Molecular Biomarkers in Fragile X Syndrome



Abstract
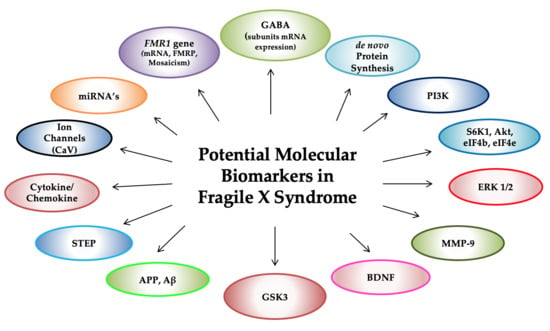
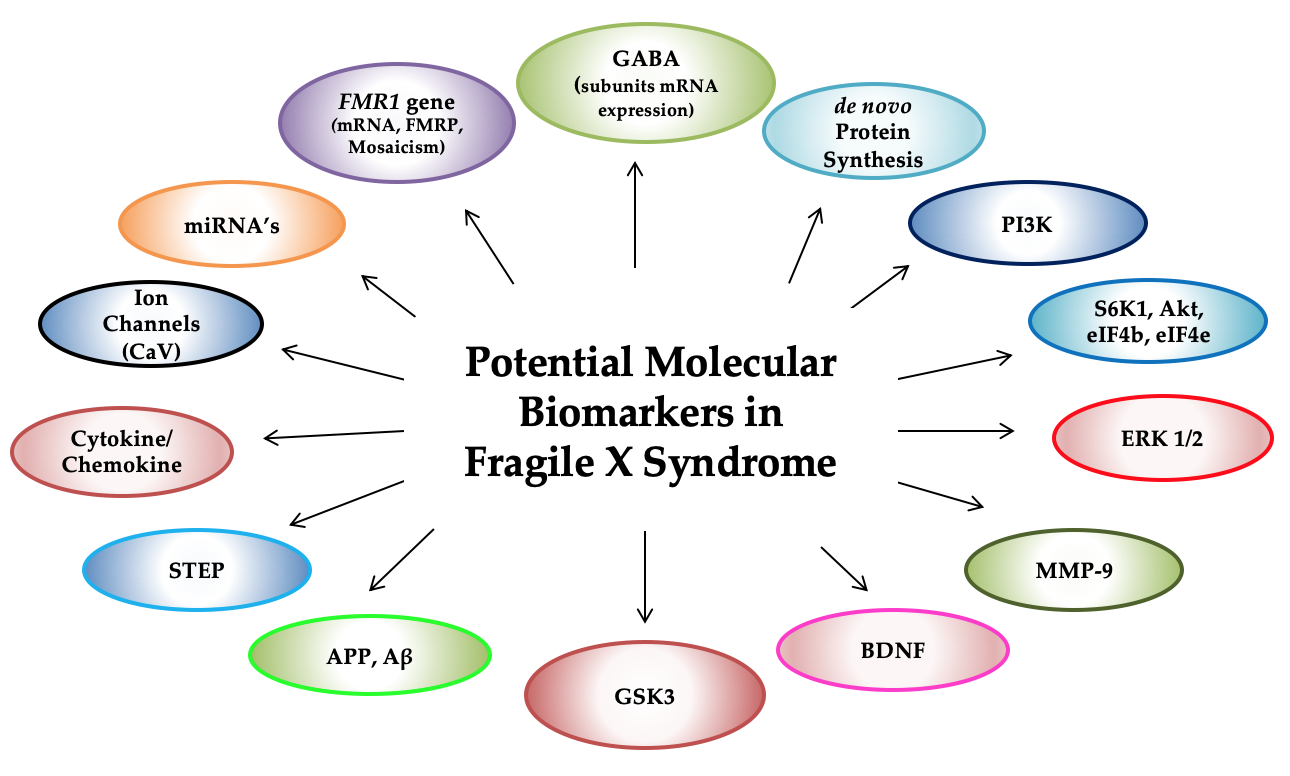
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. FMR1 Molecular Measures
3. Metabotropic Glutamate Receptors (mGluRs)
4. γ-Aminobutyric Acid (GABA) Receptors
5. De Novo Protein Synthesis
6. Phosphoinositide 3-Kinase (PI3K)
7. Mammalian Target of Rapamycin (mTOR) and Substrate p70 Ribosomal S6 Kinase (S6K1)
8. Extracellular-Regulated Kinase (ERK)
9. Matrix Metalloproteinase-9 (MMP-9)
10. Brain-Derived Neurotrophic Factor (BDNF)
11. Amyloid-β Protein Precursor and Amyloid-β (APP, Aβ)
12. Additional Potential Biomarkers
12.1. Ion Channels (CaV)
12.2. Glycogen Synthase Kinase-3 (GSK-3)
12.3. Striatal-Enriched Protein Tyrosine Phosphatase (STEP)
12.4. Plasma Cytokines and Chemokines
12.5. Diacylglycerol Kinase Kappa (Dgkκ)
12.6. MicroRNAs (miRNAs)
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Harris, S.; Goodlin-Jones, B.; Nowicki, S.; Bacalman, S.; Tassone, F.; Hagerman, R. Autism Profiles of Young Males with Fragile X Syndrome. J. Dev. Behav. Pediatr. 2005, 26, 464. [Google Scholar] [CrossRef]
- Landowska, A.; Rzonca, S.; Bal, J.; Gos, M. [Fragile X syndrome and FMR1-dependent diseases-clinical presentation, epidemiology and molecular background]. Dev. Period Med. 2018, 22, 14–21. [Google Scholar] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Oberlé, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boué, J.; Bertheas, M.; Mandel, J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Tassone, F.; Hall, D.A. FXTAS, FXPOI, and Other Premutation Disorders; Springer Nature: Basel, Switzerland, 2016. [Google Scholar]
- Hagerman, R.J.; Protic, D.; Rajaratnam, A.; Salcedo-Arellano, M.J.; Aydin, E.Y.; Schneider, A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front. Psychol. 2018, 9, 564. [Google Scholar] [CrossRef]
- Santos, A.R.; Bagni, C.; Kanellopoulos, A.K. Learning and behavioral deficits associated with the absence of the fragile X mental retardation protein: What a fly and mouse model can teach us. Learn. Mem. 2014, 21, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, G.M.; Shim, S.; Hong, D.S.; Garrett, A.S.; Quintin, E.-M.; Marzelli, M.J.; Patnaik, S.; Lightbody, A.A.; Reiss, A.L. Neuroanatomical abnormalities in fragile X syndrome during the adolescent and young adult years. J. Psychiatr. 2018, 107, 138–144. [Google Scholar] [CrossRef]
- Bostrom, C.; Yau, S.-Y.; Majaess, N.; Vetrici, M.; Gil-Mohapel, J.; Christie, B.R.; Yau, S.S.Y. Hippocampal dysfunction and cognitive impairment in Fragile-X Syndrome. Neurosci. Biobehav. Rev. 2016, 68, 563–574. [Google Scholar] [CrossRef]
- Hoeft, F.; Carter, J.C.; Lightbody, A.A.; Hazlett, H.C.; Piven, J.; Reiss, A.L. Region-specific alterations in brain development in one- to three-year-old boys with fragile X syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 9335–9339. [Google Scholar] [CrossRef]
- Harlow, E.G.; Till, S.M.; Russell, T.A.; Wijetunge, L.S.; Kind, P.; Contractor, A. Critical period plasticity is disrupted in the barrel cortex of Fmr1 knockout mice. Neuron 2010, 65, 385–398. [Google Scholar] [CrossRef]
- Lai, J.; Lerch, J.; Doering, L.; Foster, J.; Ellegood, J.; Lai, J. Regional brain volumes changes in adult male FMR1-KO mouse on the FVB strain. Neuroscience 2016, 318, 12–21. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Poe, M.D.; Lightbody, A.A.; Styner, M.; MacFall, J.R.; Reiss, A.L.; Piven, J. Trajectories of Early Brain Volume Development in Fragile X Syndrome and Autism. J. Am. Acad. Child Adolesc. Psychiatry 2012, 51, 921–933. [Google Scholar] [CrossRef]
- Measurement of Cerebral and Cerebellar Volumes in Children with Fragile X Sundrome. Available online: https://paperpile.com/app/p/2405c439-1b64-0314-96b7-37ba0f0ea488 (accessed on 12 January 2019).
