Intersection of Brain Development and Paediatric Diffuse Midline Gliomas: Potential Role of Microenvironment in Tumour Growth


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Development
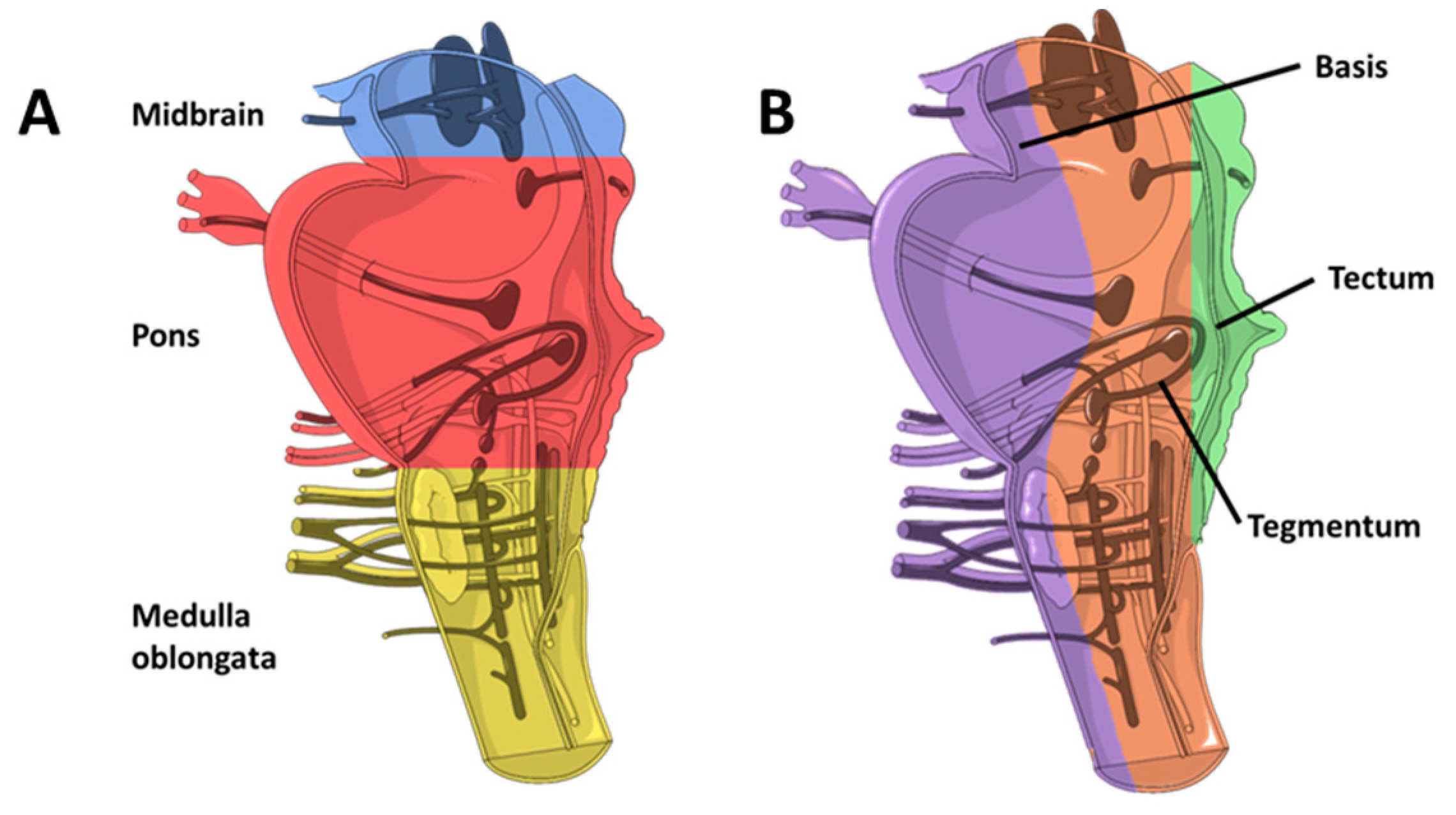
2.1. Brainstem
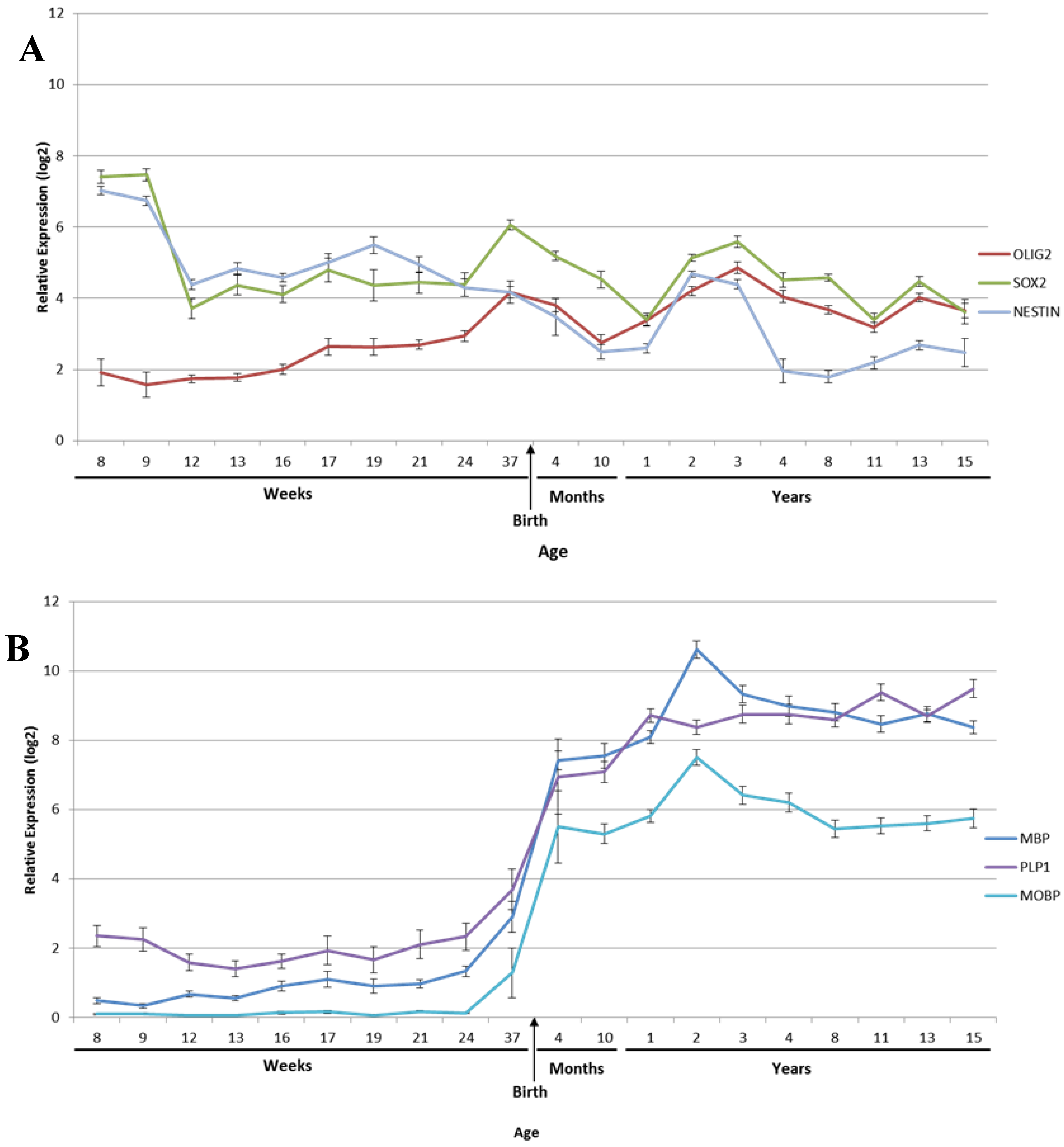
2.2. Myelination
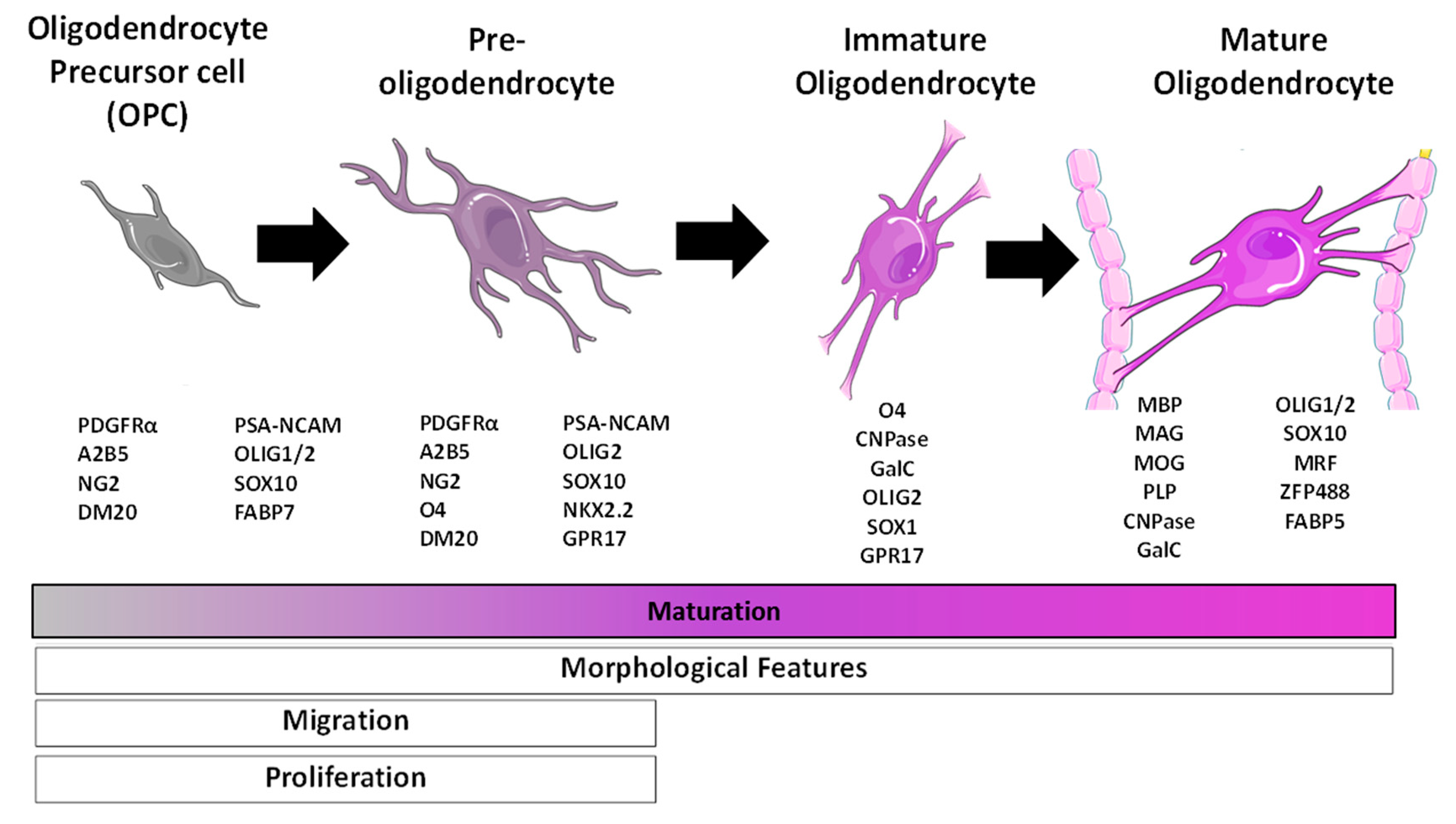
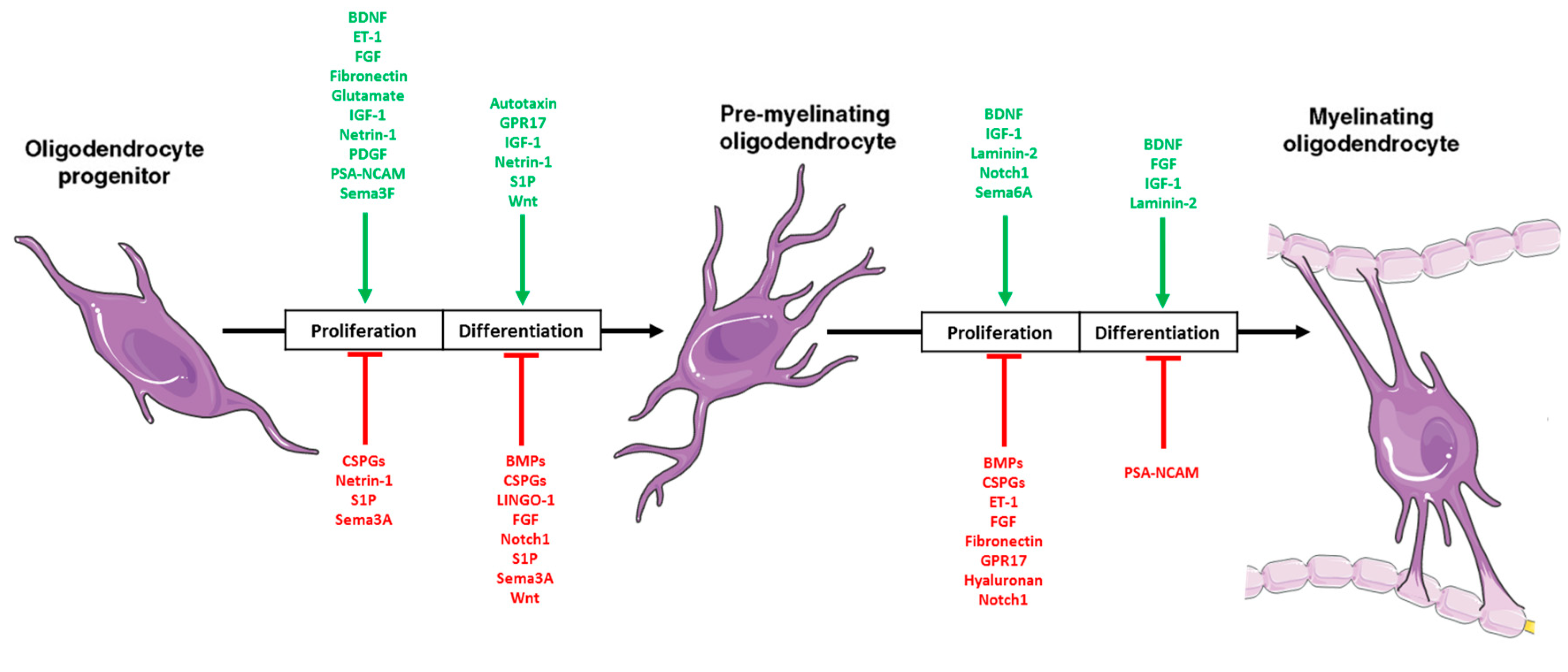
2.3. Oligodendrocytes
2.4. The Developing Microenvironment: Intersection with DIPG Microenvironment
3. Cell of Origin
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ballester, L.Y.; Wang, Z.; Shandilya, S.; Miettinen, M.; Burger, P.C.; Eberhart, C.G.; Rodriguez, F.J.; Raabe, E.; Nazarian, J.; Warren, K.; et al. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am. J. Surg. Pathol. 2013, 37, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Ellison, D.W.; Gutmann, D.H. Pediatric gliomas as neurodevelopmental disorders. Glia 2016, 64, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Sobo, M.; Lee, K.; Senthil Kumar, S.; White, A.R.; Mender, I.; Fuller, C.; Chow, L.M.; Fouladi, M.; Shay, J.W.; et al. Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Mol. Cancer Ther. 2018, 17, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, P.M.; Ariza, C.B.; Ishibashi, C.M.; Fujita, T.C.; Banin-Hirata, B.K.; Oda, J.M.; Watanabe, M.A. Role of CXCL12 and CXCR4 in normal cerebellar development and medulloblastoma. Int. J. Cancer 2016, 138, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Garancher, A.; Ramaswamy, V.; Wechsler-Reya, R.J. Medulloblastoma: From Molecular Subgroups to Molecular Targeted Therapies. Annu. Rev. Neurosci. 2018, 41, 207–232. [Google Scholar] [CrossRef] [PubMed]
- Van Zanten, S.E.M.V.; Baugh, J.; Chaney, B.; de Jongh, D.; Aliaga, E.S.; Barkhof, F.; Noltes, J.; De Wolf, R.; Van Dijk, J.; Cannarozzo, A.; et al. Development of the SIOPE DIPG network, registry and imaging repository: A collaborative effort to optimize research into a rare and lethal disease. J. Neurooncol. 2017, 1–12. [Google Scholar] [CrossRef]
