Proximity Ligation Assay Detection of Protein–DNA Interactions—Is There a Link between Heme Oxygenase-1 and G-quadruplexes?


 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Mice
2.3. Isolation of Cells from the Bone Marrow
2.4. Hmox1 Genotyping
2.5. Primary Antibodies
2.6. Fluorescence-Activated Cell Sorting (FACS)
2.7. Transfection
2.8. N-methylomesoporphyrin IX Staining
2.9. Immunocytochemistry
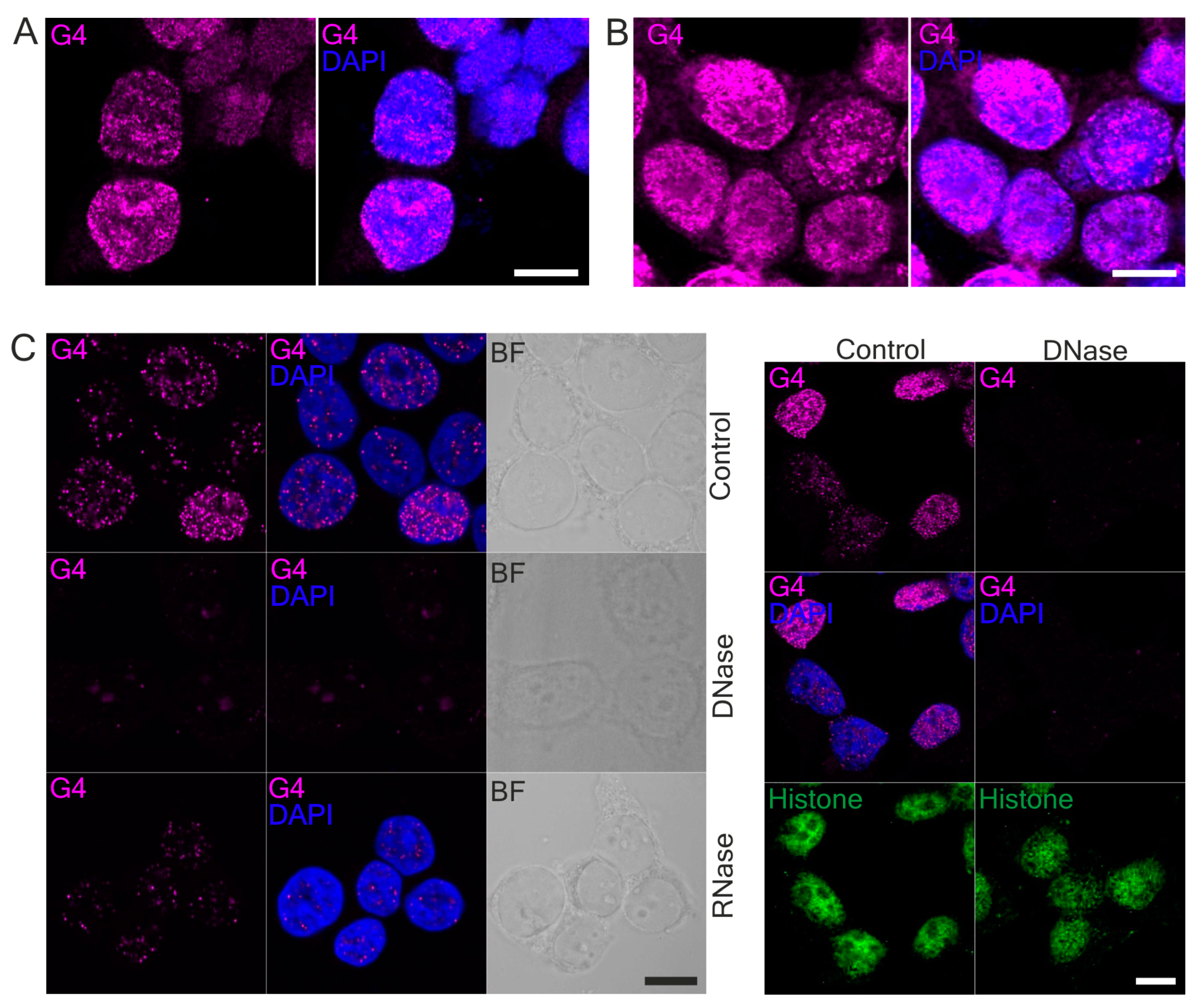
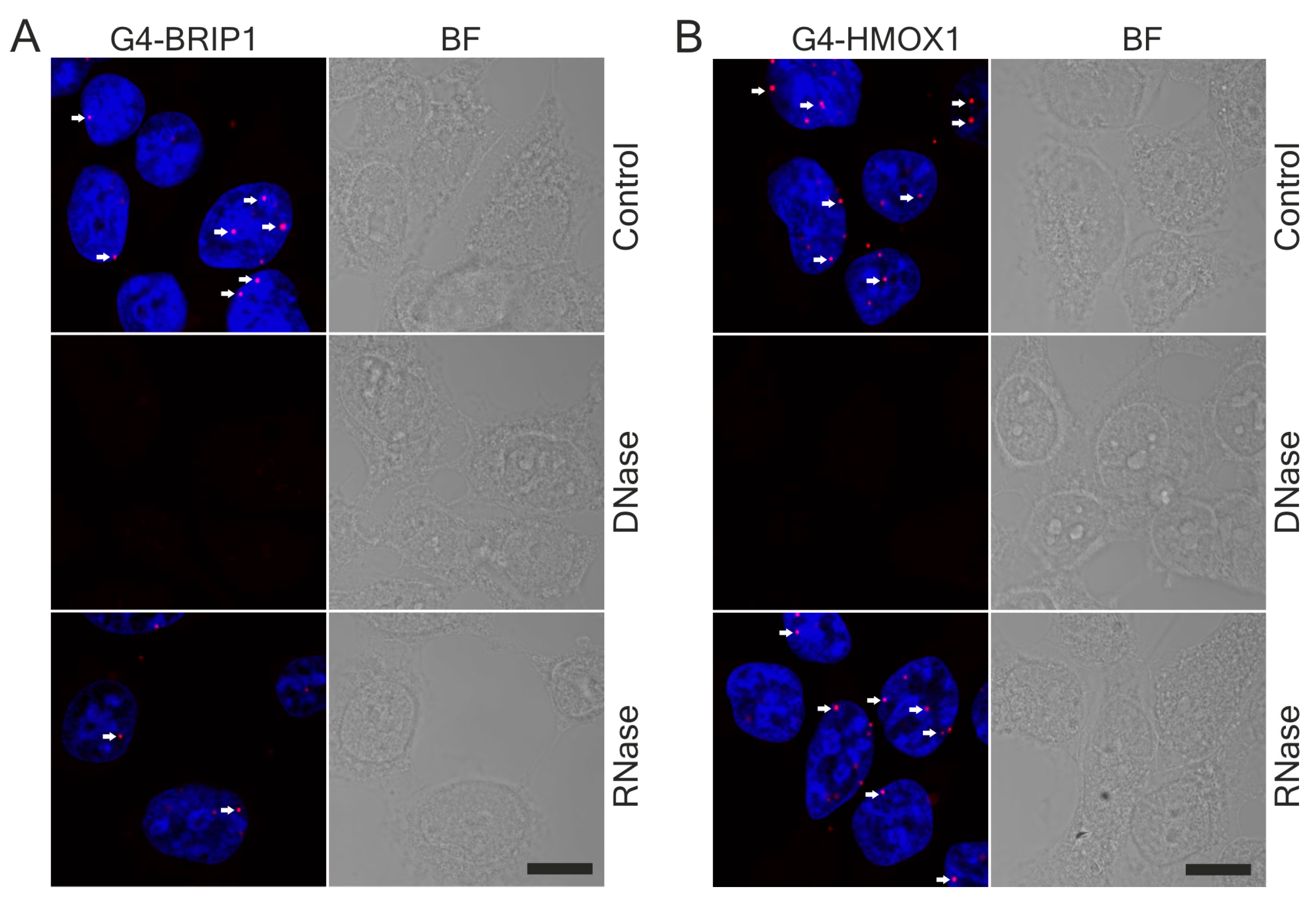
2.10. DNase and RNase Treatment
2.11. In Situ PLA
2.12. Confocal Microscopy
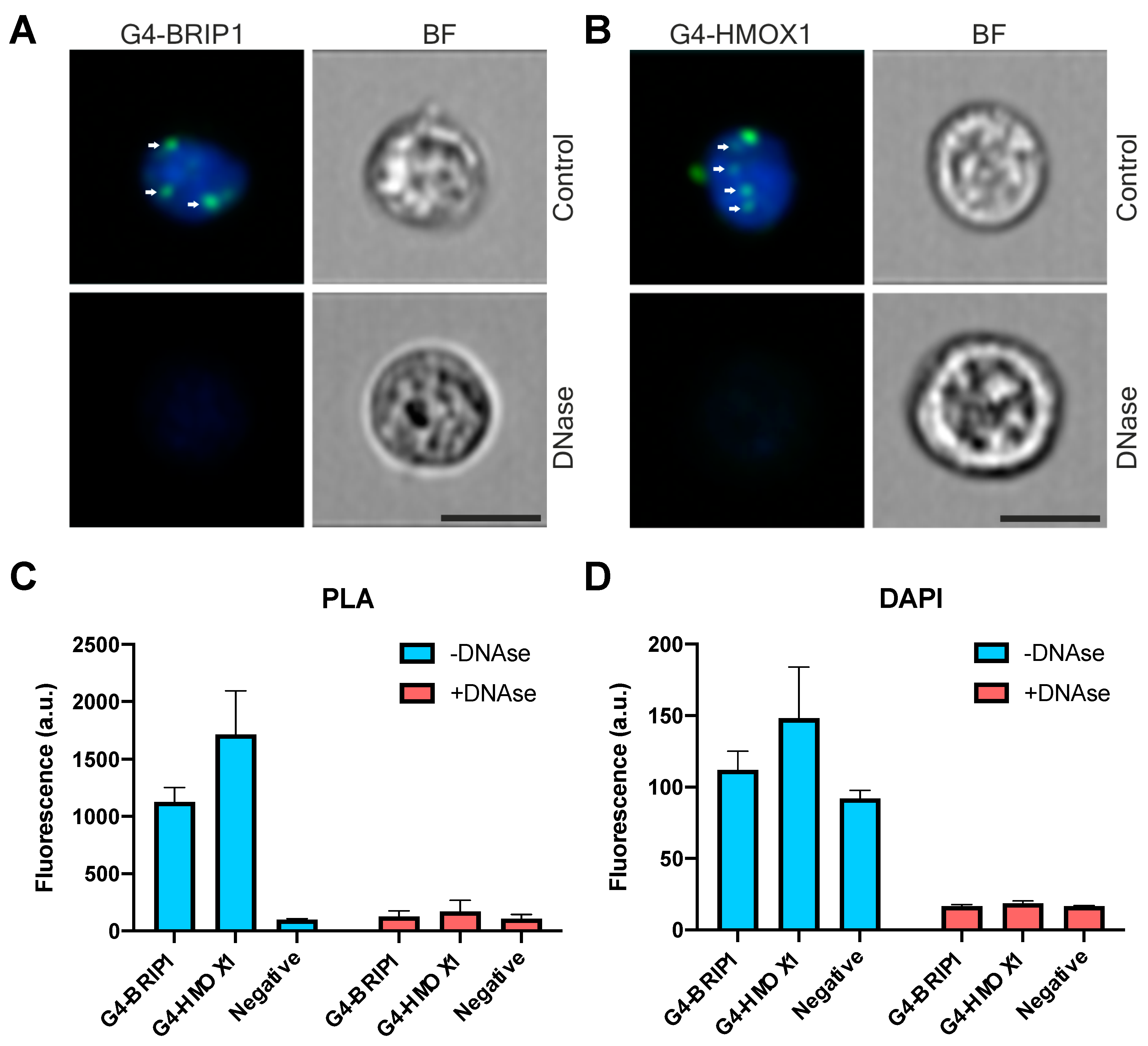
2.13. Flow Cytometry PLA
2.14. RNA-Seq
2.15. Reverse Transcription and Real-Time PCR
2.16. Western Blotting
2.17. Chromatin Immunoprecipitation (ChIP)
2.18. Statistical Analysis
3. Results
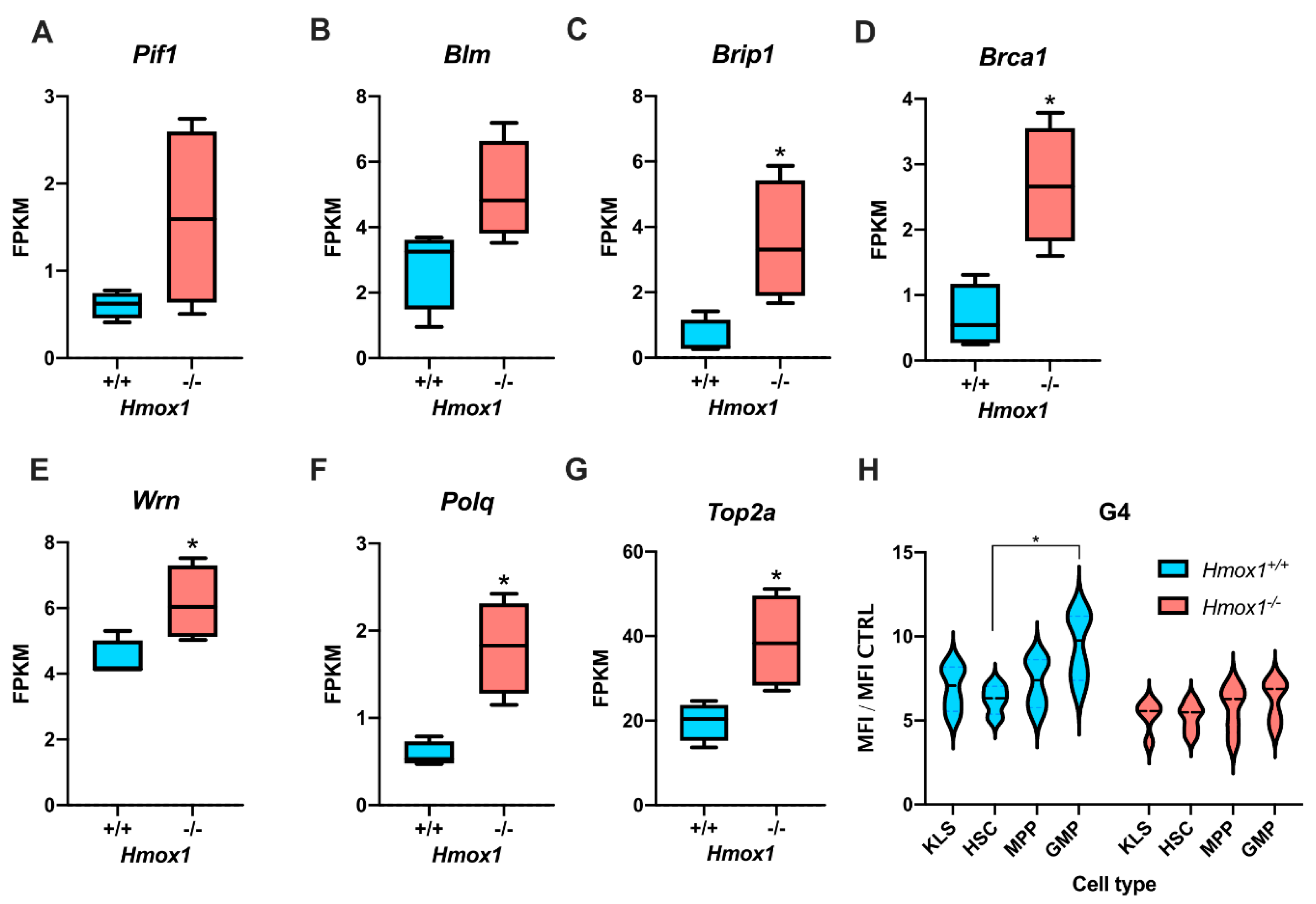
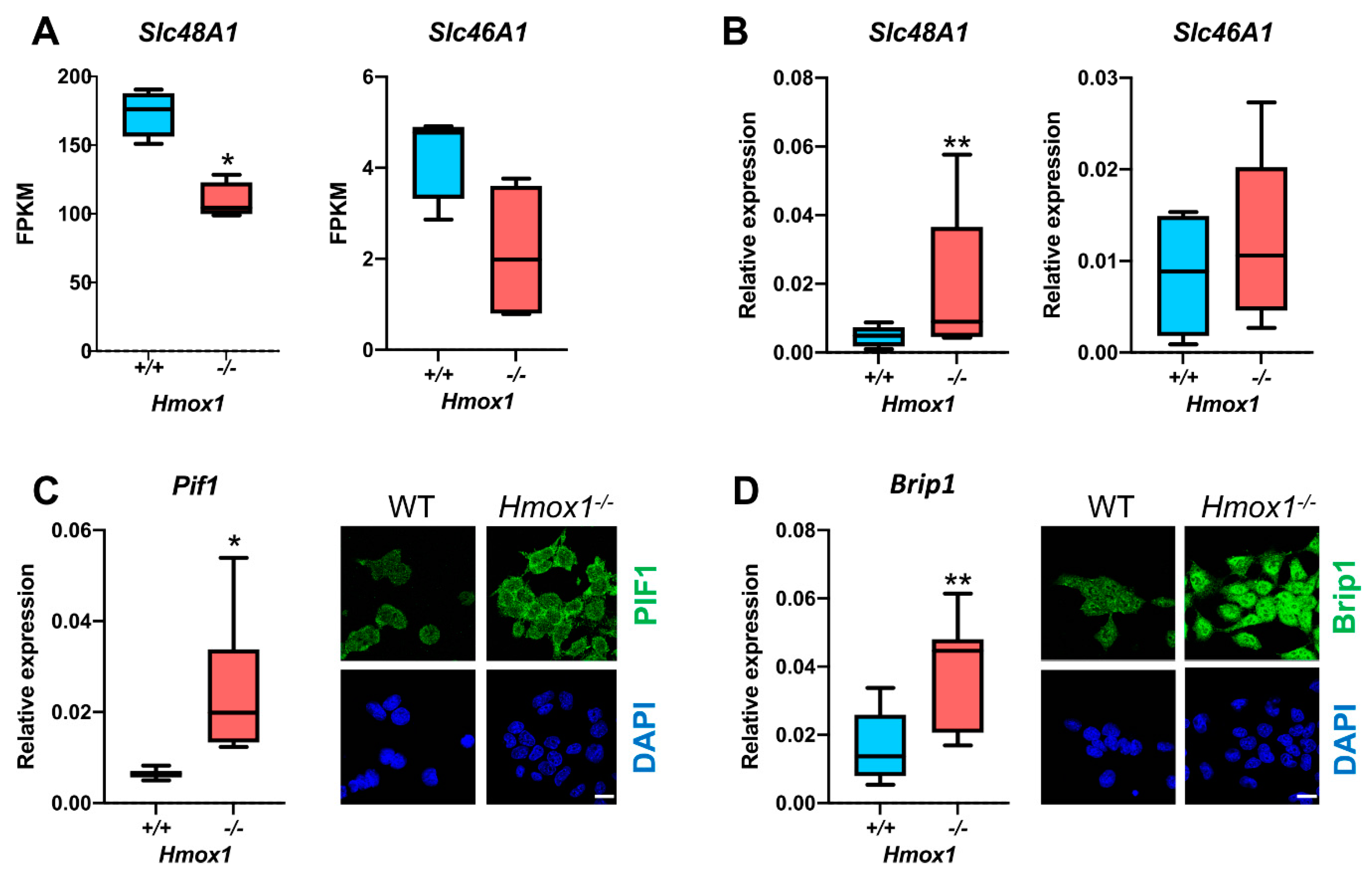
3.1. Hmox1−/− Hematopoietic Stem Cells Enhanced the Expression of G-quadruplex Helicases and Had Fewer G-quadruplexes
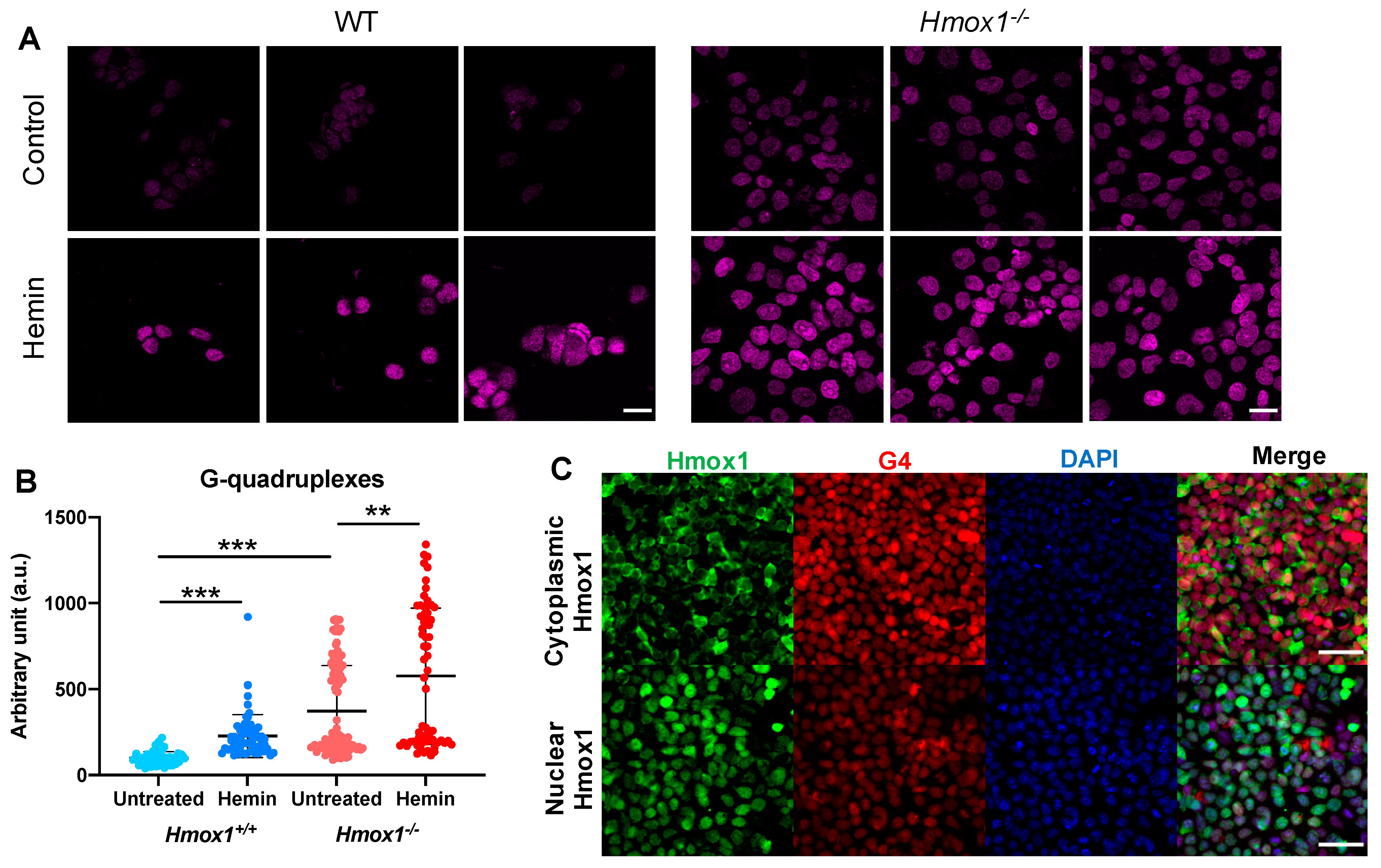
3.2. The Effect of Hmox1 Deficiency on G-quadruplexes Was Cell-Type Specific
3.3. HMOX1 Localized Close to G-quadruplexes in DNA
3.4. G4-HMOX1 Interaction Could Be Detected Using Flow Cytometry PLA
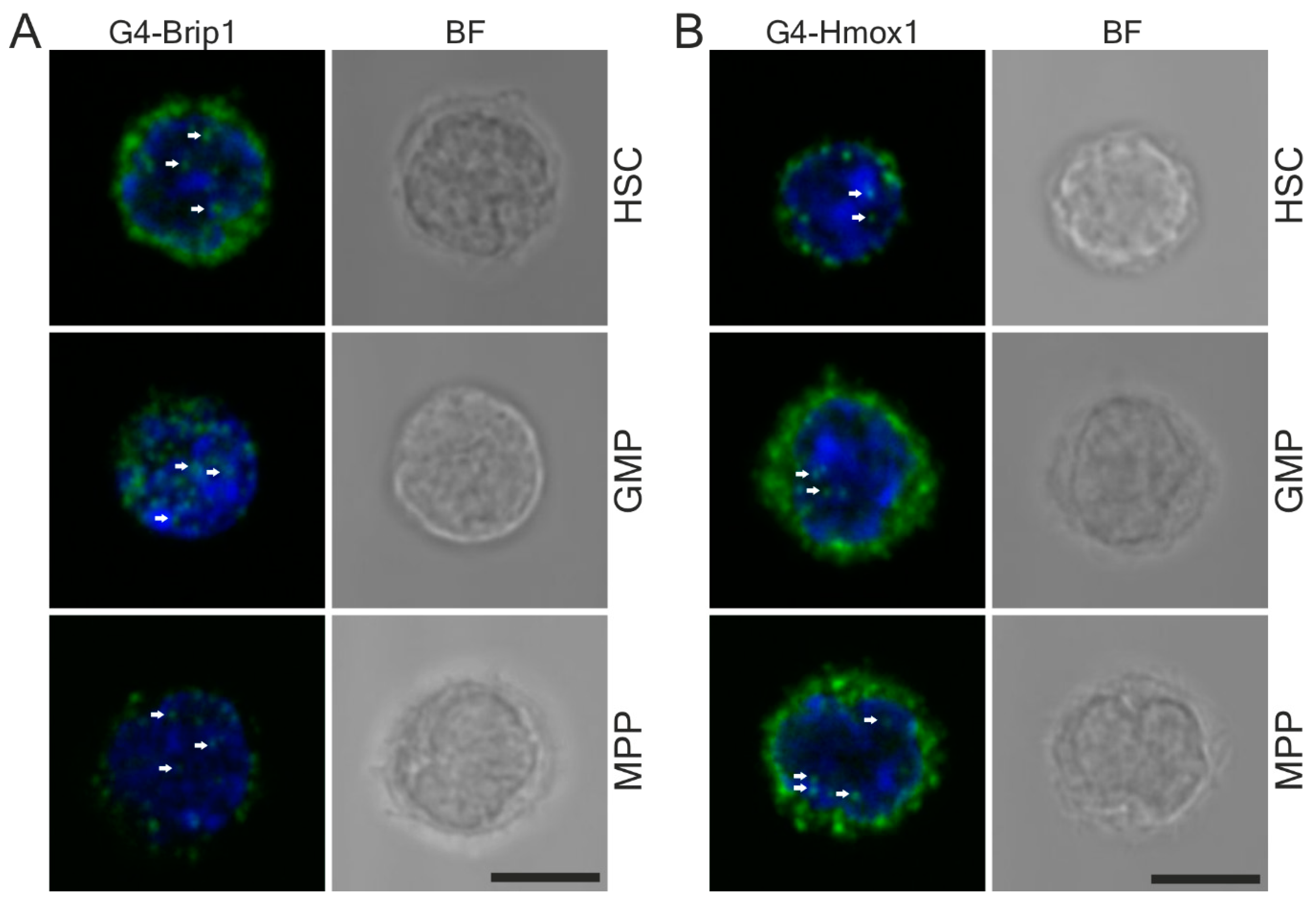
3.5. G-quadruplex and Hmox1 Interaction Could Be Detected in Primary Mouse Hematopoietic Stem Cells
3.6. Potential Interaction between Heme and G-quadruplexes Did Not Affect the Hmox1 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, J.T. G-Quartets 40 Years Later: From 5′-GMP to Molecular Biology and Supramolecular Chemistry. Angew. Chemie Int. Ed. 2004, 43, 668–698. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedrat, A.; Lacroix, L.; Mergny, J.