- Sunamura, N.; Iwashita, S.; Enomoto, K.; Kadoshima, T.; Isono, F. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci. Rep. 2018, 8, 11585. [Google Scholar] [CrossRef]
- Yang, Y.-M.; Arsenault, J.; Bah, A.; Krzeminski, M.; Fekete, A.; Chao, O.Y.; Pacey, L.K.; Wang, A.; Forman-Kay, J.; Hampson, D.R.; et al. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol. Psychiatry 2018. [Google Scholar] [CrossRef]
- Hanson, A.C.; Hagerman, R.J. Serotonin dysregulation in Fragile X Syndrome: Implications for treatment. Intractable Rare Dis. 2014, 3, 110–117. [Google Scholar] [CrossRef]
- The Dutch-Belgian Fragile X Consorthium; Bakker, C.E.; Verheij, C.; Willemsen, R.; van der Helm, R.; Oerlemans, F.; Vermey, M.; Bygrave, A.; Hoogeveen, A.; Oostra, B.A.; et al. Fmr1 Knockout Mice: A Model to Study Fragile X Mental Retardation. Cell 1994, 78, 22–23. [Google Scholar]
- Dahlhaus, R. Of Men and Mice: Modeling the Fragile X Syndrome. Front. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Rais, M.; Binder, D.K.; Razak, K.A.; Ethell, I.M. Sensory Processing Phenotypes in Fragile X Syndrome. ASN Neuro 2018, 10, 1759091418801092. [Google Scholar] [CrossRef]
- Greco, C.M.; Berman, R.F.; Martin, R.M.; Tassone, F.; Schwartz, P.H.; Chang, A.; Trapp, B.D.; Iwahashi, C.; Brunberg, J.; Grigsby, J.; et al. Neuropathology of Fragile X-Associated Tremor/ataxia Syndrome (FXTAS). Brain 2006, 129, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Bernardet, M.; Crusio, W.E. Fmr1 KO Mice as a Possible Model of Autistic Features. Sci. World J. 2006, 6, 1164–1176. [Google Scholar] [CrossRef]
- Wright, J. Questions for Elizabeth Berry-Kravis: Dodging mouse traps | Spectrum | Autism Research News. Available online: https://www.spectrumnews.org/opinion/q-and-a/questions-for-elizabeth-berry-kravis-dodging-mouse-traps/ (accessed on 9 January 2019).
- Berry-Kravis, E.M.; Lindemann, L.; Jønch, A.E.; Apostol, G.; Bear, M.F.; Carpenter, R.L.; Crawley, J.N.; Curie, A.; Des Portes, V.; Hossain, F.; et al. Drug Development for Neurodevelopmental Disorders: Lessons Learned from Fragile X Syndrome. Nat. Rev. Drug Discov. 2018, 17, 280–299. [Google Scholar] [CrossRef]
- Scharf, S.H.; Jaeschke, G.; Wettstein, J.G.; Lindemann, L. Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharmacol. 2015, 20, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Fragile X drug development flounders. Nat. Rev. Drug Discov. 2016, 15, 77. [Google Scholar] [CrossRef]
- De Caro, J.J.; Dominguez, C.; Sherman, S.L. Reproductive Health of Adolescent Girls Who Carry the FMR1 Premutation: Expected Phenotype Based on Current Knowledge of Fragile X-Associated Primary Ovarian Insufficiency. Ann. N. Y. Acad. Sci. 2008, 1135, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.E.; Bailey, D.B.; Mankowski, J.; Ford, A.; Sideris, J.; Weisenfeld, L.A.; Heath, T.M.; Golden, R.N. Mood and anxiety disorders in females with the FMR1 premutation. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150, 130–139. [Google Scholar] [CrossRef]
- Hamlin, A.; Liu, Y.; Nguyen, D.V.; Tassone, F.; Zhang, L.; Hagerman, R.J. Sleep apnea in fragile X premutation carriers with and without FXTAS. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 923–928. [Google Scholar] [CrossRef]
- Hamlin, A.A.; Sukharev, D.; Campos, L.; Mu, Y.; Tassone, F.; Hessl, D.; Nguyen, D.V.; Loesch, D.; Hagerman, R.J. Hypertension in FMR1 Premutation Males with and without Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Am. J. Med. Genet. Part A 2012, 158, 1304–1309. [Google Scholar] [CrossRef]
- Bailey, D.B.; Raspa, M.; Bishop, E.; Mitra, D.; Martin, S.; Wheeler, A.; Sacco, P. Health and Economic Consequences of Fragile X Syndrome for Caregivers. J. Dev. Behav. Pediatr. 2012, 33, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.Y.; Findling, R.L. Pharmacotherapy for mental health problems in people with intellectual disability. Curr. Opin. 2016, 29, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Lieb-Lundell, C.C.E. Three Faces of Fragile X. Phys. Ther. 2016, 96, 1782–1790. [Google Scholar] [CrossRef]
- Hoyos, L.R.; Thakur, M. Fragile X Premutation in Women: Recognizing the Health Challenges beyond Primary Ovarian Insufficiency. J. Assist. Reprod. Genet. 2017, 34, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Schneider, A.; Hagerman, R.; Song, G.; Wong, S.; Tassone, F.; Giulivi, C. Impact of FMR1 Premutation on Neurobehavior and Bioenergetics in Young Monozygotic Twins. Front. Genet. 2018, 9, 9. [Google Scholar] [CrossRef]
- Ligsay, A.; Hagerman, R.J. Review of targeted treatments in fragile X syndrome. Intractable Rare Dis. 2016, 5, 158–167. [Google Scholar] [CrossRef]
- Hagerman, P.J. The Fragile X Prevalence Paradox. J. Med. Genet. 2008, 45, 498–499. [Google Scholar] [CrossRef]
- Stembalska, A.; Łaczmańska, I.; Gil, J.; Pesz, K.A. Fragile X Syndrome in Females—A Familial Case Report and Review of the Literature. Dev. Period. Med. 2016, 20, 99–104. [Google Scholar]
- Berry-Kravis, E.; Potanos, K.; Weinberg, D.; Zhou, L.; Goetz, C.G. Fragile X-Associated Tremor/ataxia Syndrome in Sisters Related to X-Inactivation. Ann. Neurol. 2005, 57, 144–147. [Google Scholar] [CrossRef]
- Van Esch, H. The Fragile X premutation: New insights and clinical consequences. Eur. J. Med Genet. 2006, 49, 1–8. [Google Scholar] [CrossRef]
- Seltzer, M.M.; Abbeduto, L.; Greenberg, J.S.; Almeida, D.; Hong, J.; Witt, W. Chapter 7 Biomarkers in the Study of Families of Children with Developmental Disabilities. Families 2009, 37, 213–249. [Google Scholar]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef]
- Dölen, G.; Bear, M.F. Role for Metabotropic Glutamate Receptor 5 (mGluR5) in the Pathogenesis of Fragile X Syndrome. J. Physiol. 2008, 586, 1503–1508. [Google Scholar] [CrossRef]
- Kim, M.; Ceman, S. Fragile X Mental Retardation Protein: Past, Present and Future. Curr. Pept. Sci. 2012, 13, 358–371. [Google Scholar] [CrossRef]