- El-Khouly, F.E.; van Vuurden, D.G.; Stroink, T.; Hulleman, E.; Kaspers, G.J.L.; Hendrikse, N.H.; Veldhuijzen van Zanten, S.E.M. Effective drug delivery in diffuse intrinsic pontine glioma: A theoretical model to identify potential candidates. Front. Oncol. 2017, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Karajannis, M.A.; Jones, D.T.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro. Oncol. 2016, 19, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J. Pediatric high grade glioma: A review and update on tumor clinical characteristics and biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Johung, T.B.; Monje, M. Diffuse Intrinsic Pontine Glioma: New Pathophysiological Insights and Emerging Therapeutic Targets. Curr. Neuropharmacol. 2017, 15, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Karajannis, M.A. Pediatric Brain Tumors: An Update. Curr Probl. Pediatr. Adolesc. Heal. Care 2016, 46, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E. Diffuse intrinsic pontine glioma: Poised for progress. Front. Oncol. 2012, 2, 205. [Google Scholar] [CrossRef] [PubMed]
- Vanan, M.I.; Eisenstat, D.D. DIPG in Children—What Can We Learn from the Past? Front. Oncol. 2015, 5, 237. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, K.M.; Hoeman, C.M.; Becher, O.J. Children are not just little adults: Recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr. Res. 2014, 75, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.J.; Jabado, N.; Grill, J. Diffuse intrinsic pontine gliomas—current management and new biologic insights. Is there a glimmer of hope? Neuro. Oncol. 2017, 19, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavle, A.; Chintagumpala, M. Pediatric high-grade glioma: A review of biology, prognosis, and treatment. J. Radiat. Oncol. 2018, 7, 7–15. [Google Scholar] [CrossRef]
- Creasy, C.L. Untangling the role of mutant histone H3 in diffuse intrinsic pontine glioma. Nat. Med. 2017, 23, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; David James, C.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buczkowicz, P.; Hawkins, C. Pathology, Molecular Genetics, and Epigenetics of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.R.; Vinci, M.; Bullock, A.N.; Jones, C. ACVR1 mutations in DIPG: Lessons learned from FOP. Cancer Res. 2014, 74, 4565–4570. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.; et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl. Acad. Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-L.; Neilsen, C.V.; Bryce, P.J. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE 2010, 5, e11944. [Google Scholar] [CrossRef] [PubMed]