-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef] [PubMed]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Chen, L.; Liu, W.; Yang, D. Molecular Recognition of the Hybrid-Type G-Quadruplexes in Human Telomeres. Molecules 2019, 24, 1578. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.-M.; Lemaitre, J.-M. Unraveling cell type–specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef]
- Kostadinov, R.; Malhotra, N.; Viotti, M.; Shine, R.; D’Antonio, L.; Bagga, P. GRSDB: A database of quadruplex forming G-rich sequences in alternatively processed mammalian pre-mRNA sequences. Nucleic Acids Res. 2006, 34, D119–D124. [Google Scholar] [CrossRef] [Green Version]
- Canesin, G.; Di Ruscio, A.; Li, M.; Ummarino, S.; Hedblom, A.; Choudhury, R.; Krzyzanowska, A.; Csizmadia, E.; Palominos, M.; Stiehm, A.; et al. Scavenging of Labile Heme by Hemopexin Is a Key Checkpoint in Cancer Growth and Metastases. Cell Rep. 2020, 32, 108181. [Google Scholar] [CrossRef] [PubMed]
- Andrushchenko, V.; Tsankov, D.; Krasteva, M.; Wieser, H.; Bouř, P. Spectroscopic detection of DNA quadruplexes by vibrational circular dichroism. J. Am. Chem. Soc. 2011, 133, 15055–15064. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.J.; Terentis, A.C. Analysis of G-quadruplex conformations using Raman and polarized Raman spectroscopy. J. Raman Spectrosc. 2016, 47, 259–268. [Google Scholar] [CrossRef]
- Adrian, M.; Heddi, B.; Phan, A.T. NMR spectroscopy of G-quadruplexes. Methods 2012, 57, 11–24. [Google Scholar] [CrossRef]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Kuo, I.C.; Ling, I.F.; Chen, C.T.; Chen, H.C.; Lou, P.J.; Lin, J.J.; Chang, T.C. Detection of quadruplex DNA structures in human telomeres by a fluorescent carbazole derivative. Anal. Chem. 2004, 76, 4490–4494. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Henderson, A.; Wu, Y.; Huang, Y.C.; Chavez, E.A.; Platt, J.; Johnson, F.B.; Brosh, R.M.; Sen, D.; Lansdorp, P.M.; Lansdorp, P.M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014, 42, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, O.; Bourdoncle, A.; Boulé, J.B.; Brosh, R.M.; Mergny, J.L. G-quadruplexes and helicases. Nucleic Acids Res. 2016, 44, 1989–2006. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Ribeyre, C.; Lopes, J.; Boulé, J.-B.; Piazza, A.; Guédin, A.; Zakian, V.A.; Mergny, J.-L.; Nicolas, A. The Yeast Pif1 Helicase Prevents Genomic Instability Caused by G-Quadruplex-Forming CEB1 Sequences In Vivo. PLoS Genet. 2009, 5, e1000475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brázda, V.; Hároníková, L.; Liao, J.C.C.; Fojta, M. DNA and RNA quadruplex-binding proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell 2016, 61, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Harvey, R.M.; Martinez-Guzman, O.; Yuan, X.; Chandrasekharan, B.; Raju, G.; Outten, F.W.; Hamza, I.; Reddi, A.R. Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. USA 2016, 113, 7539–7544. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, K.T.; Chang, H.C.; Ardehali, H. Role of heme in cardiovascular physiology and disease. J. Am. Heart Assoc. 2015, 4, e001401. [Google Scholar] [CrossRef] [Green Version]
- Travascio, P.; Li, Y.; Sen, D. DNA-enhanced peroxidase activity of a DNA aptamer-hemin complex. Chem. Biol. 1998, 5, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Grigg, J.C.; Shumayrikh, N.; Sen, D. G-quadruplex structures formed by expanded hexanucleotide repeat RNA and DNA from the neurodegenerative disease-linked C9orf72 gene efficiently sequester and activate heme. PLoS ONE 2014, 9, e106449. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Tai, H.; Fukaya, M.; Shibata, T.; Nishimura, R.; Neya, S.; Yamamoto, Y. Structural characterization of a carbon monoxide adduct of a heme-DNA complex. J. Biol. Inorg. Chem. 2012, 17, 437–445. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kinoshita, M.; Katahira, Y.; Shimizu, H.; Di, Y.; Shibata, T.; Tai, H.; Suzuki, A.; Neya, S. Characterization of Heme-DNA Complexes Composed of Some Chemically Modified Hemes and Parallel G-Quadruplex DNAs. Biochemistry 2015, 54, 7168–7177. [Google Scholar] [CrossRef]
- Shumayrikh, N.; Huang, Y.C.; Sen, D. Heme activation by DNA: Isoguanine pentaplexes, but not quadruplexes, bind heme and enhance its oxidative activity. Nucleic Acids Res. 2015, 43, 4191–4201. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 overexpression and underexpression: Clinical implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weibrecht, I.; Leuchowius, K.-J.; Clausson, C.-M.; Conze, T.; Jarvius, M.; Howell, W.M.; Kamali-Moghaddam, M.; Söderberg, O. Proximity ligation assays: A recent addition to the proteomics toolbox. Expert Rev. Proteomics 2010, 7, 401–409. [Google Scholar] [CrossRef]
- Gustafsdottir, S.M.; Schallmeiner, E.; Fredriksson, S.; Gullberg, M.; Söderberg, O.; Jarvius, M.; Jarvius, J.; Howell, M.; Landegren, U. Proximity ligation assays for sensitive and specific protein analyses. Anal. Biochem. 2005, 345, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Stepniewski, J.; Kachamakova-Trojanowska, N.; Ogrocki, D.; Szopa, M.; Matlok, M.; Beilharz, M.; Dyduch, G.; Malecki, M.T.; Jozkowicz, A.; Dulak, J. Induced pluripotent stem cells as a model for diabetes investigation. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepniewski, J.; Pacholczak, T.; Skrzypczyk, A.; Ciesla, M.; Szade, A.; Szade, K.; Bidanel, R.; Langrzyk, A.; Grochowski, R.; Vandermeeren, F.; et al. Heme oxygenase-1 affects generation and spontaneous cardiac differentiation of induced