High and Low Levels of an NTRK2-Driven Genetic Profile Affect Motor- and Cognition-Associated Frontal Gray Matter in Prodromal Huntington’s Disease

 , ,
, ,  and
and
Abstract
:
1. Introduction
1.1. Huntington’s Disease
1.2. Effects of Multiple Genes and Variants
1.3. Benefits of Multivariate Methods
1.4. Parallel Independent Component Analysis (pICA)
1.5. Brain-Derived Neurotrophic Factor (BDNF)-Signaling Genes (a Candidate Pathway)
1.6. The Present Study
2. Materials and Methods
2.1. Participants
2.2. Data Availability
2.3. Cognitive and Motor Variables
2.4. Genomic Data Preprocessing
2.5. Imaging Data Collection
2.6. Imaging Data Preprocessing
2.7. Parallel ICA with Reference (pICAr)
2.8. SNP and GMC Correlations with Clinical Variables
2.9. Associations of Top-Weighted Component SNPs with Clinical Variables
2.10. Confirmation of Significant Results
2.11. Regression Influence Plot
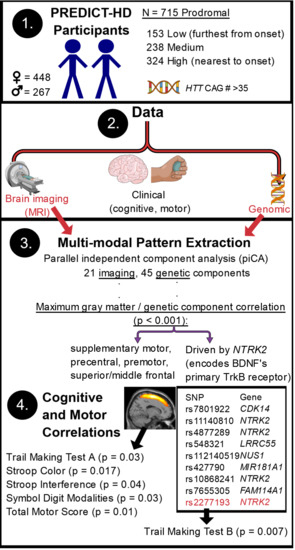
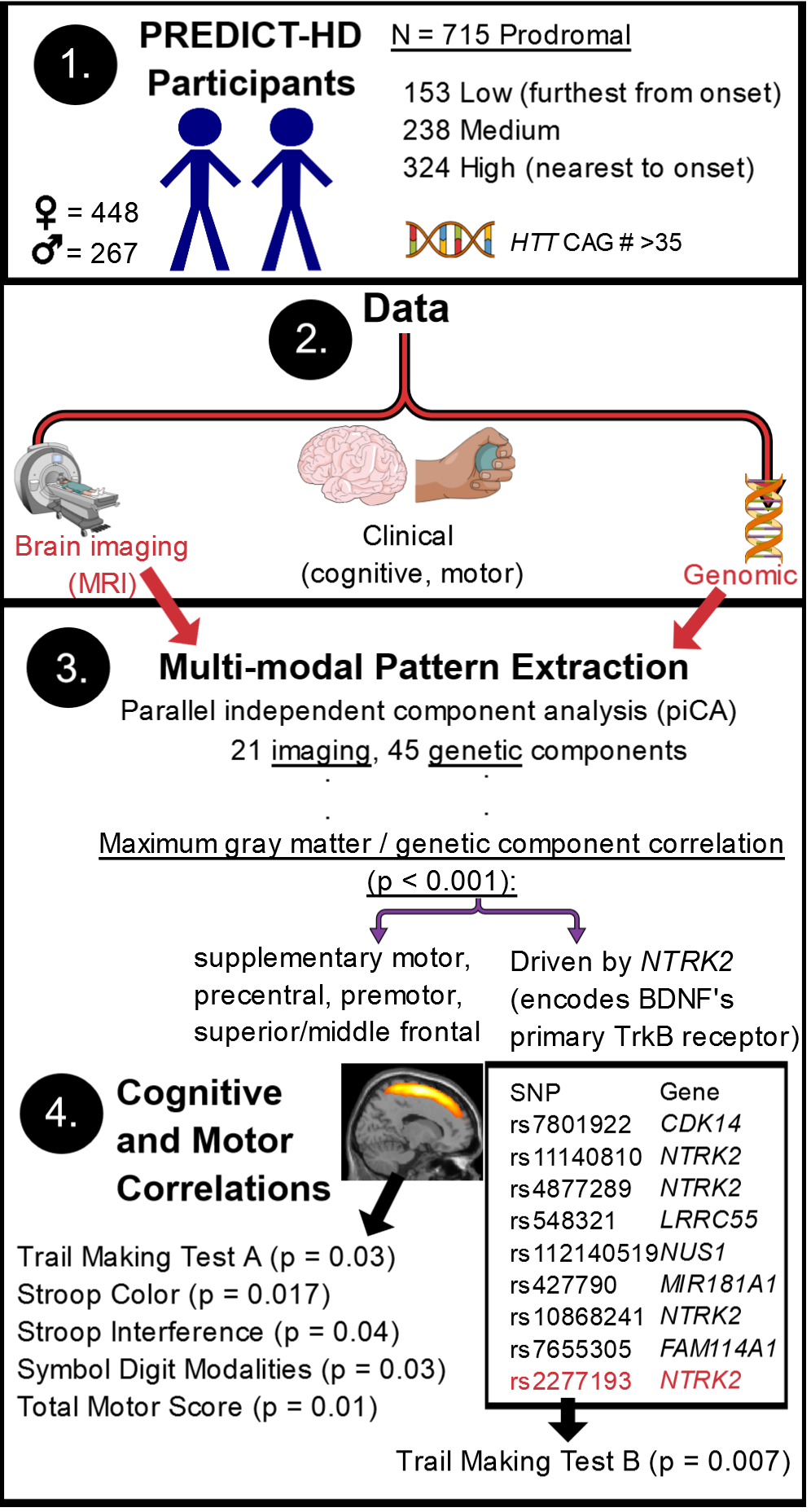
3. Results
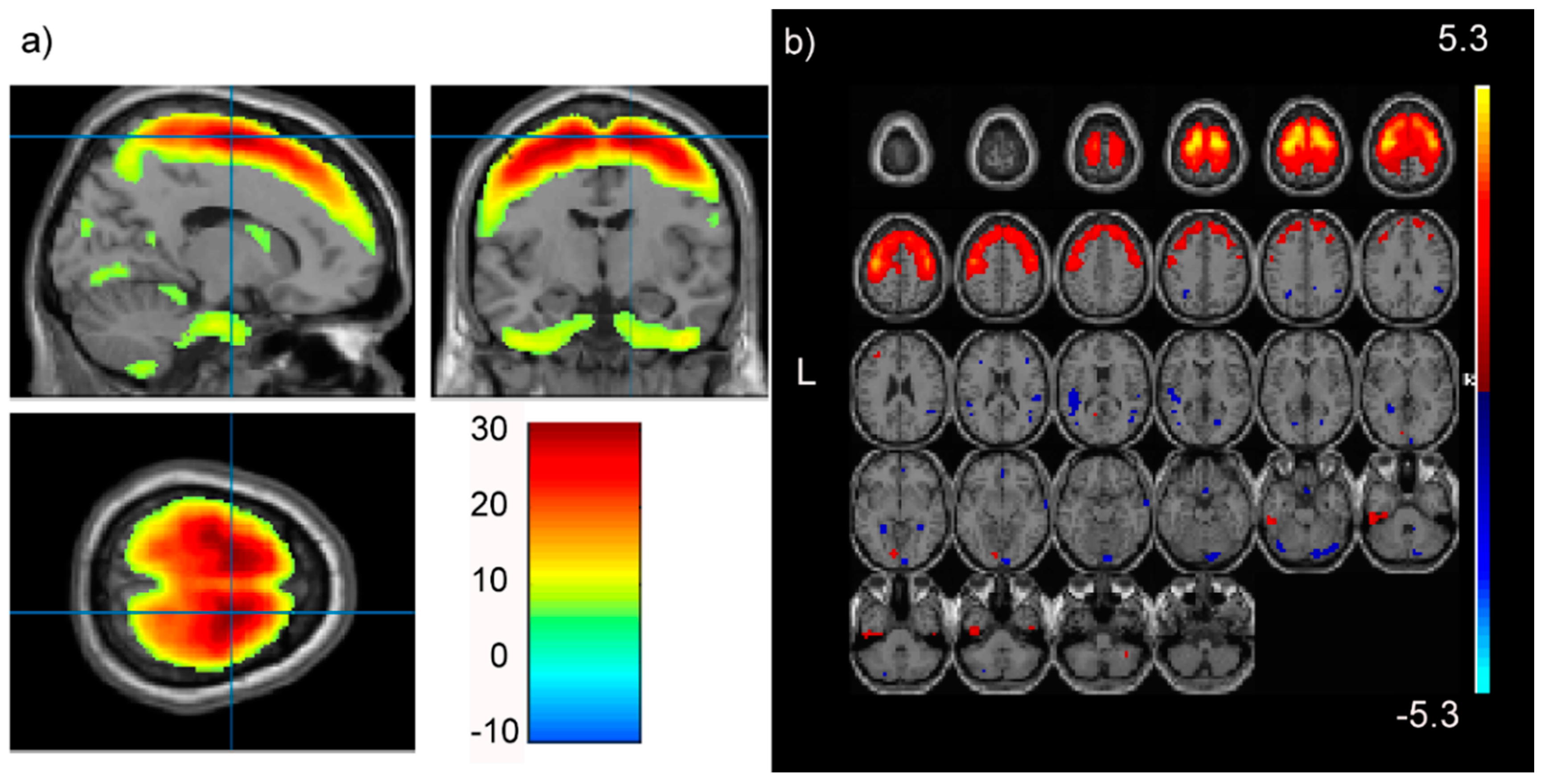
3.1. pICAr
3.2. SNP and GMC Correlations with Clinical Variables
3.3. Confirmation of Significant Results
3.4. Associations of Top-Weighted Component SNPs with Clinical Variables
4. Discussion
4.1. High or Low Levels of the NTRK2 SNP Profile Affect Prodromal Frontal GMC