- Berlow, N.E.; Svalina, M.N.; Quist, M.J.; Settelmeyer, T.P.; Zherebitskiy, V.; Kogiso, M.; Qi, L.; Du, Y.; Hawkins, C.E.; Hulleman, E.; et al. IL-13 receptors as possible therapeutic targets in diffuse intrinsic pontine glioma. PLoS ONE 2018, 13, e0193565. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales La Madrid, A.; Hashizume, R.; Kieran, M.W. Future Clinical Trials in DIPG: Bringing Epigenetics to the Clinic. Front. Oncol. 2015, 5, 148. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, M.; Dubois, J.; Yu, Q.; Mukherjee, P.; Huang, H. Delineation of early brain development from fetuses to infants with diffusion MRI and beyond. Neuroimage 2018. [Google Scholar] [CrossRef] [PubMed]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef] [PubMed]
- Laug, D.; Glasgow, S.M.; Deneen, B. A glial blueprint for gliomagenesis. Nat Rev Neurosci 2018, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Rowitch, D.H.; Kriegstein, A.R. Developmental genetics of vertebrate glial-cell specification. Nature 2010, 468, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.C.; Lindquist, R.A.; Nguyen, T.; Sanai, N.; Barkovich, A.J.; Huang, E.J.; Rowitch, D.H.; Alvarez-Buylla, A. Postnatal growth of the human pons: A morphometric and immunohistochemical analysis. J. Comp. Neurol. 2015, 523, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Mishra, V.; Ouyang, M.; Peng, Q.; Slinger, M.; Liu, S.; Huang, H. Human Fetal Brain Connectome: Structural Network Development from Middle Fetal Stage to Birth. Front. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tamnes, C.K.; Roalf, D.R.; Goddings, A.-L.; Lebel, C. Diffusion MRI of white matter microstructure development in childhood and adolescence: Methods, challenges and progress. Dev. Cogn. Neurosci. 2017, 33, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, L.M.; van den Heuvel, M.P.; Oranje, B.; Giedd, J.N.; Durston, S.; Peper, J.S.; Brown, T.T.; Crone, E.A. A multisample study of longitudinal changes in brain network architecture in 4–13-year-old children. Hum. Brain Mapp. 2018, 39, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Rollins, N.K.; Booth, T.N.; Chahrour, M.H. Variability of Ponto-cerebellar Fibers by Diffusion Tensor Imaging in Diverse Brain Malformations. J. Child Neurol. 2017, 32, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, J.H.; Knickmeyer, R.C.; Gao, W. Imaging structural and functional brain development in early childhood. Nat. Rev. Neurosci. 2018, 19, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Dale, P.; George, J.A.; David, F.; William, C.H.; Anthony-Samuel, L.; Leonard, E.W. Neuroscience, 5th ed.; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