- Darnell, J.C.; Klann, E. The Translation of Translational Control by FMRP: Therapeutic Targets for FXS. Nat. Neurosci. 2013, 16, 1530–1536. [Google Scholar] [CrossRef]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered Synaptic Plasticity in a Mouse Model of Fragile X Mental Retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of fragile X syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.D.; Bear, M.F. Toward Fulfilling the Promise of Molecular Medicine in Fragile X Syndrome. Annu. Med. 2011, 62, 411–429. [Google Scholar] [CrossRef]
- Bhakar, A.L.; Dölen, G.; Bear, M.F. The Pathophysiology of Fragile X (and What It Teaches Us about Synapses). Annu. Neurosci. 2012, 35, 417–443. [Google Scholar] [CrossRef] [PubMed]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic Pharmacological mGlu5 Inhibition Corrects Fragile X in Adult Mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef]
- McBride, S.M.; Choi, C.H.; Wang, Y.; Liebelt, D.; Braunstein, E.; Ferreiro, D.; Sehgal, A.; Siwicki, K.K.; Dockendorff, T.C.; Nguyen, H.T.; et al. Pharmacological Rescue of Synaptic Plasticity, Courtship Behavior, and Mushroom Body Defects in a Drosophila Model of Fragile X Syndrome. Neuron 2005, 45, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Broadie, K.S. Drosophila Fragile X Mental Retardation Protein and Metabotropic Glutamate Receptor A Convergently Regulate the Synaptic Ratio of Ionotropic Glutamate Receptor Subclasses. J. Neurosci. 2007, 27, 12378–12389. [Google Scholar] [CrossRef]
- Chang, S.; Bray, S.M.; Li, Z.; Zarnescu, D.C.; He, C.; Jin, P.; Warren, S.T. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Methods 2008, 4, 256–263. [Google Scholar] [CrossRef]
- Pan, L.; Woodruff, E., 3rd; Liang, P.; Broadie, K. Mechanistic Relationships between Drosophila Fragile X Mental Retardation Protein and Metabotropic Glutamate Receptor A Signaling. Mol. Cell. Neurosci. 2008, 37, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Repicky, S.; Broadie, K. Metabotropic Glutamate Receptor-Mediated Use-Dependent down-Regulation of Synaptic Excitability Involves the Fragile X Mental Retardation Protein. J. Neurophysiol. 2009, 101, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; McBride, S.M.J.; Schoenfeld, B.P.; Liebelt, D.A.; Ferreiro, D.; Ferrick, N.J.; Hinchey, P.; Kollaros, M.; Rudominer, R.L.; Terlizzi, A.M.; et al. Age-dependent cognitive impairment in a Drosophila Fragile X model and its pharmacological rescue. Biogerontology 2009, 11, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Kanellopoulos, A.K.; Semelidou, O.; Kotini, A.G.; Anezaki, M.; Skoulakis, E.M.C. Learning and Memory Deficits Consequent to Reduction of the Fragile X Mental Retardation Protein Result from Metabotropic Glutamate Receptor-Mediated Inhibition of cAMP Signaling in Drosophila. J. Neurosci. 2012, 32, 13111–13124. [Google Scholar] [CrossRef]
- Tessier, C.R.; Broadie, K. Molecular and Genetic Analysis of the Drosophila Model of Fragile X Syndrome. Results Probl. Cell Differ. 2012, 54, 119–156. [Google Scholar]
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling Fragile X Syndrome in Drosophila. Front. Neurosci. 2018, 11, 124. [Google Scholar] [CrossRef]
- Yan, Q.; Rammal, M.; Tranfaglia, M.; Bauchwitz, R.; Bauchwitz, R. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 2005, 49, 1053–1066. [Google Scholar] [CrossRef]
- De Vrij, F.M.; Levenga, J.; Van Der Linde, H.C.; Koekkoek, S.K.; De Zeeuw, C.I.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis. 2008, 31, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.M.; Kogan, C.S.; Messier, C.; Gandhi, R.M. 2-Methyl-6-(phenylethynyl) pyridine (MPEP) reverses maze learning and PSD-95 deficits in Fmr1 knock-out mice. Front. Cell. Neurosci. 2014, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Achuta, V.S.; Grym, H.; Putkonen, N.; Louhivuori, V.; Kärkkäinen, V.; Koistinaho, J.; Roybon, L.; Castrén, M.L. Metabotropic Glutamate Receptor 5 Responses Dictate Differentiation of Neural Progenitors to NMDA-Responsive Cells in Fragile X Syndrome. Dev. Neurobiol. 2017, 77, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Ronesi, J.A.; Collins, K.A.; Hays, S.A.; Tsai, N.-P.; Guo, W.; Birnbaum, S.G.; Hu, J.-H.; Worley, P.F.; Gibson, J.R.; Huber, K.M. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci. 2012, 15, 431–440. [Google Scholar] [CrossRef]
- Guo, W.; Molinaro, G.; Collins, K.A.; Hays, S.A.; Paylor, R.; Worley, P.F.; Szumlinski, K.K.; Huber, K.M. Selective Disruption of Metabotropic Glutamate Receptor 5-Homer Interactions Mimics Phenotypes of Fragile X Syndrome in Mice. J. Neurosci. 2016, 36, 2131–2147. [Google Scholar] [CrossRef]
- Bogdanik, L.; Mohrmann, R.; Ramaekers, A.; Bockaert, J.; Grau, Y.; Broadie, K.; Parmentier, M.-L. The Drosophila Metabotropic Glutamate Receptor DmGluRA Regulates Activity-Dependent Synaptic Facilitation and Fine Synaptic Morphology. J. Neurosci. 2004, 24, 9105–9116. [Google Scholar] [CrossRef]
- Michel, C.I.; Kraft, R.; Restifo, L.L. Defective Neuronal Development in the Mushroom Bodies of Drosophila Fragile X Mental Retardation 1 Mutants. J. Neurosci. 2004, 24, 5798–5809. [Google Scholar] [CrossRef]
- Porter, R.H.P.; Jaeschke, G.; Spooren, W.; Ballard, T.M.; Büttelmann, B.; Kolczewski, S.; Peters, J.-U.; Prinssen, E.; Wichmann, J.; Vieira, E.; et al. Fenobam: A Clinically Validated Nonbenzodiazepine Anxiolytic Is a Potent, Selective, and Noncompetitive mGlu5 Receptor Antagonist with Inverse Agonist Activity. J. Pharmacol. Exp. Ther. 2005, 315, 711–721. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hessl, D.; Coffey, S.; Hervey, C.; Schneider, A.; Yuhas, J.; Hutchison, J.; Snape, M.; Tranfaglia, M.; Nguyen, D.V.; et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J. Med Genet. 2009, 46, 266–271. [Google Scholar] [CrossRef]
- Vranesic, I.; Ofner, S.; Flor, P.J.; Bilbe, G.; Bouhelal, R.; Enz, A.; Desrayaud, S.; McAllister, K.; Kuhn, R.; Gasparini, F. AFQ056/mavoglurant, a Novel Clinically Effective mGluR5 Antagonist: Identification, SAR and Pharmacological Characterization. Bioorg. Med. Chem. 2014, 22, 5790–5803. [Google Scholar] [CrossRef]