4.2. The SNP-GMC Component Correlation is Not an Aggregate Effect of the Entire SNP Component
4.3. The NTRK2-Associated Frontal Gray Matter Profile Is Related to Prodromal Cognitive and Motor Functioning
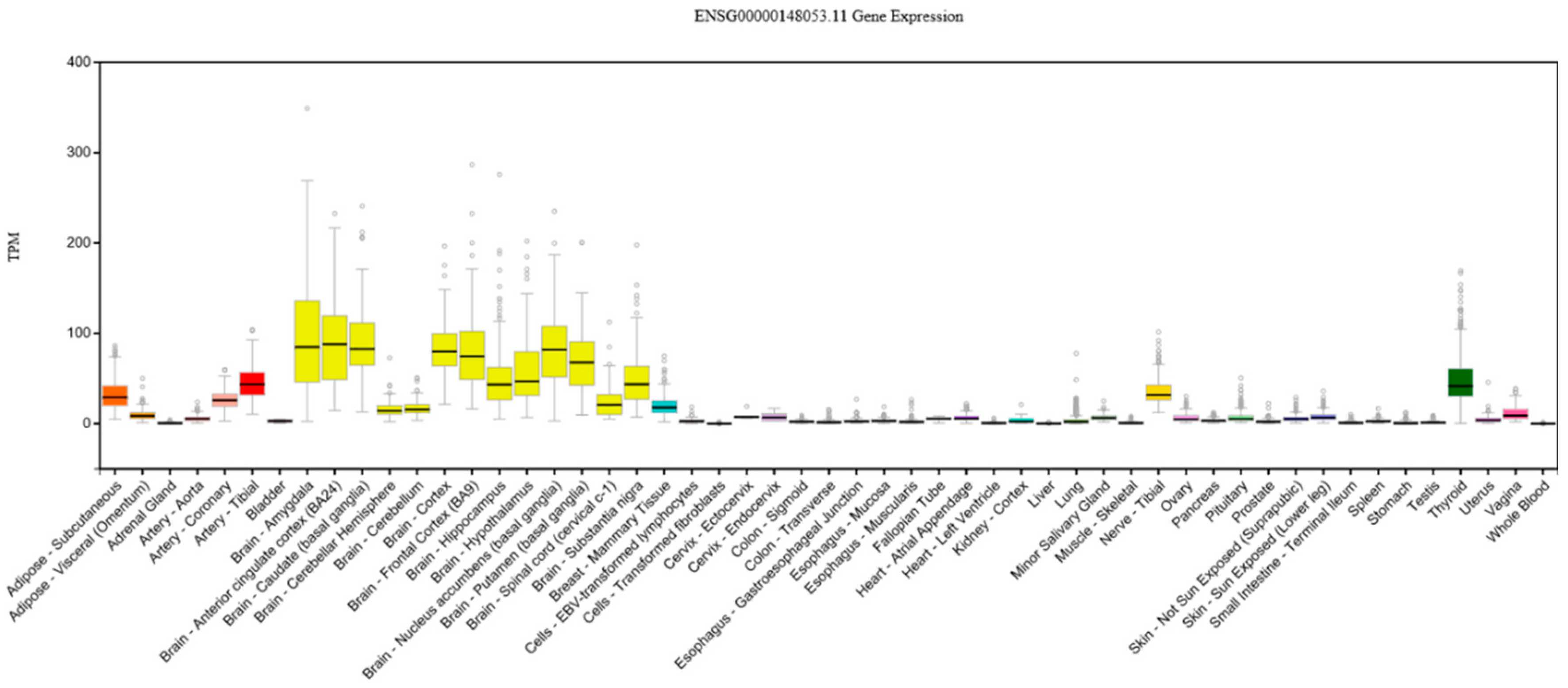
4.4. Top Contributing NTRK2 SNPs
4.5. Top Contributing SNPs Outside of NTRK2
4.6. Influence of HTT CAG-Repeats
5. Conclusions
- In this PREDICT-HD prodromal cohort, high or low levels of an SNP profile with substantial contributions from NTRK2 were associated with a GMC profile representing the supplementary and primary motor cortex, as well as other frontal regions (positive correlation).