pluripotent stem cells. IUBMB Life 2018, 70, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Ema, H.; Morita, Y.; Yamazaki, S.; Matsubara, A.; Seita, J.; Tadokoro, Y.; Kondo, H.; Takano, H.; Nakauchi, H. Adult mouse hematopoietic stem cells: Purification and single-cell assays. Nat. Protoc. 2007, 1, 2979–2987. [Google Scholar] [CrossRef]
- Shivalingam, A.; Izquierdo, M.A.; Le Marois, A.; Vyšniauskas, A.; Suhling, K.; Kuimova, M.K.; Vilar, R. The interactions between a small molecule and G-quadruplexes are visualized by fluorescence lifetime imaging microscopy. Nat. Commun. 2015, 6, 8178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szade, K.; Zukowska, M.; Szade, A.; Nowak, W.; Skulimowska, I.; Ciesla, M.; Bukowska-Strakova, K.; Gulati, G.S.; Kachamakova-Trojanowska, N.; Kusienicka, A.; et al. Heme oxygenase-1 deficiency triggers exhaustion of hematopoietic stem cells. EMBO Rep. 2020, 21, e47895. [Google Scholar] [CrossRef] [PubMed]
- Pek, R.H.; Yuan, X.; Rietzschel, N.; Zhang, J.; Jackson, L.; Nishibori, E.; Ribeiro, A.; Simmons, W.; Jagadeesh, J.; Sugimoto, H.; et al. Hemozoin produced by mammals confers heme tolerance. Elife 2019, 8, e49503. [Google Scholar] [CrossRef]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [Green Version]
- Sacca, P.; Meiss, R.; Casas, G.; Mazza, O.; Calvo, J.C.; Navone, N.; Vazquez, E. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. Br. J. Cancer 2007, 97, 1683–1689. [Google Scholar] [CrossRef]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- London, T.B.C.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ Is a Structure-specific DNA Helicase Associated with the Maintenance of Genomic G/C Tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.T.; Puig Lombardi, E.; Verga, D.; Nicolas, A.; Teulade-Fichou, M.