- Jacquemont, S.; Curie, A.; Portes, V.D.; Torrioli, M.G.; Berry-Kravis, E.; Hagerman, R.J.; Ramos, F.J.; Cornish, K.; He, Y.; Paulding, C.; et al. Epigenetic Modification of the FMR1 Gene in Fragile X Syndrome Is Associated with Differential Response to the mGluR5 Antagonist AFQ056. Sci. Transl. Med. 2011, 3, 64ra1. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hagerman, R.; Jacquemont, S.; Charles, P.; Visootsak, J.; Brinkman, M.; Rerat, K.; Koumaras, B.; Zhu, L.; Barth, G.M.; et al. Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med. 2016, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, G.; Kolczewski, S.; Spooren, W.; Vieira, E.; Bitter-Stoll, N.; Boissin, P.; Borroni, E.; Büttelmann, B.; Ceccarelli, S.; Clemann, N.; et al. Metabotropic Glutamate Receptor 5 Negative Allosteric Modulators: Discovery of 2-Chloro-4-[1-(4-fluorophenyl)-2,5-dimethyl-1H-imidazol-4-ylethynyl]pyridine (Basimglurant, RO4917523), a Promising Novel Medicine for Psychiatric Diseases. J. Med. Chem. 2015, 58, 1358–1371. [Google Scholar] [CrossRef]
- Lindemann, L.; Porter, R.H.; Scharf, S.H.; Kuennecke, B.; Bruns, A.; Von Kienlin, M.; Harrison, A.C.; Paehler, A.; Funk, C.; Gloge, A.; et al. Pharmacology of Basimglurant (RO4917523, RG7090), a Unique Metabotropic Glutamate Receptor 5 Negative Allosteric Modulator in Clinical Development for Depression. J. Pharmacol. Exp. Ther. 2015, 353, 213–233. [Google Scholar] [CrossRef] [PubMed]
- Gantois, I.; Pop, A.S.; De Esch, C.E.; Buijsen, R.A.; Pooters, T.; Gomez-Mancilla, B.; Gasparini, F.; Oostra, B.A.; D’Hooge, R.; Willemsen, R. Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav. Brain 2013, 239, 72–79. [Google Scholar] [CrossRef]
- Pop, A.S.; Levenga, J.; de Esch, C.E.F.; Buijsen, R.A.M.; Nieuwenhuizen, I.M.; Li, T.; Isaacs, A.; Gasparini, F.; Oostra, B.A.; Willemsen, R. Rescue of Dendritic Spine Phenotype in Fmr1 KO Mice with the mGluR5 Antagonist AFQ056/Mavoglurant. Psychopharmacology 2014, 231, 1227–1235. [Google Scholar] [CrossRef]
- Levenga, J.; Hayashi, S.; De Vrij, F.M.; Koekkoek, S.K.; Van Der Linde, H.C.; Nieuwenhuizen, I.; Song, C.; Buijsen, R.A.; Pop, A.S.; GomezMancilla, B.; et al. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol. Dis. 2011, 42, 311–317. [Google Scholar] [CrossRef]
- Tranfaglia, M. Roche Reports Fragile X Clinical Trial Negative Results. Available online: https://www.fraxa.org/roche-reports-clinical-trial-negative-results/ (accessed on 10 January 2019).
- FRAXA Research Foundation. Novartis Discontinues Development of mavoglurant (AFQ056) for Fragile X Syndrome. Fragile X Research—FRAXA Research Foundation. Available online: https://www.fraxa.org/novartis-discontinues-development-mavoglurant-afq056-fragile-x-syndrome/ (accessed on 10 January 2019).
- A Youssef, E.; Berry-Kravis, E.; Czech, C.; Hagerman, R.J.; Hessl, D.; Wong, C.Y.; Rabbia, M.; Deptula, D.; John, A.; Kinch, R.; et al. Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacol 2017, 43, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Jacquemont, S.; Berry-Kravis, E.; Des Portes, V.; Stanfield, A.; Koumaras, B.; Rosenkranz, G.; Murgia, A.; Wolf, C.; Apostol, G.; et al. Mavoglurant in Fragile X Syndrome: Results of Two Open-Label, Extension Trials in Adults and Adolescents. Sci. Rep. 2018, 8, 16970. [Google Scholar] [CrossRef] [PubMed]
- D’Hulst, C.; Heulens, I.; Brouwer, J.R.; Willemsen, R.; De Geest, N.; Reeve, S.P.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009, 1253, 176–183. [Google Scholar] [CrossRef]
- Braat, S.; D’Hulst, C.; Heulens, I.; De Rubeis, S.; Mientjes, E.; Nelson, D.L.; Willemsen, R.; Bagni, C.; Van Dam, D.; De Deyn, P.P.; et al. The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle 2015, 14, 2985–2995. [Google Scholar] [CrossRef]
- Zhang, N.; Peng, Z.; Tong, X.; Lindemeyer, A.K.; Cetina, Y.; Huang, C.S.; Olsen, R.W.; Otis, T.S.; Houser, C.R. Decreased Surface Expression of the δ Subunit of the GABAA Receptor Contributes to Reduced Tonic Inhibition in Dentate Granule Cells in a Mouse Model of Fragile X Syndrome. Exp. Neurol. 2017, 297, 168–178. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Nomura, T.; Xu, J.; Contractor, A. The Developmental Switch in GABA Polarity Is Delayed in Fragile X Mice. J. Neurosci. 2014, 34, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Tyzio, R.; Nardou, R.; Ferrari, D.C.; Tsintsadze, T.; Shahrokhi, A.; Eftekhari, S.; Khalilov, I.; Tsintsadze, V.; Brouchoud, C.; Chazal, G.; et al. Oxytocin-Mediated GABA Inhibition During Delivery Attenuates Autism Pathogenesis in Rodent Offspring. Science 2014, 343, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Segal, M.; Ben-Yosef, D. Immature Responses to GABA in Fragile X Neurons Derived from Human Embryonic Stem Cells. Front. Cell. Neurosci. 2016, 10, 167. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Corbin, J.G.; Burns, M.P. The GABAA Receptor Agonist THIP Ameliorates Specific Behavioral Deficits in the Mouse Model of Fragile X Syndrome. Dev. Neurosci. 2011, 33, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.; Featherstone, R.; Naschek, M.; Nam, J.; Du, A.; Wright, S.; Pance, K.; Melnychenko, O.; Weger, R.; Akuzawa, S.; et al. GABA-B Agonist Baclofen Normalizes Auditory-Evoked Neural Oscillations and Behavioral Deficits in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. Eneuro 2017, 4, ENEURO.0380–16.2017. [Google Scholar] [CrossRef]
- Qin, M.; Huang, T.; Kader, M.; Krych, L.; Xia, Z.; Burlin, T.; Zeidler, Z.; Zhao, T.; Smith, C.B. R-Baclofen Reverses a Social Behavior Deficit and Elevated Protein Synthesis in a Mouse Model of Fragile X Syndrome. Int. J. Neuropsychopharmacol. 2015, 18, 18. [Google Scholar] [CrossRef]
- Henderson, C.; Wijetunge, L.; Kinoshita, M.N.; Shumway, M.; Hammond, R.S.; Postma, F.R.; Brynczka, C.; Rush, R.; Thomas, A.; Paylor, R.; et al. Reversal of Disease-Related Pathologies in the Fragile X Mouse Model by Selective Activation of GABAB Receptors with Arbaclofen. Sci. Transl. Med. 2012, 4, 152. [Google Scholar] [CrossRef]
- Kang, J.-Y.; Chadchankar, J.; Vien, T.N.; Mighdoll, M.I.; Hyde, T.M.; Mather, R.J.; Deeb, T.Z.; Pangalos, M.N.; Brandon, N.J.; Dunlop, J.; et al. Deficits in the activity of presynaptic γ-aminobutyric acid type B receptors contribute to altered neuronal excitability in fragile X syndrome. J. Boil. Chem. 2017, 292, 6621–6632. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Zhou, S.; Yang, L.; Shi, Q.; Li, Y.; Zhang, K.; Yang, L.; Zhao, M.; Yang, Q.; et al. Imbalance between Glutamate and GABA in Fmr1 Knockout Astrocytes Influences Neuronal Development. Genes 2016, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, J.; Song, S.; Li, F.; Yuan, F. Reduction of α1GABAA receptor mediated by tyrosine kinase C (PKC) phosphorylation in a mouse model of fragile X syndrome. Int. J. Clin. Exp. Med. 2015, 8, 13219–13226. [Google Scholar]
