five pts. No tremor present. Supranuclear gaze palsy in 4/5, poor L-dopa response | |
| Bozi, M. et al., 2009. [23] | Case report | PKAN | 1M | 15 | NA | NA | ✓ | Mildly affected but associated with pyramidal signs. | |
| Chinnery, P.F. et al., 2007. [50] | Cross-sectional study | Neuroferritinopathy | 3F | NA | 2 English, 1 French | NA | ✓ | Associated with dystonia in all three. No tremor present. | |
| Costello, D.J. et al., 2004. [31] | Case report | Hereditary Haemochromatosis | 3M/1F | 53 | NA | NA | ✓ | Four pts all with HH and IPD diagnoses, classical signs. Good L-dopa response. | |
| Crosiers, D. et al., 2011. [51] | case report | Kufor-Rakeb syndrome | 1M | 10 | Afghan | NA | ✓ | Associated with dystonia. | |
| Czlonkowska, A. et al., 2018. [40] | Cross-sectional study | Wilson’s disease | NA | NA | Polish | NA | ✓ | Parkinsonism found in 11.3% (6/53 pts). | |
| Darling, A. et al., 2017. [21] | Cross-sectional study | PKAN | 22M/25F | NA | NA | NA | ✓ | Features of Parkinsonism displayed in all 47 pts. Associated with Dystonia. | |
| Demarquay, G. et al., 2000. [35] | Case report | Hereditary Haemochromatosis | 2M/1F | 56 | NA | NA | ✓ | Bradykinesia and rigidity on left side. Poor L-dopa response. | |
| Di Fonzo, A. et al., 2007. [52] | Cross-sectional study | Kufor-Rakeb Syndrome | 3M | NA | NA | NA | ✓ | ✓ | Features of Parkinsonism in all three pts. Supranuclear gaze palsy and hallucinations/psychotic episodes in 1/3, psychotic episodes in 1/3, and typical features in 1/3. |
| Diaz, N., 2013. [13] | Case report | PKAN | 1F | NA | NA | NA | ✓ | L-dopa unresponsive, symmetrical features. | |
| Eiberg, H. et al., 2012. [53] | Case report | Kufor-Rakeb Syndrome | 1M | 12 | NA | NA | ✓ | Supranuclear gaze palsy, cognitive impairment, and hallucinations. | |
| Evans, B.K. & Donley, D.K., 1988. [54] | Case report | Pseudohypoparathyroidism | 1F | 20 | NA | NA | ✓ | Rest tremor and bradykinesia with mental retardation. | |
| Fekete, R., 2012. [55] | Case report | NBIA, unknown type | 1M | 73 | NA | NA | ✓ | Typical features. Poor L-dopa response but dystonia present upon removal of L-dopa. | |
| Fonderico, M. et al., 2017. [56] | Case report | BPAN | 1F | 26 | NA | NA | ✓ | Mild typical parkinsonism. | |
| Gasca-Salas, C. et al., 2017. [44] | Case report | Wilson’s Disease | 1F | 38 | NA | NA | ✓ | Tremor, clumsiness, rigidity, and dystonia in left arm. Good L-dopa response. | |
| Giri, A. et al., 2016. [28] | Case report | PLAN | 1F | 27 | NA | NA | ✓ | Typical Features, PD diagnosis. | |
| Girotra, T., Mahajan, A. & Sidiropoulos, C., 2017. [32] | Case report | Hereditary Haemochromatosis | 1M | 41 | Caucasian | NA | ✓ | Typical features, mild but clear response to L-dopa. | |
| Gondim, F. de A.A. et al., 2014. [41] | Case Series | Wilson’s disease | 2M/2F | 28 | Brazil | NA | ✓ | Four pts with typical features, all responded well to L-dopa. | |
| Gore, E. et al., 2016. [57] | Case report | MPAN | 1M | 35 | Kuwaiti | NA | ✓ | Early behavioural change. | |
| Hayflick, S.J. et al., 2013. [58] | Cohort study | BPAN | 3M/18F | 25 | NA | NA | ✓ | Developmental delay, dystonia, and parkinsonism. L-dopa responsive. | |
| Hermann, A. et al., 2017. [59] | Case report | BPAN | 1F | 24 | German | NA | ✓ | Supranuclear gaze palsy, dystonia, and no L-dopa response. | |
| Ichinose, Y. et al., 2014. [60] | Case report | BPAN | 1F | 30 | NA | NA | ✓ | Associated with dystonia. | |
| Kim, Y.J. et al., 2015. [30] | Case Series | PLAN | 1M/1F | 14 | Korean | NA | ✓ | Associated with dystonia in 2/2 pts. | |
| Klysz, B., Skowronska, M. & Kmiec, T., 2014. [61] | Case report | MPAN | 1F | 15 | NA | NA | ✓ | Chorea, dystonia, and psychological manifestations. | |