- Ngeles Fernández-Gil, M.; Palacios-Bote, R.; Leo-Barahona, M.; Mora-Encinas, J.P. Anatomy of the brainstem: A gaze into the stem of life. Semin. Ultrasound, CT MRI. 2010, 31, 196–219. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Millen, K.J.; Dobyns, W.B. A developmental and genetic classification for midbrain-hindbrain malformations. Brain 2009, 132, 3199–3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jane, B.R.; Lisa, A.U.; Michael, L.C.; Steven, A.W.; Peter, V.; Minorsky, R.B.J. Campbell Biology, 11th ed.; Pearson: London, UK, 2011. [Google Scholar]
- Hatta, T.; Satow, F.; Hatta, J.; Hashimoto, R.; Udagawa, J.; Matsumoto, A.; Otani, H. Development of the pons in human fetuses. Congenit. Anom. 2007, 47, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Yung, A.R.; Druckenbrod, N.R.; Cloutier, J.-F.; Wu, Z.; Tessier-Lavigne, M.; Goodrich, L.V. Netrin-1 Confines Rhombic Lip-Derived Neurons to the CNS. Cell Rep. 2018, 22, 1666–1680. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.F.; Maheshwari, U.; Rijli, F.M. The Long Journey of Pontine Nuclei Neurons: From Rhombic Lip to Cortico-Ponto-Cerebellar Circuitry. Front. Neural Circuits 2017, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Snaidero, N.; Simons, M. The logistics of myelin biogenesis in the central nervous system. GLIA 2017, 65, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.-A.; Werner, H.B. Myelination of the Nervous System: Mechanisms and Functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Moon, W.J.; Han, J.J. Quantification of myelin in children using multiparametric quantitative MRI: A pilot study. Neuroradiology 2017, 59, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Deoni, S.C.L.; Mercure, E.; Blasi, A.; Gasston, D.; Thomson, A.; Johnson, M.; Williams, S.C.R.; Murphy, D.G.M. Mapping infant brain myelination with magnetic resonance imaging. J. Neurosci. 2011, 31, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Purger, D.; Gibson, E.M.; Monje, M. Myelin plasticity in the central nervous system. Neuropharmacology 2016, 110, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Jahn, O.; Tenzer, S.; Werner, H.B. Myelin proteomics: Molecular anatomy of an insulating sheath. Mol. Neurobiol. 