- Fatemi, S.H.; Folsom, T.D. GABA receptor subunit distribution and FMRP–mGluR5 signaling abnormalities in the cerebellum of subjects with schizophrenia, mood disorders, and autism. Schizophr. Res. 2015, 167, 42–56. [Google Scholar] [CrossRef]
- Braat, S.; Kooy, R.F. Fragile X syndrome neurobiology translates into rational therapy. Drug Discov. Today 2014, 19, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Hare, E.B.; Hagerman, R.J. Modulation of the GABAergic pathway for the treatment of fragile X syndrome. Neuropsychiatr. Dis. Treat. 2014, 10, 1769–1779. [Google Scholar] [PubMed]
- Olmos-Serrano, J.L.; Paluszkiewicz, S.M.; Martin, B.S.; Kaufmann, W.E.; Corbin, J.G.; Huntsman, M.M. Defective GABAergic Neurotransmission and Pharmacological Rescue of Neuronal Hyperexcitability in the Amygdala in a Mouse Model of Fragile X Syndrome. J. Neurosci. 2010, 30, 9929–9938. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Papouin, T.; Séguéla, P.; Avoli, M. Downregulation of Tonic GABAergic Inhibition in a Mouse Model of Fragile X Syndrome. Cereb. Cortex 2008, 19, 1515–1520. [Google Scholar] [CrossRef]
- Centonze, D.; Rossi, S.; Mercaldo, V.; Napoli, I.; Ciotti, M.T.; De Chiara, V.; Musella, A.; Prosperetti, C.; Calabresi, P.; Bernardi, G.; et al. Abnormal Striatal GABA Transmission in the Mouse Model for the Fragile X Syndrome. Boil. Psychiatry 2008, 63, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Hagerman, R.; Visootsak, J.; Budimirovic, D.; Kaufmann, W.E.; Cherubini, M.; Zarevics, P.; Walton-Bowen, K.; Wang, P.; Bear, M.F.; et al. Arbaclofen in fragile X syndrome: Results of phase 3 trials. J. Neurodev. Disord. 2017, 9, 3. [Google Scholar] [CrossRef]
- Berry-Kravis, E.M.; Hessl, D.; Rathmell, B.; Zarevics, P.; Cherubini, M.; Walton-Bowen, K.; Mu, Y.; Nguyen, D.V.; Gonzalez-Heydrich, J.; Wang, P.P.; et al. Effects of STX209 (Arbaclofen) on Neurobehavioral Function in Children and Adults with Fragile X Syndrome: A Randomized, Controlled, Phase 2 Trial. Sci. Transl. Med. 2012, 4, 152ra127. [Google Scholar] [CrossRef]
- Schaefer, T.L.; Davenport, M.H.; Grainger, L.M.; Robinson, C.K.; Earnheart, A.T.; Stegman, M.S.; Lang, A.L.; Ashworth, A.A.; Molinaro, G.; Huber, K.M.; et al. Acamprosate in a Mouse Model of Fragile X Syndrome: Modulation of Spontaneous Cortical Activity, ERK1/2 Activation, Locomotor Behavior, and Anxiety. J. Neurodev. Disord. 2017, 9, 6. [Google Scholar] [CrossRef]
- Erickson, C.A.; Wink, L.K.; Ray, B.; Early, M.C.; Stiegelmeyer, E.; Mathieu-Frasier, L.; Patrick, V.; Lahiri, D.K.; McDougle, C.J. Impact of acamprosate on behavior and brain-derived neurotrophic factor: An open-label study in youth with fragile X syndrome. Psychopharmacology 2013, 228, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ligsay, A.; Van Dijck, A.; Nguyen, D.V.; Lozano, R.; Chen, Y.; Bickel, E.S.; Hessl, D.; Schneider, A.; Angkustsiri, K.; Tassone, F.; et al. A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J. Neurodev. Disord. 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Yoon, B.C.; Holt, C.E. Axonal mRNA Localization and Local Protein Synthesis in Nervous System Assembly, Maintenance and Repair. Nat. Rev. Neurosci. 2012, 13, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Kang, J.; Burlin, T.V.; Jiang, C.; Smith, C.B. Postadolescent Changes in Regional Cerebral Protein Synthesis: An In Vivo Study in the Fmr1 Null Mouse. J. Neurosci. 2005, 25, 5087–5095. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Bhattacharya, A.; Nadel, J.; Moulton, K.; Zeak, N.M.; Glicksman, A.; Dobkin, C.; Brick, D.J.; Schwartz, P.H.; Smith, C.B.; et al. Identification of Fragile X Syndrome-Specific Molecular Markers in Human Fibroblasts: A Useful Model to Test the Efficacy of Therapeutic Drugs. Hum. Mutat. 2014, 35, 1485–1494. [Google Scholar] [CrossRef]
- Jacquemont, S.; Pacini, L.; E Jønch, A.; Cencelli, G.; Rozenberg, I.; He, Y.; D’Andrea, L.; Pedini, G.; Eldeeb, M.; Willemsen, R.; et al. Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum. Mol. Genet. 2018, 27, 2039–2051. [Google Scholar] [CrossRef]
- Qin, M.; Schmidt, K.C.; Zametkin, A.J.; Bishu, S.; Horowitz, L.M.; Burlin, T.V.; Xia, Z.; Huang, T.; Quezado, Z.M.; Smith, C.B. Altered Cerebral Protein Synthesis in Fragile X Syndrome: Studies in Human Subjects and Knockout Mice. J. Cereb. Blood Flow Metab. 2013, 33, 499–507. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Huang, T.; Smith, C.B. Lithium Reverses Increased Rates of Cerebral Protein Synthesis in a Mouse Model of Fragile X Syndrome. Neurobiol. Dis. 2012, 45, 1145. [Google Scholar] [CrossRef]
- Gross, C.; Hoffmann, A.; Bassell, G.J.; Berry-Kravis, E.M. Therapeutic Strategies in Fragile X Syndrome: From Bench to Bedside and Back. Neurotherapeutics 2015, 12, 584–608. [Google Scholar] [CrossRef]
- Jacquemont, S.; Berry-Kravis, E.; Hagerman, R.; von Raison, F.; Gasparini, F.; Apostol, G.; Ufer, M.; Des Portes, V.; Gomez-Mancilla, B. The Challenges of Clinical Trials in Fragile X Syndrome. Psychopharmacology 2014, 231, 1237–1250. [Google Scholar] [CrossRef]
- Pop, A.S.; Gomez-Mancilla, B.; Neri, G.; Willemsen, R.; Gasparini, F. Fragile X Syndrome: A Preclinical Review on Metabotropic Glutamate Receptor 5 (mGluR5) Antagonists and Drug Development. Psychopharmacology 2014, 231, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Osterweil, E.K.; Chuang, S.-C.; Chubykin, A.A.; Sidorov, M.; Bianchi, R.; Wong, R.K.S.; Bear, M.F. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 2013, 77, 243–250. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic Removal of p70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef]
- Reversal of Activity-Mediated Spine Dynamics and Learning Impairment in a Mouse Model of Fragile X Syndrome. Available online: https://www.ncbi.nlm.nih.gov/pubmed/24712992 (accessed on 11 January 2019).