-P.; Londoño-Vallejo, A.; Maizels, N. G-quadruplexes Sequester Free Heme in Living Cells. Cell Chem. Biol. 2019, 26, 1681–1691.e5. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.L.; Zhang, Z.; Wang, M.; Lu, L.; Zhong, H.J.; Leung, C.H. Recent Developments in G-Quadruplex Probes. Chem. Biol. 2015, 22, 812–828. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.E.; Cao, K.; Ryvkin, P.; Wang, L.-S.; Johnson, F.B. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res. 2010, 38, 1114–1122. [Google Scholar] [CrossRef]
- Saha, A.; Duchambon, P.; Masson, V.; Loew, D.; Bombard, S.; Teulade-Fichou, M.-P. Nucleolin Discriminates Drastically between Long-Loop and Short-Loop Quadruplexes. Biochemistry 2020, 59, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Dong, S.; Wang, E. G-Quadruplex Aptamers with Peroxidase-Like DNAzyme Functions: Which Is the Best and How Does it Work? Chem. Asian J. 2009, 4, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Canale, T.D.; Sen, D. Hemin-utilizing G-quadruplex DNAzymes are strongly active in organic co-solvents. Biochim. Biophys. Acta 2017, 1861, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Geyer, C.R.; Sen, D. Recognition of Anionic Porphyrins by DNA Aptamers. Biochemistry 1996, 35, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Grochot-Przeczek, A.; Dulak, J.; Jozkowicz, A. Haem oxygenase-1: Non-canonical roles in physiology and pathology. Clin. Sci. 2012, 122, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Eelen, G.; Vanden Bempt, I.; Verlinden, L.; Drijkoningen, M.; Smeets, A.; Neven, P.; Christiaens, M.R.; Marchal, K.; Bouillon, R.; Verstuyf, A. Expression of the BRCA1-interacting protein Brip1/BACH1/FANCJ is driven by E2F and correlates with human breast cancer malignancy. Oncogene 2008, 27, 4233–4241. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Shin-ya, K.; Brosh, R.M.J. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [Green Version]