| Kumar, N. et al., 2016. [33] | Case Series | Hereditary Haemochromatosis | 2M/1F | 59 | 1 Irish-Portuguese, 1 Scottish, 1 unknown | NA | ✓ | Parkinsonian signs in three pts. One responded well to L-dopa, one not treated. | |
| Lee, C.-H. et al., 2013. [17] | Case report | PKAN | 2M | 20 | Taiwanese | NA | ✓ | ✓ | Typical parkinsonism in one pt though onset at 18. Bilateral features in the other. |
| Lee, J.-H. et al., 2016. [14] | Cross-sectional study | PKAN | 6M | 36 | NA | NA | ✓ | Poor response to L-dopa in all. Associated with dystonia in 4/6 pts, isolated parkinsonism in 2/6 pts. | |
| Mak, C.M. et al., 2011. [18] | Case report | PKAN | 1M | 27 | Hong Kong | NA | ✓ | Bilateral features. | |
| Ni, W. et al., 2016. [62] | Case report | Neuroferritinopathy | 1F | 44 | NA | NA | ✓ | No response to L-dopa, pyramidal signs. | |
| Nielsen, J.E., Jensen, L.N. & Krabbe, K., 1995. [34] | Case report | Hereditary Haemochromatosis | 1M | 29 | NA | NA | ✓ | Typical PD features, immediate improvement with L-dopa. | |
| Nishioka, K. et al., 2015. [63] | Cross-sectional study | BPAN | 7F | 32 | NA | NA | ✓ | Cognitive dysfunction as presenting symptom in all seven. Otherwise typical parkinsonism. L-dopa responsive. | |
| Oder, W. et al., 1991. [42] | Cross-sectional study | Wilson’s Disease | NA | NA | NA | NA | ✓ | 8/25 pts with parkinsonian features. Bradykinesia, resting tremor present. | |
| Olgiati, S. et al., 2017. [64] | Cross-sectional study | MPAN | NA | NA | NA | NA | ✓ | 9/15 pts with parkinsonian features. Cognitive impairment and pyramidal signs seen. | |
| Pearson, D.W. et al., 1981. [65] | Case report | Pseudohypoparathyroidism | 1M | 58 | NA | NA | ✓ | Typical PD features. Very fast disease progression. | |
| Pestana Knight, E.M., Gilman, S. & Selwa, L., 2009. [45] | Case report | Wilson’s Disease | 1M | 55 | NA | NA | ✓ | Typical PD features associated with epilepsy. | |
| Racette, B.A. et al., 2001. [15] | Case report | PKAN | 1F | 60 | NA | NA | ✓ | Bilateral features, no response to L-dopa. | |
| Rohani, M. et al., 2017. [66] | Case report | Fahr disease | 1F | 50 | NA | NA | ✓ | Typical L-dopa responsive parkinsonism. | |
| Rosana, A. & La Rosa, L., 2007. [36] | Case report | Hereditary Haemochromatosis | 1M | 58 | NA | NA | ✓ | No response to L-dopa. | |
| Sakarya, A., Oncu, B. & Elibol, B., 2012. [19] | Case report | PKAN | 1M | 16 | NA | NA | ✓ | Early severe cognitive impairment, bilateral onset, pyramidal features. | |
| Scale, T. et al., 2014. [67] | Case report | Fahr Disease | 1M | 62 | NA | NA | ✓ | No response to L-dopa. | |
| Schneider, S.A. et al., 2010. [12] | Case report | Kufor-Rakeb syndrome | 1M | 16 | Pakistan | NA | ✓ | Associated with dystonia. | |
| Sechi, G. et al., 2007. [43] | Case report | Wilson’s disease | 3F | 70 | NA | NA | ✓ | Very late onset L-dopa responsive parkinsonism. | |
| Seo, J.-H., Song, S.-K. & Lee, P.H., 2009. [16] | Case report | PKAN | 1M | 35 | NA | NA | ✓ | No response to L-dopa. | |
| Song, C.-Y. et al., 2017. [68] | Case report | Pseudohypoparathyroidism | 1F | 52 | NA | NA | ✓ | Very fast disease progression. | |
| Thomas, M., Hayflick, S.J. & Jankovic, J., 2004. [22] | Cross-sectional study | PKAN | 14M/8F | 35 | NA | NA | ✓ | ✓ | Typical parkinsonism seen, though clinical features not defined. Associated with dystonia in 4/22 pts. |
| Vroegindeweij, L.H.P. et al., 2017. [69] | Case Series | Aceruloplasminemia | 4M/1F | NA | 4 Dutch, 1 Italian | NA | ✓ | Parkinsonian features in all pts. Associated with cognitive decline and cerebellar features in all pts. | |
| Williams, S. et al., 2013. [37] | Case report | Hereditary Haemochromatosis | 1F | 60 | Caucasian | NA | ✓ | Short disease course, early autonomic involvement, no L-dopa response. | |