- This frontal gray matter profile was associated with cognitive and motor performance in this population.
- The SNP component was not significantly associated with clinical functioning, but one of its top NTRK2 SNPs had a protective association with performance on TMTB, a measure of task switching and visual attention, indicating some influence on cognition.
- Correlations between the SNP component and clinical/GMC variables were mainly due to top contributing SNPs, rather than being an aggregate effect of the entire SNP component.
- Top component SNPs have been associated with active histone modifications in the brain (cingulate gyrus, inferior temporal, angular gyrus, DLPFC, caudate, hippocampus, and substantia nigra) and altered regulatory motifs (especially the glucocorticoid receptor (GR) and zinc finger protein (Zec)).
- Top NTRK2 SNPs in the component were close to alternative stop codons and reportedly regulated genes implicated in diverse functions (especially in the frontal cortex, thalamus, putamen, and cerebellum).
6. Limitations
7. Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Paulsen, J.S.; Long, J.D.; Johnson, H.J.; Aylward, E.H.; Ross, C.A.; Williams, J.K.; Nance, M.A.; Erwin, C.J.; Westervelt, H.J.; Harrington, D.L. Clinical and biomarker changes in premanifest huntington disease show trial feasibility: A decade of the predict-hd study. Front. Aging Neurosci. 2014, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Limas, D.E.; Jensen, K.; Fernandez-Funez, P. Drosophila models of proteinopathies: The little fly that could. Curr. Pharm. Des. 2012, 18, 1108–1122. [Google Scholar] [PubMed]
- Long, J.D.; Paulsen, J.S. PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Multivariate prediction of motor diagnosis in huntington’s disease: 12 years of predict-hd. Mov. Disord. 2015, 30, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Scharfman, H.E. Brain-derived neurotrophic factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Terauchi, A.; Nakamura, K.; Higo, T.; Nukina, N.; Matsumoto, N.; Hisatsune, C.; Nakamura, T.; Mikoshiba, K. Aberrant calcium signaling by transglutaminase-mediated posttranslational modification of inositol 1,4,5-trisphosphate receptors. Proc. Natl. Acad. Sci. USA 2014, 111, E3966–E3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Cowan, C.M.; Zhang, L.Y.; Hayden, M.R.; Raymond, L.A. Interaction of postsynaptic density protein-95 with nmda receptors influences excitotoxicity in the yeast artificial chromosome mouse model of huntington’s disease. J. Neurosci. 2009, 29, 10928–10938. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.J.; Johnson, R.; Zuccato, C.; Bithell, A.; Cattaneo, E. The role of rest in transcriptional and epigenetic dysregulation in huntington’s disease. Neurobiol. Dis. 2010, 39, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Mao, P.; Manczak, M. Mitochondrial structural and functional dynamics in huntington’s disease. Brain Res. Rev. 2009, 61, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef] [PubMed]
- Pearlson, G.D.; Liu, J.; Calhoun, V.D. An introductory review of parallel independent component analysis (p-ica) and a guide to applying p-ica to genetic data and imaging phenotypes to identify disease-associated biological pathways and systems in common complex disorders. Front. Genet. 2015, 6, 276. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Calhoun, V.D.; Ulloa, A.E.; Liu, J. Parallel ica with multiple references: A semi-blind multivariate approach. In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Chicago, IL, USA, 26–30 August 2014; pp. 6659–6662. [Google Scholar]
- Gupta, C.N.; Chen, J.; Liu, J.; Damaraju, E.; Wright, C.; Perrone-Bizzozero, N.I.; Pearlson, G.; Luo, L.; Michael, A.M.; Turner, J.A. Genetic markers of white matter integrity in schizophrenia revealed by parallel ica. Front. Hum. Neurosci. 2015, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Laforce, R.; Tosun, D.; Ghosh, P.; Lehmann, M.; Madison, C.M.; Weiner, M.W.; Miller, B.L.; Jagust, W.J.; Rabinovici, G.D. Parallel ica of fdg-pet and pib-pet in three conditions with underlying alzheimer’s pathology. Neuroimage Clin. 2014, 4, 508–516. [Google Scholar] [CrossRef] [PubMed]
- BDNF in Huntington’s Disease: Role in Pathogenesis and Treatment. Available online: http://cdn.intechopen.com/pdfswm/28345.pdf (accessed on 22 June 2018).