- Dolan, B.M.; Duron, S.G.; Campbell, D.A.; Vollrath, B.; Rao, B.S.S.; Ko, H.-Y.; Lin, G.G.; Govindarajan, A.; Choi, S.-Y.; Tonegawa, S.; et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 2013, 110, 5671–5676. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Mamcarz, M.; Mullins, C.; Choudhury, A.; Boyle, R.G.; Smith, D.G.; Walker, D.W.; Klann, E. Targeting Translation Control with p70 S6 Kinase 1 Inhibitors to Reverse Phenotypes in Fragile X Syndrome Mice. Neuropsychopharmacology 2016, 41, 1991–2000. [Google Scholar] [CrossRef]
- Gross, C.; Nakamoto, M.; Yao, X.; Chan, C.-B.; Yim, S.Y.; Ye, K.; Warren, S.T.; Bassell, G.J. Excess Phosphoinositide 3-Kinase Subunit Synthesis and Activity as a Novel Therapeutic Target in Fragile X Syndrome. J. Neurosci. 2010, 30, 10624–10638. [Google Scholar] [CrossRef]
- Hayashi, M.L.; Rao, B.S.S.; Seo, J.-S.; Choi, H.-S.; Dolan, B.M.; Choi, S.-Y.; Chattarji, S.; Tonegawa, S.; Rao, B.S.S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11489–11494. [Google Scholar] [CrossRef]
- Tian, M.; Zeng, Y.; Hu, Y.; Yuan, X.; Liu, S.; Li, J.; Lu, P.; Sun, Y.; Gao, L.; Fu, D.; et al. 7, 8-Dihydroxyflavone induces synapse expression of AMPA GluA1 and ameliorates cognitive and spine abnormalities in a mouse model of fragile X syndrome. Neuropharmacology 2015, 89, 43–53. [Google Scholar] [CrossRef]
- Sawicka, K.; Pyronneau, A.; Chao, M.; Bennett, M.V.L.; Zukin, R.S. Elevated ERK/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in fragile X mice. Proc. Natl. Acad. Sci. USA 2016, 113, E6290–E6297. [Google Scholar] [CrossRef]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Bassell, G.J. Neuron-specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front. Neurosci. 2014, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Bassell, G.J. Excess Protein Synthesis in FXS Patient Lymphoblastoid Cells Can Be Rescued with a p110β-Selective Inhibitor. Mol. Med. 2012, 18, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Raj, N.; Molinaro, G.; Allen, A.G.; Whyte, A.J.; Gibson, J.R.; Huber, K.M.; Gourley, S.L.; Bassell, G.J. Selective role of the catalytic PI3K subunit p110β in impaired higher-order cognition in Fragile X syndrome. Cell Rep. 2015, 11, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Chang, C.-W.; Kelly, S.M.; Bhattacharya, A.; McBride, S.M.; Danielson, S.W.; Jiang, M.Q.; Chan, C.B.; Ye, K.; Gibson, J.R.; et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in Fragile X syndrome. Cell Rep. 2015, 11, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Klann, E.; Dever, T.E. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 931–942. [Google Scholar] [CrossRef]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nat. Cell Boil. 2005, 433, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Legius, E.; Bagni, C.; Borrie, S.C. Cognitive Dysfunctions in Intellectual Disabilities: The Contributions of the Ras-MAPK and PI3K-AKT-mTOR Pathways. Annu. Genom. Hum. Genet. 2017, 18, 115–142. [Google Scholar]
- Zhu, P.J.; Chen, C.-J.; Mays, J.; Stoica, L.; Costa-Mattioli, M. mTORC2, but not mTORC1, is required for hippocampal mGluR-LTD and associated behaviors. Nat. Neurosci. 2018, 21, 799–802. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Sanchez, E.; Hagerman, R.J.; Mu, Y.; Nguyen, D.V.; Wong, H.; Whelan, A.M.; Zukin, R.S.; Klann, E.; Tassone, F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with Fragile X syndrome. Genes Brain 2012, 11, 332–341. [Google Scholar] [CrossRef]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR Signaling in Fragile X Syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef]
- Monyak, R.E.; Emerson, D.; Schoenfeld, B.P.; Zheng, X.; Chambers, D.B.; Rosenfelt, C.; Langer, S.; Hinchey, P.; Choi, C.H.; McDonald, T.V.; et al. Insulin Signaling Misregulation Underlies Circadian and Cognitive Deficits in a Drosophila Fragile X Model. Mol. Psychiatry 2017, 22, 1140–1148. [Google Scholar] [CrossRef]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef]
- Kim, S.H.; Markham, J.A.; Weiler, I.J.; Greenough, W.T. Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 4429–4434. [Google Scholar] [CrossRef]
- Hou, L.; Antion, M.D.; Hu, D.; Spencer, C.M.; Paylor, R.; Klann, E. Dynamic Translational and Proteasomal Regulation of Fragile X Mental Retardation Protein Controls mGluR-Dependent Long-Term Depression. Neuron 2006, 51, 441–454. [Google Scholar] [CrossRef]
- Weng, N.; Weiler, I.J.; Sumis, A.; Greenough, W.T.; Berry-Kravis, E.; Berry-Kravis, E. Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147, 1253–1257. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Sumis, A.; Hervey, C.; Nelson, M.; Porges, S.W.; Weng, N.; Weiler, I.J.; Greenough, W.T. Open-Label Treatment Trial of Lithium to Target the Underlying Defect in Fragile X Syndrome. J. Dev. Behav. Pediatr. 2008, 29, 293–302. [Google Scholar] [CrossRef]
- Erickson, C.A.; Weng, N.; Weiler, I.J.; Greenough, W.T.; Stigler, K.A.; Wink, L.K.; McDougle, C.J. Open-label riluzole in fragile X syndrome. Brain Res. 2011, 1380, 264–270. [Google Scholar] [CrossRef]
- Pellerin, D.; Çaku, A.; Fradet, M.; Bouvier, P.; Dubé, J.; Corbin, F. Lovastatin corrects ERK pathway hyperactivation in fragile X syndrome: Potential of platelet’s signaling cascades as new outcome measures in clinical trials. Biomarkers 2016, 21, 1–12. [Google Scholar] [CrossRef]
- Zamzow, R. Drug Duo Delivers Brain, Behavioral Benefits for Fragile X Syndrome | Spectrum | Autism Research News. Available online: https://www.spectrumnews.org/news/drug-duo-delivers-brain-behavioral-benefits-fragile-x-syndrome/ (accessed on 11 January 2019).