| Xie, F. et al., 2015. [29] | Case report | PLAN | 2M | 34 | NA | NA | ✓ | Typical features, good L-dopa response. |
References
- Lubbe, S.; Morris, H.R. Recent advances in Parkinson’s disease genetics. J. Neurol. 2014, 261, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet (Lond. Engl.) 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieburtz, K.; Wunderle, K.B. Parkinson’s disease: Evidence for environmental risk factors. Mov. Disord. 2013, 28, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Deutschlander, A.B.; Ross, O.A.; Dickson, D.W.; Wszolek, Z.K. Atypical parkinsonian syndromes: A general neurologist’s perspective. Eur. J. Neurol. 2018, 25, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, S.; Green, A.; Preece, M.A.; Burton, H. The incidence of inherited metabolic disorders in the West Midlands, UK. Arch. Dis. Child. 2006, 91, 896–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waber, L. Inborn errors of metabolism. Pediatr. Ann. 1990, 19, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Pietracupa, S.; Martin-Bastida, A.; Piccini, P. Iron metabolism and its detection through MRI in parkinsonian disorders: A systematic review. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2017, 38, 2095–2101. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Yasuda, T. Iron accumulation in Parkinson’s disease. J. Neural Transm. 2012, 119, 1511–1514. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, T.P. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLOS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Gahl, W.A. Disorders of metal metabolism. Transl. Sci. Rare Dis. 2017, 2, 101–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, S.; Paisan-Ruiz, C.; Quinn, N.; Lees, A.; MD, F.; Houlden, H.; Hardy, J.; Bhatia, K.P. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov. Disord. 2010, 25, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Diaz, N. Late onset atypical pantothenate-kinase-associated neurodegeneration. Case Rep. Neurol. Med. 2013, 2013, 860201. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Park, J.; Ryu, H.-S.; Park, H.; Kim, Y.E.; Hong, J.Y.; Nam, S.O.; Sung, Y.-H.; Lee, S.-H.; Lee, J.-Y.; et al. Clinical Heterogeneity of Atypical Pantothenate Kinase-Associated Neurodegeneration in Koreans. J. Mov. Disord. 2016, 9, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racette, B.A.; Perry, A.; D’Avossa, G.; Perlmutter, J.S. Late-onset neurodegeneration with brain iron accumulation type 1: Expanding the clinical spectrum. Mov. Disord. 2001, 16, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-H.; Song, S.-K.; Lee, P.H. A Novel PANK2 Mutation in a Patient with Atypical Pantothenate-Kinase-Associated Neurodegeneration Presenting with Adult-Onset Parkinsonism. J. Clin. Neurol. 2009, 5, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Lu, C.-S.; Chuang, W.-L.; Yeh, T.-H.; Jung, S.-M.; Huang, C.-L.; Lai, S.-C. Phenotypes and genotypes of patients with pantothenate kinase-associated neurodegeneration in Asian and Caucasian populations: 2 cases and literature review. Sci. World J. 2013, 2013, 860539. [Google Scholar] [CrossRef] [PubMed]
- Mak, C.M.; Sheng, B.; Lee, H.H.; Lau, K.; Chan, W.; Lam, C.; Chan, Y. Young-onset parkinsonism in a Hong Kong Chinese man with adult-onset Hallervorden-Spatz syndrome. Int. J. Neurosci. 2011, 121, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Sakarya, A.; Oncu, B.; Elibol, B. Panthothenate kinase-associated neurodegeneration (PKAN) presenting with language deterioration, personality alteration, and severe parkinsonism. J. Neuropsychiatry Clin. Neurosci. 2012, 24, E13–E14. [Google Scholar] [CrossRef] [PubMed]