2009, 40, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Van Tilborg, E.; de Theije, C.G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. GLIA 2018, 66, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Budday, S.; Steinmann, P.; Kuhl, E. Physical biology of human brain development. Front. Cell Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbein, N.J.; Prados, M.D.; Wara, W.; Russo, C.; Edwards, M.S.; Barkovich, A.J. Radiologic classification of brain stem tumors: Correlation of magnetic resonance imaging appearance with clinical outcome. Pediatr. Neurosurg. 1996, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, H.; Goto, N.; Nara, T. Development of the human pontine nuclei: A morphometric study. Dev. Brain Res. 1992, 65, 13–20. [Google Scholar] [CrossRef]
- Anderson, J.L.; Muraleedharan, R.; Oatman, N.; Klotter, A.; Sengupta, S.; Waclaw, R.R.; Wu, J.; Drissi, R.; Miles, L.; Raabe, E.H.; et al. The transcription factor Olig2 is important for the biology of diffuse intrinsic pontine gliomas. Neuro. Oncol. 2017, 19, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, R.A.; Guinto, C.D.; Rodas-Rodriguez, J.L.; Fuentealba, L.C.; Tate, M.C.; Rowitch, D.H.; Alvarez-Buylla, A. Identification of proliferative progenitors associated with prominent postnatal growth of the pons. Nat Commun. 2016, 7, 11628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, C.W.; Monje, M. Wrapped to Adapt: Experience-Dependent Myelination. Neuron 2017, 95, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D. A new mechanism of nervous system plasticity: Activity-dependent myelination. Nat. Rev. Neurosci. 2015, 16, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Aktas, O.; Hartung, H.P.; Küry, P. The complex world of oligodendroglial differentiation inhibitors. Ann. Neurol. 2011, 69, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Koreman, E.; Sun, X.; Lu, Q.R. Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol. Cell Neurosci. 2018, 87, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Marie, C.; Clavairoly, A.; Frah, M.; Hmidan, H.; Yan, J.; Zhao, C.; Van Steenwinckel, J.; Daveau, R.; Zalc, B.; Hassan, B.; et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. USA 2018, 115, E8246–E8255. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Moyon, S.; Hernandez, M.; Casaccia, P. Epigenetic control of oligodendrocyte development: Adding new players to old keepers. Curr. Opin. Neurobiol. 2016, 39, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta—Mol. Cell Res. 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Ribeiro, F.; Sebastião, A.; Xapelli, S. Neuroinflammatory modulators of oligodendrogenesis. Neuroimmunol. Neuroinflammation 2015, 2, 263. [Google Scholar]
- Marques, S.; van Bruggen, D.; Vanichkina, D.P.; Floriddia, E.M.; Munguba, H.; Väremo, L.; Giacomello, S.; Falcão, A.M.; Meijer, M.; Björklund, Å.K.; et al. Transcriptional Convergence of Oligodendrocyte Lineage Progenitors during Development. Dev. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Emery, B.; Lu, Q.R. Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, C.; Bertalot, T.; Zusso, M.; Skaper, S.D.; Giusti, P. Systematic Review of Pharmacological Properties of the Oligodendrocyte Lineage. Front. Cell Neurosci. 2016, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.B.; Armstrong, R.C. Intracellular signals and cytoskeletal elements involved in oligodendrocyte progenitor migration. GLIA 1999, 26, 22–35. [Google Scholar] [CrossRef]
- Hernandez, M.; Casaccia, P. Interplay between transcriptional control and chromatin regulation in the oligodendrocyte lineage. GLIA 2015, 63, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, N.A.; Fuss, B. Extracellular cues influencing oligodendrocyte differentiation and (re)myelination. Exp. Neurol. 2016, 283, 512–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Azzarelli, R.; Simons, B.D.; Philpott, A. The developmental origin of brain tumours: A cellular and molecular framework. Development 2018, 145, dev162693. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Hubert, C.G.; Miller, T.E.; Taylor, M.D.; Rich, J.N. An epigenetic gateway to brain tumor cell identity. Nat. Neurosci. 2016, 19, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.L.; Nagaraja, S.; Filbin, M.G.; Suvà, M.L.; Vogel, H.; Monje, M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Scutti, J.A.B. Importance of immune monitoring approaches and the use of immune checkpoints for the treatment of diffuse intrinsic pontine glioma: From bench to clinic and vice versa (Review). Int. J. Oncol. 2018, 52, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial Dynamics During Human Brain Development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef] [PubMed]