- Reinhard, S.M.; Razak, K.; Ethell, I.M. A delicate balance: Role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Front. Cell. Neurosci. 2015, 9, 280. [Google Scholar] [CrossRef]
- Janusz, A.; Miłek, J.; Perycz, M.; Pacini, L.; Bagni, C.; Kaczmarek, L.; Dziembowska, M. The Fragile X Mental Retardation Protein Regulates Matrix Metalloproteinase 9 mRNA at Synapses. J. Neurosci. 2013, 33, 18234–18241. [Google Scholar] [CrossRef]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med Genet. 1999, 84, 233–239. [Google Scholar] [CrossRef]
- Sidhu, H.; Dansie, L.E.; Hickmott, P.W.; Ethell, D.W.; Ethell, I.M. Genetic Removal of Matrix Metalloproteinase 9 Rescues the Symptoms of Fragile X Syndrome in a Mouse Model. J. Neurosci. 2014, 34, 9867–9879. [Google Scholar] [CrossRef]
- Lovelace, J.W.; Wen, T.H.; Reinhard, S.; Hsu, M.S.; Sidhu, H.; Ethell, I.M.; Binder, D.K.; Razak, K.A. Matrix metalloproteinase-9 deletion rescues auditory evoked potential habituation deficit in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 2016, 89, 126–135. [Google Scholar] [CrossRef]
- Bilousova, T.V.; Dansie, L.; Ngo, M.; Aye, J.; Charles, J.R.; Ethell, D.W.; Ethell, I.M. Minocycline Promotes Dendritic Spine Maturation and Improves Behavioural Performance in the Fragile X Mouse Model. J. Med. Genet. 2009, 46, 94–102. [Google Scholar] [CrossRef]
- Utari, A.; Chonchaiya, W.; Rivera, S.M.; Schneider, A.; Hagerman, R.J.; Faradz, S.M.H.; Ethell, I.M.; Nguyen, D.V. Side Effects of Minocycline Treatment in Patients with Fragile X Syndrome and Exploration of Outcome Measures. Am. J. Intellect. Dev. Disabil. 2010, 115, 433–443. [Google Scholar] [CrossRef]
- Paribello, C.; Tao, L.; Folino, A.; Berry-Kravis, E.; Tranfaglia, M.; Ethell, I.M.; Ethell, D.W. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010, 10, 91. [Google Scholar] [CrossRef]
- Leigh, M.J.S.; Nguyen, D.V.; Mu, Y.; Winarni, T.I.; Schneider, A.; Chechi, T.; Polussa, J.; Doucet, P.; Tassone, F.; Rivera, S.M.; et al. A Randomized Double-Blind, Placebo-Controlled Trial of Minocycline in Children and Adolescents with Fragile X Syndrome. J. Dev. Behav. Pediatr. 2013, 34, 147–155. [Google Scholar] [CrossRef]
- Dziembowska, M.; Pretto, D.I.; Janusz, A.; Kaczmarek, L.; Leigh, M.J.; Gabriel, N.; Durbin-Johnson, B.; Hagerman, R.J.; Tassone, F. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am. J. Med. Genet. Part A 2013, 161, 1897–1903. [Google Scholar] [CrossRef]
- AlOlaby, R.R.; Sweha, S.R.; Silva, M.; Durbin-Johnson, B.; Yrigollen, C.M.; Pretto, D.; Hagerman, R.J.; Tassone, F. Molecular biomarkers predictive of sertraline treatment response in young children with fragile X syndrome. Brain Dev. 2017, 39, 483–492. [Google Scholar] [CrossRef]
- Kim, S.W.; Cho, K.J. Activity-dependent alterations in the sensitivity to BDNF-TrkB signaling may promote excessive dendritic arborization and spinogenesis in fragile X syndrome in order to compensate for compromised postsynaptic activity. Med Hypotheses 2014, 83, 429–435. [Google Scholar] [CrossRef]
- Louhivuori, V.; Vicario, A.; Uutela, M.; Rantamäki, T.; Louhivuori, L.M.; Castrén, E.; Tongiorgi, E.; Åkerman, K.E.; Castrén, M.L. BDNF and TrkB in neuronal differentiation of Fmr1-knockout mouse. Neurobiol. Dis. 2011, 41, 469–480. [Google Scholar] [CrossRef]
- Uutela, M.; Lindholm, J.; Louhivuori, V.; Wei, H.; Louhivuori, L.M.; Pertovaara, A.; Akerman, K.; Castrén, E.; Castrén, M.L. Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes Brain 2012, 11, 513–523. [Google Scholar] [CrossRef]
- Castrén, M.L.; Castrén, E. BDNF in Fragile X Syndrome. Neuropharmacology 2014, 76, 729–736. [Google Scholar] [CrossRef]
- Louhivuori, V.; Arvio, M.; Soronen, P.; Oksanen, V.; Paunio, T.; Castrén, M.L. The Val66Met polymorphism in the BDNF gene is associated with epilepsy in fragile X syndrome. Epilepsy Res. 2009, 85, 114–117. [Google Scholar] [CrossRef]
- Westmark, C.J.; Westmark, P.R.; O’Riordan, K.J.; Ray, B.C.; Hervey, C.M.; Salamat, M.S.; Abozeid, S.H.; Stein, K.M.; Stodola, L.A.; Tranfaglia, M.; et al. Reversal of Fragile X Phenotypes by Manipulation of AβPP/Aβ Levels in Fmr1 KO Mice. PLoS ONE 2011, 6, e26549. [Google Scholar] [CrossRef]
- Erickson, C.A.; Ray, B.; Maloney, B.; Wink, L.K.; Bowers, K.; Schaefer, T.L.; McDougle, C.J.; Sokol, D.K.; Lahiri, D.K. Impact of Acamprosate on Plasma Amyloid-β Precursor Protein in Youth: A Pilot Analysis in Fragile X Syndrome-Associated and Idiopathic Autism Spectrum Disorder Suggests a Pharmacodynamic Protein Marker. J. Psychiatr. 2014, 59, 220–228. [Google Scholar] [CrossRef]
- Lee, H.Y.; Jan, L.Y. Fragile X syndrome: Mechanistic insights and therapeutic avenues regarding the role of potassium channels. Curr. Opin. Neurobiol. 2012, 22, 887–894. [Google Scholar] [CrossRef]
- Ferron, L.; Nieto-Rostro, M.; Cassidy, J.S.; Dolphin, A.C. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat. Commun. 2014, 5, 3628. [Google Scholar] [CrossRef]
- Castagnola, S.; Delhaye, S.; Folci, A.; Paquet, A.; Brau, F.; Duprat, F.; Jarjat, M.; Grossi, M.; Béal, M.; Martin, S.; et al. New Insights Into the Role of Cav2 Protein Family in Calcium Flux Deregulation in Fmr1-KO Neurons. Front. Mol. Neurosci. 2018, 11, 11. [Google Scholar] [CrossRef]
- Castaño, Z.; Gordon-Weeks, P.R.; Kypta, R.M.; Gordon-Weeks, P.R.; Gordon-Weeks, P.R. The neuron-specific isoform of glycogen synthase kinase-3β is required for axon growth. J. Neurochem. 2010, 113, 117–130. [Google Scholar] [CrossRef]
- Min, W.W.; Yuskaitis, C.J.; Yan, Q.; Sikorski, C.; Chen, S.; Jope, R.S.; Bauchwitz, R.P. Elevated Glycogen Synthase Kinase-3 Activity in Fragile X Mice: Key Metabolic Regulator with Evidence for Treatment Potential. Neuropharmacology 2009, 56, 463–472. [Google Scholar] [CrossRef]