- Alberca, R.; Rafel, E.; Chinchon, I.; Vadillo, J.; Navarro, A. Late onset parkinsonian syndrome in Hallervorden-Spatz disease. J. Neurol. Neurosurg. Psychiatry 1987, 50, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.; Tello, C.; Marti, M.J.; Garrido, C.; Aguilera-Albesa, S.; Tomas Vila, M.; Gaston, I.; Madruga, M.; Gonzalez Gutierrez, L.; Ramos Lizana, J.; et al. Clinical rating scale for pantothenate kinase-associated neurodegeneration: A pilot study. Mov. Disord. 2017, 32, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Hayflick, S.J.; Jankovic, J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov. Disord. 2004, 19, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Bozi, M.; Matarin, M.; Theocharis, I.; Potagas, C.; Stefanis, L. A patient with pantothenate kinase-associated neurodegeneration and supranuclear gaze palsy. Clin. Neurol. Neurosurg. 2009, 111, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Dusek, P.; Hardy, J.; Westenberger, A.; Jankovic, J.; Bhatia, K.P. Genetics and Pathophysiology of Neurodegeneration with Brain Iron Accumulation (NBIA). Curr. Neuropharmacol. 2013, 11, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kawai, M.; Inoue, K.; Sasaki, R.; Arai, H.; Nanba, E.; Kuzuhara, S.; Ihara, Y.; Kanazawa, I.; Murayama, S. Widespread expression of alpha-synuclein and tau immunoreactivity in Hallervorden-Spatz syndrome with protracted clinical course. J. Neurol. Sci. 2000, 177, 48–59. [Google Scholar] [CrossRef]
- Gregory, A.; Westaway, S.K.; Holm, I.E.; Kotzbauer, P.T.; Hogarth, P.; Sonek, S.; Coryell, J.C.; Nguyen, T.M.; Nardocci, N.; Zorzi, G.; et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology 2008, 71, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Li, A.; Schneider, S.A.; Holton, J.L.; Johnson, R.; Kidd, D.; Chataway, J.; Bhatia, K.P.; Lees, A.J.; Hardy, J.; et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol. Aging 2012, 33, 814–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, A.; Guven, G.; Hanagasi, H.; Hauser, A.-K.; Erginul-Unaltuna, N.; Bilgic, B.; Gurvit, H.; Heutink, P.; Gasser, T.; Lohmann, E.; et al. PLA2G6 Mutations Related to Distinct Phenotypes: A New Case with Early-onset Parkinsonism. Tremor Other Hyperkinet. Mov. (N. Y.) 2016, 6, 363. [Google Scholar] [CrossRef]
- Xie, F.; Cen, Z.; Ouyang, Z.; Wu, S.; Xiao, J.; Luo, W. Homozygous p.D331Y mutation in PLA2G6 in two patients with pure autosomal-recessive early-onset parkinsonism: further evidence of a fourth phenotype of PLA2G6-associated neurodegeneration. Parkinsonism Relat. Disord. 2015, 21, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Lyoo, C.H.; Hong, S.; Kim, N.Y.; Lee, M.S. Neuroimaging studies and whole exome sequencing of PLA2G6-associated neurodegeneration in a family with intrafamilial phenotypic heterogeneity. Parkinsonism Relat. Disord. 2015, 21, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.J.; Walsh, S.L.; Harrington, H.J.; Walsh, C.H. Concurrent hereditary haemochromatosis and idiopathic Parkinson’s disease: A case report series. J. Neurol. Neurosurg. Psychiatry 2004, 75, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Girotra, T.; Mahajan, A.; Sidiropoulos, C. Levodopa Responsive Parkinsonism in Patients with Hemochromatosis: Case Presentation and Literature Review. Case Rep. Neurol. Med. 2017, 2017, 5146723. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Rizek, P.; Sadikovic, B.; Adams, P.C.; Jog, M. Movement Disorders Associated With Hemochromatosis. Can. J. Neurol. Sci. 2016, 43, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.E.; Jensen, L.N.; Krabbe, K. Hereditary haemochromatosis: A case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J. Neurol. Neurosurg. Psychiatry 1995, 59, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Demarquay, G.; Setiey, A.; Morel, Y.; Trepo, C.; Chazot, G.; Broussolle, E. Clinical report of three patients with hereditary hemochromatosis and movement disorders. Mov. Disord. 2000, 15, 1204–1209. [Google Scholar] [CrossRef]