- Stroik, S.; Kurtz, K.; Lin, K.; Karachenets, S.; Myers, C.L.; Bielinsky, A.-K.; Hendrickson, E.A. EXO1 resection at G-quadruplex structures facilitates resolution and replication. Nucleic Acids Res. 2020, 48, 4960–4975. [Google Scholar] [CrossRef] [Green Version]
- Dunn, L.L.; Midwinter, R.G.; Ni, J.; Hamid, H.A.; Parish, C.R.; Stocker, R. New Insights into Intracellular Locations and Functions of Heme Oxygenase-1. Antioxid. Redox Signal. 2014, 20, 1723. [Google Scholar] [CrossRef] [Green Version]
- Paez, A.V.; Pallavicini, C.; Schuster, F.; Valacco, M.P.; Giudice, J.; Ortiz, E.G.; Anselmino, N.; Labanca, E.; Binaghi, M.; Salierno, M.; et al. Heme oxygenase-1 in the forefront of a multi-molecular network that governs cell–cell contacts and filopodia-induced zippering in prostate cancer. Cell Death Dis. 2016, 7, e2570. [Google Scholar] [CrossRef]
- Mestre-Fos, S.; Ito, C.; Moore, C.M.; Reddi, A.R.; Williams, L.D. Human ribosomal G-quadruplexes regulate heme bioavailability. J. Biol. Chem. 2020, 295, 14855–14865. [Google Scholar] [CrossRef] [PubMed]
- Sweeny, E.A.; Singh, A.B.; Chakravarti, R.; Martinez-Guzman, O.; Saini, A.; Haque, M.M.; Garee, G.; Dans, P.D.; Hannibal, L.; Reddi, A.R.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 2018, 293, 14557–14568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, R.F.; Moshkin, Y.M.; Mouton, S.; Grzeschik, N.A.; Kalicharan, R.D.; Kuipers, J.; Wolters, A.H.G.; Nishida, K.; Romashchenko, A.V.; Postberg, J.; et al. Guanine quadruplex structures localize to heterochromatin. Nucleic Acids Res. 2016, 44, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamura, T.; Katsuda, Y.; Kitamura, Y.; Ihara, T. G-quadruplexes in mRNA: A key structure for biological function. Biochem. Biophys. Res. Commun. 2020, 526, 261–266. [Google Scholar] [CrossRef]