- Portis, S.; Giunta, B.; Obregon, D.; Tan, J. The role of glycogen synthase kinase-3 signaling in neurodevelopment and fragile X syndrome. Int. J. Physiol. Pathophysiol. Pharmacol. 2012, 4, 140–148. [Google Scholar]
- Yuskaitis, C.J.; Mines, M.A.; King, M.K.; Sweatt, J.D.; Miller, C.A.; Jope, R.S. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of Fragile X syndrome. Biochem. Pharmacol. 2010, 79, 632–646. [Google Scholar] [CrossRef]
- Mines, M.A.; Yuskaitis, C.J.; King, M.K.; Beurel, E.; Jope, R.S. GSK3 Influences Social Preference and Anxiety-Related Behaviors during Social Interaction in a Mouse Model of Fragile X Syndrome and Autism. PLoS ONE 2010, 5, e9706. [Google Scholar] [CrossRef]
- Guo, W.; Murthy, A.C.; Zhang, L.; Johnson, E.B.; Schaller, E.G.; Allan, A.M.; Zhao, X. Inhibition of GSK3β improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 2011, 21, 681–691. [Google Scholar] [CrossRef]
- Franklin, A.V.; King, M.K.; Palomo, V.; Martinez, A.; McMahon, L.L.; Jope, R.S. Glycogen Synthase Kinase-3 Inhibitors Reverse Deficits in Long-Term Potentiation and Cognition in Fragile X Mice. Biol. Psychiatry 2014, 75, 198–206. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Beurel, E.; Jope, R.S. Evidence of Reactive Astrocytes but Not Peripheral Immune System Activation in a Mouse Model of Fragile X Syndrome. Biochim. Biophys. Acta. 2010, 1802, 1006–1012. [Google Scholar] [CrossRef]
- Mines, M.A.; Jope, R.S.; Mines, M.M. Glycogen Synthase Kinase-3: A Promising Therapeutic Target for Fragile X Syndrome. Front. Neurosci. 2011, 4, 35. [Google Scholar] [CrossRef]
- Goebel-Goody, S.M.; Lombroso, P.J. Taking STEPs Forward to Understand Fragile X Syndrome. Results Probl. Cell Differ. 2012, 54, 223–241. [Google Scholar]
- Goebel-Goody, S.M.; Baum, M.; Paspalas, C.D.; Fernandez, S.M.; Carty, N.C.; Kurup, P.; Lombroso, P.J. Therapeutic Implications for Striatal-Enriched Protein Tyrosine Phosphatase (STEP) in Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 65–87. [Google Scholar] [CrossRef]
- Johnson, M.A.; Lombroso, P.J. A Common STEP in the Synaptic Pathology of Diverse Neuropsychiatric Disorders. Yale J. Boil. Med. 2012, 85, 481–490. [Google Scholar]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Royston, S.; Tagliatela, S.M.; Naegele, J.R.; Lombroso, P.J.; Tagliatela, S.; Goebel-Goody, S.M.; Wilson-Wallis, E.D.; Goebel-Goody, S.M.; Wilson-Wallis, E.D.; Goebel-Goody, S.M.; et al. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain 2012, 11, 586–600. [Google Scholar]
- Xu, J.; Chatterjee, M.; Baguley, T.D.; Brouillette, J.; Kurup, P.; Ghosh, D.; Kanyo, J.; Zhang, Y.; Seyb, K.; Ononenyi, C.; et al. Inhibitor of the Tyrosine Phosphatase STEP Reverses Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. PLoS Biol. 2014, 12, e1001923. [Google Scholar] [CrossRef]
- Chatterjee, M.; Kurup, P.K.; Lundbye, C.J.; Hugger Toft, A.K.; Kwon, J.; Benedict, J.; Kamceva, M.; Banke, T.G.; Lombroso, P.J. STEP Inhibition Reverses Behavioral, Electrophysiologic, and Synaptic Abnormalities in Fmr1 KO Mice. Neuropharmacology 2018, 128, 43–53. [Google Scholar] [CrossRef]
- Ashwood, P.; Nguyen, D.V.; Hessl, D.; Hagerman, R.J.; Tassone, F. Plasma Cytokine Profiles in Fragile X Subjects: Is There a Role for Cytokines in the Pathogenesis? Brain Behav. Immun. 2010, 24, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Tabet, R.; Moutin, E.; Becker, J.A.J.; Heintz, D.; Fouillen, L.; Flatter, E.; Krężel, W.; Alunni, V.; Koebel, P.; Dembélé, D.; et al. Fragile X Mental Retardation Protein (FMRP) Controls Diacylglycerol Kinase Activity in Neurons. Proc. Natl. Acad. Sci. USA. 2016, 113, E3619–E3628. [Google Scholar] [CrossRef]
- Lin, S.-L. microRNAs and Fragile X Syndrome. Adv. Exp. Med. Biol. 2015, 888, 107–121. [Google Scholar] [PubMed]
- Lin, S.-L.; Chang, S.-J.; Ying, S.-Y. First in vivo evidence of microRNA-induced fragile X mental retardation syndrome. Mol. Psychiatry 2006, 11, 616–617. [Google Scholar] [CrossRef]
- Chang, S.-J.E.; Chang-Lin, S.; Chang, D.C.; Chang, C.P.; Lin, S.-L.; Ying, S.-Y. Repeat-Associated MicroRNAs Trigger Fragile X Mental Retardation-Like Syndrome in Zebrafish~!2008-10-29~!2008-11-12~!2008-11-26~! Open Neuropsychopharmacol. J. 2008, 1, 6–18. [Google Scholar] [CrossRef]
- Lin, S.-L.; Ying, S.-Y. Role of Repeat-Associated MicroRNA (ramRNA) in Fragile X Syndrome (FXS). In Current Perspectives in microRNAs (miRNA); Springer: Dordrecht, The Netherlands, 2008; pp. 245–266. [Google Scholar]
- Muddashetty, R.S.; Nalavadi, V.C.; Gross, C.; Yao, X.; Xing, L.; Laur, O.; Warren, S.T.; Bassell, G.J. Reversible Inhibition of PSD-95 mRNA Translation by miR-125a, FMRP Phosphorylation, and mGluR Signaling. Mol. Cell 2011, 42, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef]
- Liu, T.; Wan, R.-P.; Tang, L.-J.; Liu, S.-J.; Li, H.-J.; Zhao, Q.-H.; Liao, W.-P.; Sun, X.-F.; Yi, Y.-H.; Long, Y.-S. A MicroRNA Profile in Fmr1 Knockout Mice Reveals MicroRNA Expression Alterations with Possible Roles in Fragile X Syndrome. Mol. Neurobiol. 2015, 51, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, Z.; Ghaderian, S.M.H.; Najmabadi, H.; Omrani, M.D. High expression of miR-510 was associated with CGG expansion located at upstream of FMR1 into full mutation. J. Cell. Biochem. 2018, 120, 1916–1923. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafarullah, M.; Tassone, F. Molecular Biomarkers in Fragile X Syndrome. Brain Sci. 2019, 9, 96. https://doi.org/10.3390/brainsci9050096
Zafarullah M, Tassone F. Molecular Biomarkers in Fragile X Syndrome. Brain Sciences. 2019; 9(5):96. https://doi.org/10.3390/brainsci9050096
Chicago/Turabian StyleZafarullah, Marwa, and Flora Tassone. 2019. "Molecular Biomarkers in Fragile X Syndrome" Brain Sciences 9, no. 5: 96. https://doi.org/10.3390/brainsci9050096
APA StyleZafarullah, M., & Tassone, F. (2019). Molecular Biomarkers in Fragile X Syndrome. Brain Sciences, 9(5), 96. https://doi.org/10.3390/brainsci9050096