- Rosana, A.; La Rosa, L. A case of hereditary haemochromatosis in a patient with extrapyramidal syndrome. Blood Transfus. 2007, 5, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.; Vinjam, M.R.; Ismail, A.; Hassan, A. A parkinsonian movement disorder with brain iron deposition and a haemochromatosis mutation. J. Neurol. 2013, 260, 2170–2171. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P. Pathophysiology and clinical features of Wilson disease. Metab. Brain Dis. 2004, 19, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.T. Neurologic Wilson’s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Litwin, T.; Dziezyc, K.; Karlinski, M.; Bring, J.; Bjartmar, C. Characteristics of a newly diagnosed Polish cohort of patients with neurological manifestations of Wilson disease evaluated with the Unified Wilson’s Disease Rating Scale. BMC Neurol. 2018, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- De Gondim, F.A.; Araujo, D.F.; Oliveira, I.S.; Vale, O.C. Small fiber dysfunction in patients with Wilson’s disease. Arq. Neuropsiquiatr. 2014, 72, 592–595. [Google Scholar] [CrossRef]
- Oder, W.; Grimm, G.; Kollegger, H.; Ferenci, P.; Schneider, B.; Deecke, L. Neurological and neuropsychiatric spectrum of Wilson’s disease: A prospective study of 45 cases. J. Neurol. 1991, 238, 281–287. [Google Scholar] [PubMed]
- Sechi, G.; Antonio Cocco, G.; Errigo, A.; Deiana, L.; Rosati, G.; Agnetti, V.; Stephen Paulus, K.; Mario Pes, G. Three sisters with very-late-onset major depression and parkinsonism. Parkinsonism Relat. Disord. 2007, 13, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Gasca-Salas, C.; Alonso, A.; Gonzalez-Redondo, R.; Obeso, J.A. Coexisting Parkinson’s and Wilson’s Disease: Chance or Connection? Can. J. Neurol. Sci. 2017, 44, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Pestana Knight, E.M.; Gilman, S.; Selwa, L. Status epilepticus in Wilson’s disease. Epileptic Disord. 2009, 11, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Rizek, P.; Kumar, N.; Jog, M.S. An update on the diagnosis and treatment of Parkinson disease. Can. Med. Assoc. J. 2016, 188, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxford Centre for Evidence-Based Medicine—Levels of Evidence (March 2009). 2009. Available online: https://www.cebm.net/2009/06/oxford-centre-evidence-based-medicine-levels-evidence-march-2009/ (accessed on 29 July 2018).
- Batla, A.; Adams, M.E.; Erro, R.; Ganos, C.; Balint, B.; Mencacci, N.E.; Bhatia, K.P. Cortical pencil lining in neuroferritinopathy: A diagnostic clue. Neurology 2015, 84, 1816–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, M.I.; Bruggemann, N.; Chana, P.; Venegas, P.; Kagi, M.; Parrao, T.; Orellana, P.; Garrido, C.; Rojas, C.V.; Hauke, J.; et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov. Disord. 2010, 25, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Crompton, D.E.; Birchall, D.; Jackson, M.J.; Coulthard, A.; Lombes, A.; Quinn, N.; Wills, A.; Fletcher, N.; Mottershead, J.P.; et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain 2007, 130, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Crosiers, D.; Ceulemans, B.; Meeus, B.; Nuytemans, K.; Pals, P.; van Broeckhoven, C.; Cras, P.; Theuns, J. Juvenile dystonia-parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Parkinsonism Relat. Disord. 