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J. Detection of huntington’s disease decades before diagnosis: The predict-hd study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.S.; Long, J.D.; Ross, C.A.; Harrington, D.L.; Erwin, C.J.; Williams, J.K.; Westervelt, H.J.; Johnson, H.J.; Aylward, E.H.; Zhang, Y. Prediction of manifest huntington’s disease with clinical and imaging measures: A prospective observational study. Lancet Neurol. 2014, 13, 1193–1201. [Google Scholar] [CrossRef]
- Wechsler, D. Manual for the Wechsler Adult Intelligence Scale—Revised; Psychological Corporation: New York, NY, USA, 1981. [Google Scholar]
- Lezak, M.D.; Howieson, D.; Loring, D. Neuropsychological Assessment, 4th ed.; Oxford University Press: New York, NY, USA, 2004. [Google Scholar]
- Smith, A. Symbol Digit Modalities Test (SDMT) Manual (Revised); Western Psychological Services: Los Angeles, CA, USA, 1982. [Google Scholar]
- Stroop, J.R. Studies of interference in serial verbal reactions. J. Exp. Psychol. 1935, 18, 643–662. [Google Scholar] [CrossRef]
- Golden, C. Stroop color and word test: Cat. No. 30150m. In A Manual for Clinical and Experimental Uses; Stoelting: Chicago, IL, USA, 1978. [Google Scholar]
- Reitan, R. Validity of the trail making test as an indicator of organic brain damage. Percept. Mot. Skills 1958, 8, 271–276. [Google Scholar] [CrossRef]
- O’Rourke, J.J.; Beglinger, L.J.; Smith, M.M.; Mills, J.; Moser, D.J.; Rowe, K.C.; Langbehn, D.R.; Duff, K.; Stout, J.C.; Harrington, D.L. The trail making test in prodromal huntington disease: Contributions of disease progression to test performance. J. Clin. Exp. Neuropsychol. 2011, 33, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of huntington’s disease. Cell 2015, 162, 516–526. [Google Scholar]
- Kim, E.Y.; Magnotta, V.A.; Liu, D.; Johnson, H.J. Stable atlas-based mapped prior (stamp) machine-learning segmentation for multicenter large-scale mri data. Magn. Reson. Imaging 2014, 32, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Calhoun, V.D.; Arias-Vasquez, A.; Zwiers, M.P.; van Hulzen, K.; Fernández, G.; Fisher, S.E.; Franke, B.; Turner, J.A.; Liu, J. G-protein genomic association with normal variation in gray matter density. Hum. Brain Mapp. 2015, 36, 4272–4286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corp, I. Ibm SPSS Statistics for Windows, 21.0; IBM Corp: Armonk, NY, USA, 2012. [Google Scholar]
- Chen, J.; Calhoun, V.D.; Liu, J. Ica order selection based on consistency: Application to genotype data. In Proceedings of the 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, San Diego, CA, USA, 28 August–1 September 2012; pp. 360–363. [Google Scholar]
- Fox, J. Applied Regression Analysis and Generalized Linear Models, 2nd ed.; Sage Publications: Thousand Oaks, CA, USA, 2008. [Google Scholar]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 2nd ed.; Sage Publications: Thousand Oaks, CA, USA, 2011. [Google Scholar]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y. The genecards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar]