- Blehm, B.H.; Jiang, N.; Kotobuki, Y.; Tanner, K. Deconstructing the role of the ECM microenvironment on drug efficacy targeting MAPK signaling in a pre-clinical platform for cutaneous melanoma. Biomaterials 2015, 56, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, Z.; Zhang, M.; Piao, Y.; Chen, L.; Liang, H.; Wei, Y.; Hu, Z.; Zhao, L.; Teng, L.; et al. H3 K27M-mutant diffuse midline gliomas in different anatomical locations. Hum. Pathol. 2018, 78, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Lorger, M. Tumor microenvironment in the brain. Cancers (Basel) 2012, 4, 218–243. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E. Beyond the Blood: Brain Barrier: The Importance of Central Nervous System (CNS) Pharmacokinetics for the Treatment of CNS Tumors, Including Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Plessier, A.; LeDret, L.; Varlet, P.; Beccaria, K.; Lacombe, J.; Meriaux, S.; Geffroy, F.; Fiette, L.; Flamant, P.; Chretien, F.; et al. New in vivo avatars of diffuse intrinsic pontine gliomas (DIPG) from stereotactic biopsies performed at diagnosis. Oncotarget 2017, 8, 52543–52559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, K.A.; Pollack, I.F.; Reid, J.M.; Adamson, P.C.; Ames, M.M.; Vezina, G.; Blaney, S.; Ivy, P.; Zhou, T.; Krailo, M.; et al. Motexafin gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: A Children’s Oncology Group phase I study. Neuro-Oncology 2008, 10, 752–758. [Google Scholar] [CrossRef] [PubMed]
- McCully, C.M.; Pastakia, D.; Bacher, J.; Thomas, M.L.; Steffen-Smith, E.A.; Saleem, K.; Murphy, R.F.; Walbridge, S.; Brinster, L.; Widemann, B.C.; et al. Model for concomitant microdialysis sampling of the pons and cerebral cortex in rhesus macaques (Macaca mulatta). Comp. Med. 2013, 63, 355–360. [Google Scholar] [PubMed]
- Liddelow, S.A. Development of the choroid plexus and blood-CSF barrier. Front. Neurosci. 2015, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.J.; Haines, D.E. The Ventricles, Choroid Plexus, and Cerebrospinal Fluid. In Fundamental Neuroscience for Basic and Clinical Applications, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 93–106. [Google Scholar]