- Avin, A.; Levy, M.; Porat, Z.; Abramson, J. Quantitative analysis of protein-protein interactions and post-translational modifications in rare immune populations. Nat. Commun. 2017, 8, 1524. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzeptowski, W.; Chudy, P.; Sokołowski, G.; Żukowska, M.; Kusienicka, A.; Seretny, A.; Kalita, A.; Czmoczek, A.; Gubała, J.; Baran, S.; et al. Proximity Ligation Assay Detection of Protein–DNA Interactions—Is There a Link between Heme Oxygenase-1 and G-quadruplexes? Antioxidants 2021, 10, 94. https://doi.org/10.3390/antiox10010094
Krzeptowski W, Chudy P, Sokołowski G, Żukowska M, Kusienicka A, Seretny A, Kalita A, Czmoczek A, Gubała J, Baran S, et al. Proximity Ligation Assay Detection of Protein–DNA Interactions—Is There a Link between Heme Oxygenase-1 and G-quadruplexes? Antioxidants. 2021; 10(1):94. https://doi.org/10.3390/antiox10010094
Chicago/Turabian StyleKrzeptowski, Wojciech, Patryk Chudy, Grzegorz Sokołowski, Monika Żukowska, Anna Kusienicka, Agnieszka Seretny, Agata Kalita, Alicja Czmoczek, Jakub Gubała, Sonia Baran, and et al. 2021. "Proximity Ligation Assay Detection of Protein–DNA Interactions—Is There a Link between Heme Oxygenase-1 and G-quadruplexes?" Antioxidants 10, no. 1: 94. https://doi.org/10.3390/antiox10010094
APA StyleKrzeptowski, W., Chudy, P., Sokołowski, G., Żukowska, M., Kusienicka, A., Seretny, A., Kalita, A., Czmoczek, A., Gubała, J., Baran, S., Klóska, D., Jeż, M., Stępniewski, J., Szade, K., Szade, A., Grochot-Przęczek, A., Józkowicz, A., & Nowak, W. N. (2021). Proximity Ligation Assay Detection of Protein–DNA Interactions—Is There a Link between Heme Oxygenase-1 and G-quadruplexes? Antioxidants, 10(1), 94. https://doi.org/10.3390/antiox10010094