- Zakzanis, K.K.; Mraz, R.; Graham, S.J. An fmri study of the trail making test. Neuropsychologia 2005, 43, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Moll, J.; de Oliveira-Souza, R.; Moll, F.T.; Bramati, I.E.; Andreiuolo, P.A. The cerebral correlates of set-shifting: An fmri study of the trail making test. Arq. Neuropsiquiatr. 2002, 60, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Galer, S.; Op De Beeck, M.; Urbain, C.; Bourguignon, M.; Ligot, N.; Wens, V.; Marty, B.; Van Bogaert, P.; Peigneux, P.; De Tiège, X. Investigating the neural correlates of the stroop effect with magnetoencephalography. Brain Topogr. 2015, 28, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.C.; Skudlarski, P.; Gatenby, J.C.; Peterson, B.S.; Gore, J.C. An event-related functional mri study of the stroop color word interference task. Cereb. Cortex 2000, 10, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, E.; Yeh, A.; Benedict, R.; Parrish, J.; Weinstock-Guttman, B. Cognitive dysfunction in ms: Bridging the gap between neurocognitive deficits, neuropsychological batteries and mri. Future Neurol. 2008, 3, 49–59. [Google Scholar] [CrossRef]
- Nachev, P.; Kennard, C.; Husain, M. Functional role of the supplementary and pre-supplementary motor areas. Nat. Rev. Neurosci. 2008, 9, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, P.; Castren, E.; Stamm, S. Analysis of the human trkb gene genomic organization reveals novel trkb isoforms, unusual gene length, and splicing mechanism. Biochem. Biophys. Res. Commun. 2002, 290, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- BrainSeq: A Human Brain Genomics Consortium. Brainseq: Neurogenomics to drive novel target discovery for neuropsychiatric disorders. Neuron 2015, 88, 1078–1083. [Google Scholar]
- Ramasamy, A.; Trabzuni, D.; Guelfi, S.; Varghese, V.; Smith, C.; Walker, R.; De, T.; Coin, L.; de Silva, R.; Cookson, M.R. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 2014, 17, 1418–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, L.D.; Kellis, M. Haploreg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- GTEx-Consortium. Human genomics. The genotype-tissue expression (gtex) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar]
- Andrade, M.A.; Bork, P. Heat repeats in the huntington’s disease protein. Nat. Genet. 1995, 11, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hwang, Y.J.; Kim, K.Y.; Kowall, N.W.; Ryu, H. Epigenetic mechanisms of neurodegeneration in huntington’s disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Gene(s) | pICA Reference SNPs (#) | Full Name | Function |
|---|---|---|---|---|
| REST/NRSF | REST, RCOR1, RCOR3 † | 4 | RE1 silencing transcription factor/neuron-restrictive silencer factor | Transcriptional repression |
| Sin3A | SIN3A | 2 | SIN3 transcription regulator family member A | Part of co-repressor complex with REST and coREST |
| CoREST | RCOR1 | 9 | REST co-repressor | Part of co-repressor complex with REST and Sin3A |
| HAP1 | HAP1 | 7 | Huntingtin-associated protein 1 | Binds to huntingtin, facilitates brain-derived neurotrophic factor (BDNF) transcription and transport |
| TrkB | NTRK2 | 52 | Tropomyosin receptor kinase B | BDNF high-affinity receptor |
| P75 | NGFR | 26 | Low-affinity nerve growth factor receptor | BDNF low-affinity receptor |