2011, 17, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Di Fonzo, A.; Chien, H.F.; Socal, M.; Giraudo, S.; Tassorelli, C.; Iliceto, G.; Fabbrini, G.; Marconi, R.; Fincati, E.; Abbruzzese, G.; et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007, 68, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Eiberg, H.; Hansen, L.; Korbo, L.; Nielsen, I.M.; Svenstrup, K.; Bech, S.; Pinborg, L.H.; Friberg, L.; Hjermind, L.E.; Olsen, O.R.; et al. Novel mutation in ATP13A2 widens the spectrum of Kufor-Rakeb syndrome (PARK9). Clin. Genet. 2012, 82, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.K.; Donley, D.K. Pseudohypoparathyroidism, parkinsonism syndrome, with no basal ganglia calcification. J. Neurol. Neurosurg. Psychiatry 1988, 51, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Fekete, R. Late onset neurodegeneration with brain-iron accumulation presenting as parkinsonism. Case Rep. Neurol. Med. 2012, 2012, 387095. [Google Scholar] [CrossRef] [PubMed]
- Fonderico, M.; Laudisi, M.; Andreasi, N.G.; Bigoni, S.; Lamperti, C.; Panteghini, C.; Garavaglia, B.; Carecchio, M.; Emanuele, E.A.; Gian, L.F.; et al. Patient Affected by Beta-Propeller Protein-Associated Neurodegeneration: A Therapeutic Attempt with Iron Chelation Therapy. Front. Neurol. 2017, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Gore, E.; Appleby, B.S.; Cohen, M.L.; DeBrosse, S.D.; Leverenz, J.B.; Miller, B.L.; Siedlak, S.L.; Zhu, X.; Lerner, A.J. Clinical and imaging characteristics of late onset mitochondrial membrane protein-associated neurodegeneration (MPAN). Neurocase 2016, 22, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kruer, M.C.; Gregory, A.; Haack, T.B.; Kurian, M.A.; Houlden, H.H.; Anderson, J.; Boddaert, N.; Sanford, L.; Harik, S.I.; et al. Beta-Propeller protein-associated neurodegeneration: A new X-linked dominant disorder with brain iron accumulation. Brain 2013, 136, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Kitzler, H.H.; Pollack, T.; Biskup, S.; Kruger, S.; Funke, C.; Terrile, C.; Haack, T.B. A Case of Beta-propeller Protein-associated Neurodegeneration due to a Heterozygous Deletion of WDR45. Tremor Other Hyperkinet. Mov. (N. Y.) 2017, 7, 465. [Google Scholar] [CrossRef]
- Ichinose, Y.; Miwa, M.; Onohara, A.; Obi, K.; Shindo, K.; Saitsu, H.; Matsumoto, N.; Takiyama, Y. Characteristic MRI findings in beta-propeller protein-associated neurodegeneration (BPAN). Neurol. Clin. Pract. 2014, 4, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Klysz, B.; Skowronska, M.; Kmiec, T. Mitochondrial protein associated neurodegeneration—Case report. Neurol. Neurochir. Pol. 2014, 48, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Li, H.-F.; Zheng, Y.-C.; Wu, Z.-Y. FTL mutation in a Chinese pedigree with neuroferritinopathy. Neurol. Genet. 2016, 2, e74. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Oyama, G.; Yoshino, H.; Li, Y.; Matsushima, T.; Takeuchi, C.; Mochizuki, Y.; Mori-Yoshimura, M.; Murata, M.; Yamasita, C.; et al. High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol. Aging 2015, 36, 2004.e9–2004.e15. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Dogu, O.; Tufekcioglu, Z.; Diler, Y.; Saka, E.; Gultekin, M.; Kaleagasi, H.; Kuipers, D.; Graafland, J.; Breedveld, G.J.; et al. The p.Thr11Met mutation in c19orf12 is frequent among adult Turkish patients with MPAN. Parkinsonism Relat. Disord. 2017, 39, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Pearson, D.W.; Durward, W.F.; Fogelman, I.; Boyle, I.T.; Beastall, G. Pseudohypoparathyroidism presenting as severe Parkinsonism. Postgrad. Med. J. 1981, 57, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.; Poon, Y.-Y.; Naranian, T.; Fasano, A. SCL20A2 mutation mimicking fluctuating Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 39, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Scale, T.; Lewis, C.; Hedayat, A.; Bilal, M.; Wani, M. Cerebral calcification from Fahr’s disease with co-existing haemochromatosis. Prog. Neurol. Psychiatry 2014, 18, 14–16. [Google Scholar] [CrossRef]