- League-Pascual, J.C.; Lester-McCully, C.M.; Shandilya, S.; Ronner, L.; Rodgers, L.; Cruz, R.; Peer, C.J.; Figg, W.D.; Warren, K.E. Plasma and cerebrospinal fluid pharmacokinetics of select chemotherapeutic agents following intranasal delivery in a non-human primate model. J. Neurooncol. 2017, 132, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Piunti, A.; Lulla, R.R.; Qi, J.; Horbinski, C.M.; Tomita, T.; James, C.D.; Shilatifard, A.; Saratsis, A.M. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol. Commun. 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.; Monje, M. An Active Role for Neurons in Glioma Progression: Making Sense of Scherer’s Structures. Neuro. Oncol. 2018, 20, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Ligon, K.L.; Alberta, J.A.; Kho, A.T.; Weiss, J.; Kwaan, M.R.; Nutt, C.L.; Louis, D.N.; Stiles, C.D.; Rowitch, D.H. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J. Neuropathol. Exp. Neurol. 2004, 63, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Wang, H.; Liu, S.; Niu, J.; Wang, L.; He, Y.; Etxeberria, A.; Chan, J.R.; Xiao, L. Stage-Specific Deletion of Olig2 Conveys Opposing Functions on Differentiation and Maturation of Oligodendrocytes. J. Neurosci. 2013, 33, 8454–8462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castel, D.; Philippe, C.; Calmon, R.; Ludivine, Dret L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loveson, K.F.; Fillmore, H.L. Intersection of Brain Development and Paediatric Diffuse Midline Gliomas: Potential Role of Microenvironment in Tumour Growth. Brain Sci. 2018, 8, 200. https://doi.org/10.3390/brainsci8110200
Loveson KF, Fillmore HL. Intersection of Brain Development and Paediatric Diffuse Midline Gliomas: Potential Role of Microenvironment in Tumour Growth. Brain Sciences. 2018; 8(11):200. https://doi.org/10.3390/brainsci8110200
Chicago/Turabian StyleLoveson, Katie F., and Helen L. Fillmore. 2018. "Intersection of Brain Development and Paediatric Diffuse Midline Gliomas: Potential Role of Microenvironment in Tumour Growth" Brain Sciences 8, no. 11: 200. https://doi.org/10.3390/brainsci8110200
APA StyleLoveson, K. F., & Fillmore, H. L. (2018). Intersection of Brain Development and Paediatric Diffuse Midline Gliomas: Potential Role of Microenvironment in Tumour Growth. Brain Sciences, 8(11), 200. https://doi.org/10.3390/brainsci8110200