| RILP | RILP | 5 | REST-interacting LIM domain protein | REST nuclear receptor |
| Sortilin | SORT1 | 12 | Sortilin 1 | Suggested apoptotic function with p75 and pro-BDNF |
| BDNF | BDNF | 6 | Brain-derived neurotrophic factor | Neuronal growth, survival, differentiation |
| SNP | Weight (||) | +/− | Ranking | Gene | Minor Allele Frequency | Class |
|---|---|---|---|---|---|---|
| rs7801922 | 1.29 | + | 1 | CDK14 | T = 0.34/1704 (1000 Genomes) T = 0.38/11000 (TOPMED) | SNV |
| rs11140810 | 1.20 | + | 2 | NTRK2 | G = 0.42/2104 (1000 Genomes) G = 0.42/12329 (TOPMED) | SNV |
| rs4877289 | 1.08 | + | 3 | NTRK2 | G = 0.38/1926 (1000 Genomes) G = 0.38/11160 (TOPMED) | SNV |
| rs548321 | 1.03 | + | 4 | 70 kb 5′ of LRRC55 | G = 0.41/2055 (1000 Genomes) G = 0.38/11140 (TOPMED) | SNV |
| rs112140519 | 1.01 | + | 5 | 53 kb 3′ of NUS1 | -=0.33/1652 (1000 Genomes) | DIV |
| rs427790 | 1.00 | + | 6 | MIR181A1, NR5A2 | C = 0.33/1658 (1000 Genomes) C = 0.38/10948 (TOPMED) | SNV |
| rs10868241 | 1.0 | + | 7 | NTRK2 | A = 0.32/1614 (1000 Genomes) A = 0.24/6986 (TOPMED) | SNV |
| rs7655305 | 0.99 | + | 8 | FAM114A1 | G = 0.43/2140 (1000 Genomes) G = 0.43/12519 (TOPMED) | SNV |
| rs2277193 | 0.98 | + | 9 | NTRK2 | C = 0.34/1679 (1000 Genomes) C = 0.41/11827 (TOPMED) | SNV |
| rs8012614 | 0.98 | + | 10 | HEATR4 | C = 0.29/1442 (1000 Genomes) C = 0.37/10860 (TOPMED) | SNV |
| SNP (rs) | Occ. | Thal. | Temp. | WM | Put. | Hipp. | Fron. | Cereb. | SNigra | Med. | DLPFC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 8012614 HEATR4 (I) | NUMB; TMEM90A | HEATR4 * | ACOT4 * | RBM25 *; ACOT4 * | RBM25 * | RBM25 * | RBM25 *; ACOT4 *; HEATR4 *; DNAL1 | ||||
| 7801922 CDK14 (I) | STEAP1 | C7orf63; STEAP2 * | STEAP2 * | C7orf63 * | STEAP2 * | DPY19L2P4; CDK14 * | C7orf63 * | ||||
| 7655305 FAM114A1 (I) | RPL6 | PDS5A | FAM114A1 * | TLR6 | PTTG2; FLJ13197; UGDH | FAM114A1 * | FAM114A1 * | TLR1 | |||
| 548321 LRRC55 (IG) | UBE2L6; ZDHHC5 | TIMM10 | |||||||||
| 427790 MIR181A1, NR5A2 (IG) | NEK7 | NR5A2 *; PTPRC; ATP6V1G3 | NR5A2 *; MIR181A1 * | MIR181A1 * | MIR181A1 * | NR5A2 * | |||||
| 11140810 NTRK2 (I) | NTRK2 † | NTRK2 †; HNRNPK †; MIR7-1 † | NTRK2 †; SLC28A3 † | NAA35 † | NTRK2 † | NTRK2 † | HNRNPK †; MIR7-1 † | SLC28A3 † | NTRK2 † | ||
| 2277193 NTRK2 (I) | SLC28A3 †; HNRNPK †; MIR7-1 † | SLC28A3 † | NAA35 † | SLC28A3 †; AGTPBP1 † | SLC28A3 †; HNRNPK †; MIR7-1 †; NAA35 †; AGTPBP1 † | NAA35 † | SLC28A3 † | ||||
| 4877289 NTRK2 (I) | AGTPBP1 † | HNRNPK †; MIR7-1 † | SLC28A3 † | RMI1 †; SLC28A3 †; AGTPBP1 † | |||||||
| 10868241 NTRK2 (I) | HNRNPK †; MIR7-1 † | SLC28A3 † | RMI1 † | SLC28A3 † | SLC28A3 †; NTRK2 † | HNRNPK †; MIR7-1 †; AGTPBP1 † | NTRK2 † |
| Gene Name | Full Name | Associated NTRK2 SNP(s) | Description | Type | Related Pathways |
|---|---|---|---|---|---|
| SLC28A3 | Solute Carrier Family 28 Member 3 | rs1114081, rs2277193, rs4877289, rs10868241 | Neurotransmission, vascular tone, adenosine concentration near cell surface receptors, transport/metabolism of nucleoside drugs | Protein coding, nucleoside transporter | Vitamin and nucleoside transport, thiopurine pathway, pharmacokinetics/pharmacodynamics |