- Song, C.-Y.; Zhao, Z.-X.; Li, W.; Sun, C.-C.; Liu, Y.-M. Pseudohypoparathyroidism with basal ganglia calcification: A case report of rare cause of reversible parkinsonism. Medicine (Baltim.) 2017, 96, e6312. [Google Scholar] [CrossRef] [PubMed]
- Vroegindeweij, L.H.P.; Langendonk, J.G.; Langeveld, M.; Hoogendoorn, M.; Kievit, A.J.A.; Di Raimondo, D.; Wilson, J.H.P.; Boon, A.J.W. New insights in the neurological phenotype of aceruloplasminemia in Caucasian patients. Parkinsonism Relat. Disord. 2017, 36, 33–40. [Google Scholar] [CrossRef] [PubMed]

| Year of Publication Range | 1981–2018 |
|---|---|
| Number of Publications per Decade | |
| Before 1990 | 3 |
| 1991–2000 | 3 |
| 2001–2010 | 12 |
| 2011–2018 | 32 |
| Type of Study | |
| Cohort study | 1 |
| Cross-sectional study | 9 |
| Case reports/series | 40 |
| Condition | Metal Involved | Brain Region Implicated | Total No. of Papers (No. Typical, No. Atypical) | No. (% Total) of Male and Female Patients Described | Average Age of Patients (Years, Mean ± Standard Error) |
|---|---|---|---|---|---|
| Panthonase Kinase associated Neurodegeneration (PKAN) | Iron | Basal ganglia (GP, SN) | 11 (3;11) | 49 M (57.6%) 36 F (42.4%) | 33 ± 3.8 |
| Hereditary Haemochromatosis | Iron | - | 7 (4;3) | 10 M (71.4%) 4 F (28.6%) | 53 ± 3.3 |
| Wilson’s Disease | Copper | Basal ganglia (PMN, GP) | 6 (6;4) | 3 M (33.3%) 6 F (66.6%) | 46 ± 6.8 |
| Beta-Propeller Protein-Associated Neurodegeneration (BPAN) | Iron | Basal ganglia (SN, GP) | 5 (1,4) | 3 M (9.7%) 28 F (90.3%) | 27 ± 1.1 |
| Kufor-Rakeb Syndrome | Iron | Basal Ganglia (SN, GP) | 5 (1;5) | 10 M (90.9%) 1 F (9.1%) | 13 ± 0.7 |
| Mitochondrial-Membrane Protein-Associated Neurodegeneration (MPAN) | Iron | Basal Ganglia (SN, GP) | 3 (0;3) | 1 M (50.0%) 1 F (50.0%) | 25 ± 10.0 |
| Neuroferritinopathy | Iron | Cerebellum, Basal ganglia, motor cortex | 3 (0;3) | 5 F (100.0%) | 61± 17.5 |
| PLA2G6-Associated Neurodegeneration (PLAN) | Iron | Basal ganglia (SN, GP) | 3 (2;1) | 3 M (60.0%) 2 F (40.0%) | 24 ± 5.2 |
| Pseudohypoparathyroidism | Calcium | Basal ganglia, deep white matter | 3 (0;3) | 1 M (33.3%) 2 F (66.6%) | 43 ± 11.8 |
| Fahr Disease | Calcium | Basal ganglia, deep white matter, cerebellum | 2 (1;1) | 1 M (50.0%) 1 F (50.0%) | 56 ± 6.0 |
| Aceruloplasminemia | Iron | Basal ganglia | 1 (0;1) | 4 M (80.0%) 1 F (20.0%) | NA |
| Neurodegenerative Brain Iron Accumulation (NBIA), Unknown Type | Iron | - | 1 (0;1) | 1 M (100.0%) | 73 ± 0.0 |
| Total | - | - | 50 (16;38) | 86 M (49.7%) 87 F (50.3%) | 35 ± 1.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botsford, E.; George, J.; Buckley, E.E. Parkinson’s Disease and Metal Storage Disorders: A Systematic Review. Brain Sci. 2018, 8, 194. https://doi.org/10.3390/brainsci8110194
Botsford E, George J, Buckley EE. Parkinson’s Disease and Metal Storage Disorders: A Systematic Review. Brain Sciences. 2018; 8(11):194. https://doi.org/10.3390/brainsci8110194
Chicago/Turabian StyleBotsford, Edward, Jayan George, and Ellen E. Buckley. 2018. "Parkinson’s Disease and Metal Storage Disorders: A Systematic Review" Brain Sciences 8, no. 11: 194. https://doi.org/10.3390/brainsci8110194
APA StyleBotsford, E., George, J., & Buckley, E. E. (2018). Parkinson’s Disease and Metal Storage Disorders: A Systematic Review. Brain Sciences, 8(11), 194. https://doi.org/10.3390/brainsci8110194