| AGTPBP1 | ATP/GTP Binding Protein 1 | rs10868241 | Contains nuclear localization signals and an ATP/GTP-binding motif, involved in the deglutamylation of protein polyglutamate side chains, removal of gene-encoded polyglutamates from protein carboxy-terminus, and shortening of long polyglutamate chains | Protein coding, zinc carboxypeptidase, metallocarboxypeptidase | Neuroscience |
| HNRNPK | Heterogeneous Nuclear Ribonucleoprotein K | rs1114081, rs2277193, rs4877289, rs10868241 | Major pre-mRNA-binding protein, binds to poly(C) sequences, involved in nuclear metabolism of hnRNAs, and p53/TP53 response to DNA damage (transcriptional activation and repression) | Protein coding, heterogeneous nuclear ribonucleoprotein (hnRNP) | Translational control and mRNA splicing |
| MIR7-1 | MicroRNA 7-1 | rs1114081, rs2277193, rs4877289, rs10868241 | Affiliated with an undefined RNA class | RNA gene | mRNA splicing, SUMOylation |
| NAA35 | N(Alpha)-Acetyltransferase 35, NatC Auxiliary Subunit | rs1114081, rs2277193 | Involved in the regulation of apoptosis, and the proliferation of smooth muscle cells | Protein coding | Golgi-to-endoplasmic reticulum (ER), trans-Golgi-network retrograde transport |
| NTRK2 SNP | CG | IT | AG | DLPFC | Ant. Caud. | MHipp | SNigra | Regulatory Motifs Altered |
|---|---|---|---|---|---|---|---|---|
| rs11140810 | H3K27ac | H3K27ac | H3K27ac | H3K27ac | Foxa, GLI, Hic1, Zec | |||
| rs4877289 | H3K4me1 | H3K4me1 | ||||||
| rs10868241 | H3K27ac, H3K4me1 | H3K27ac, H3K4me1 | H3K27ac | H3K27ac, H3K4me1 | H3K27ac | H3K27ac, H3K4me1 | H3K27ac, H3K4me1 | |
| rs2277193 | H3K27ac | H3K27ac | H3K27ac | H3K27ac | GR, Pax-6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciarochi, J.A.; Liu, J.; Calhoun, V.; Johnson, H.; Misiura, M.; Bockholt, H.J.; Espinoza, F.A.; Caprihan, A.; Plis, S.; Turner, J.A.; et al. High and Low Levels of an NTRK2-Driven Genetic Profile Affect Motor- and Cognition-Associated Frontal Gray Matter in Prodromal Huntington’s Disease. Brain Sci. 2018, 8, 116. https://doi.org/10.3390/brainsci8070116
Ciarochi JA, Liu J, Calhoun V, Johnson H, Misiura M, Bockholt HJ, Espinoza FA, Caprihan A, Plis S, Turner JA, et al. High and Low Levels of an NTRK2-Driven Genetic Profile Affect Motor- and Cognition-Associated Frontal Gray Matter in Prodromal Huntington’s Disease. Brain Sciences. 2018; 8(7):116. https://doi.org/10.3390/brainsci8070116
Chicago/Turabian StyleCiarochi, Jennifer A., Jingyu Liu, Vince Calhoun, Hans Johnson, Maria Misiura, H. Jeremy Bockholt, Flor A. Espinoza, Arvind Caprihan, Sergey Plis, Jessica A. Turner, and et al. 2018. "High and Low Levels of an NTRK2-Driven Genetic Profile Affect Motor- and Cognition-Associated Frontal Gray Matter in Prodromal Huntington’s Disease" Brain Sciences 8, no. 7: 116. https://doi.org/10.3390/brainsci8070116
APA StyleCiarochi, J. A., Liu, J., Calhoun, V., Johnson, H., Misiura, M., Bockholt, H. J., Espinoza, F. A., Caprihan, A., Plis, S., Turner, J. A., Paulsen, J. S., & The PREDICT-HD Investigators and Coordinators of the Huntington Study Group. (2018). High and Low Levels of an NTRK2-Driven Genetic Profile Affect Motor- and Cognition-Associated Frontal Gray Matter in Prodromal Huntington’s Disease. Brain Sciences, 8(7), 116. https://doi.org/10.3390/brainsci8070116