
ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression

 ,
,
Abstract
:1. Introduction
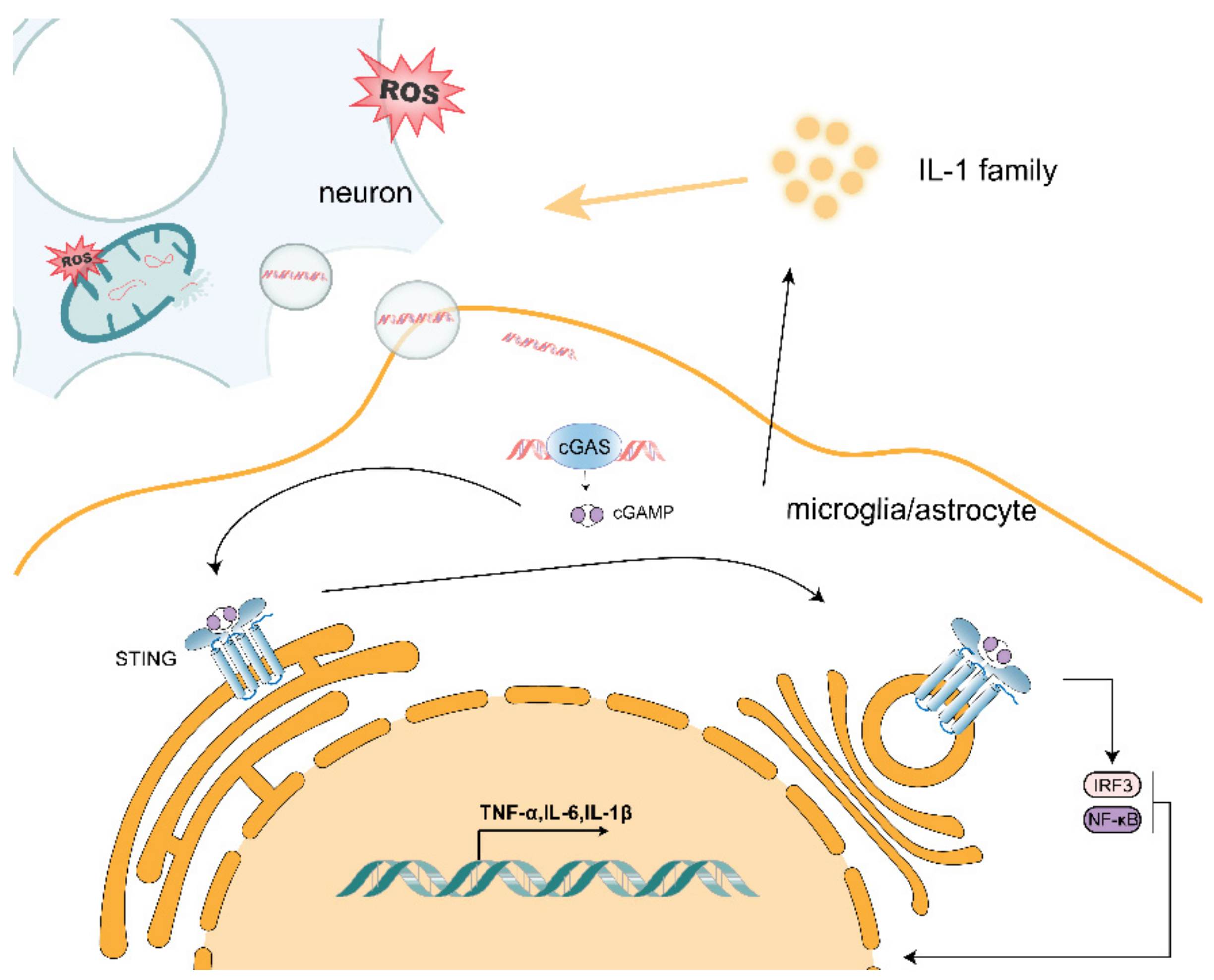
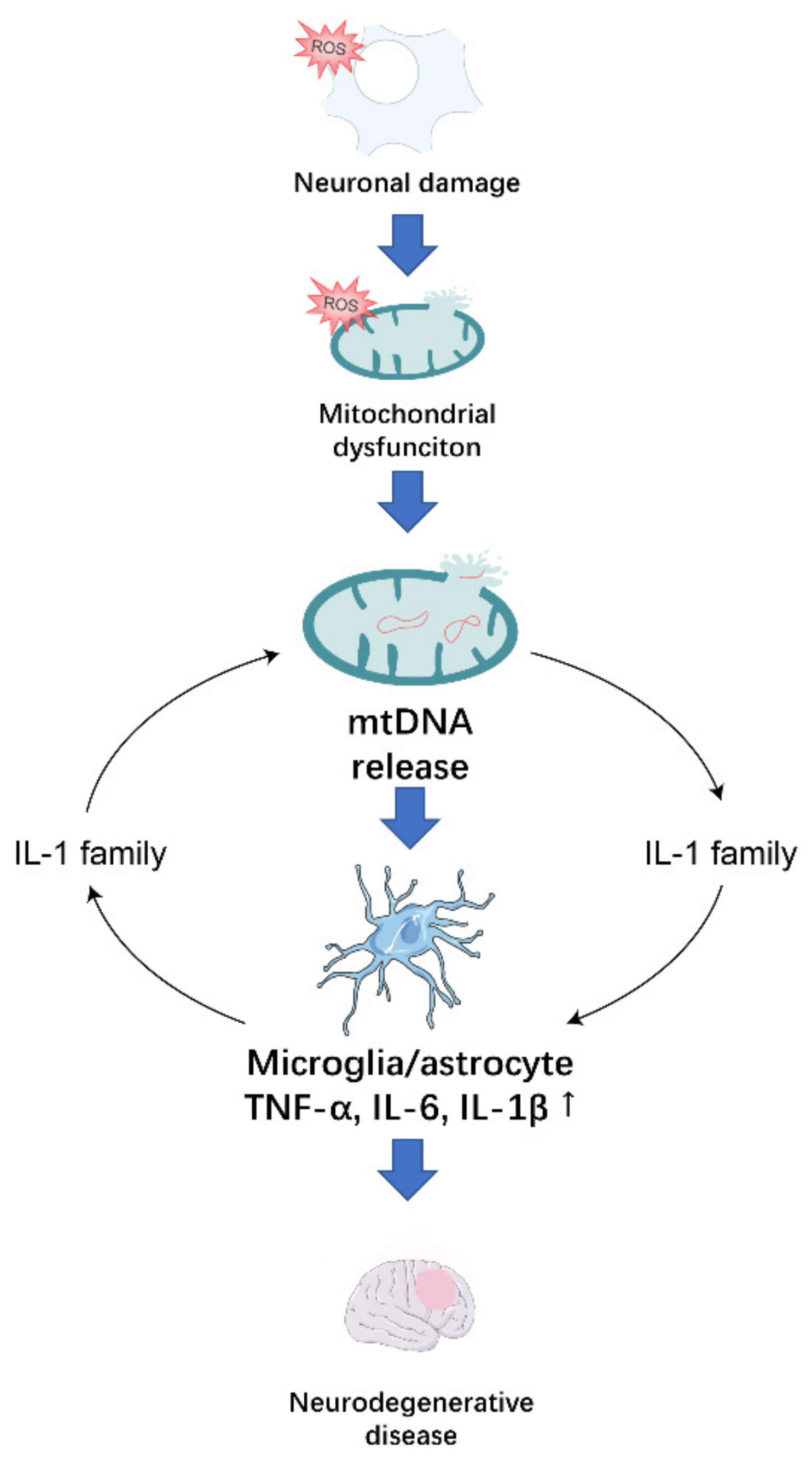
2. ROS-Induced Mitochondrial Dysfunction Promotes Neurodegeneration through the Release of mtDNA
2.1. Possible Mechanisms of mtDNA Release
2.2. Effect of mtDNA on Microglia
2.3. Effect of mtDNA on Astrocytes
2.4. mtDNA Promotes Activation of the cGAS/STING Pathway
2.5. mtDNA/cGAS/STING Pathway Exacerbates Neurodegenerative Disease
3. The IL-1 Family Plays an Important Role in the Formation of the Inflammatory Environment in Neurodegenerative Diseases
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2019, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Heemels, M.-T. Neurodegenerative diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, H.; Iguchi, Y.; Sahashi, K.; Katsuno, M. Significance of Oligomeric and Fibrillar Species in Amyloidosis: Insights into Pathophysiology and Treatment. Molecules 2021, 26, 5091. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.; Mercado-Ayon, E.; Mercado-Ayon, Y.; Na Dong, Y.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2020, 702, 108698. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S210–S212. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2020, 36, 16–24. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Bonte, M.-A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.-C. Glycosphingolipids and neuroinflammation in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 1–16. [Google Scholar] [CrossRef]
- Yun, Y.; Ha, Y. CRISPR/Cas9-Mediated Gene Correction to Understand ALS. Int. J. Mol. Sci. 2020, 21, 3801. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, Z.; Alagöz, M.A.; Bahçecioğlu, F.; Gök, S. Monoamine Oxidase-B (MAO-B) Inhibitors in the Treatment of Alzheimer’s and Parkinson’s Disease. Curr. Med. Chem. 2021, 28, 6045–6065. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Srivastav, S.; Castellani, R.J.; Plascencia-Villa, G.; Perry, G. Neuroprotective and Antioxidant Effect of Ginkgo biloba Extract Against AD and Other Neurological Dis-orders. Neurotherapeutics 2019, 16, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Coley, N.; Ousset, P.J.; Berrut, G.; Dartigues, J.F.; Dubois, B.; Grandjean, H.; Pasquier, F.; Piette, F.; Robert, P.; et al. Long-term use of standardised Ginkgo biloba extract for the prevention of Alzheimer’s disease (GuidAge): A randomised placebo-controlled trial. Lancet Neurol. 2012, 11, 851–859. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Johnson, S.L.; Liu, W.; DaSilva, N.A.; Meschwitz, S.; Dain, J.A.; Seeram, N.P. Evaluation of Polyphenol Anthocyanin-Enriched Extracts of Blackberry, Black Raspberry, Blueberry, Cranberry, Red Raspberry, and Strawberry for Free Radical Scavenging, Reactive Carbonyl Species Trapping, Anti-Glycation, Anti-β-Amyloid Aggregation, and Microglial Neuroprotective Effects. Int. J. Mol. Sci. 2018, 19, 461. [Google Scholar] [CrossRef] [Green Version]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A Focus on Several Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2015, 2015, 392169. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.; Matté, A.; Pagnussat, A.S.; Netto, C.A.; Salbego, C.G. Resveratrol prevents CA1 neurons against ischemic injury by parallel modulation of both GSK-3beta and CREB through PI3-K/Akt pathways. Eur. J. Neurosci. 2012, 36, 2899–2905. [Google Scholar] [CrossRef]
- Attia, H.; Maklad, Y.A. Neuroprotective effects of coenzyme Q10 on paraquat-induced Parkinson’s disease in experimental animals. Behav. Pharmacol. 2018, 29, 79–86. [Google Scholar] [CrossRef]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2013, 2, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.; Yadav, R.K.; Minj, E.; Tiwari, A.; Mehan, S. Exploring Molecular Approaches in Amyotrophic Lateral Sclerosis: Drug Targets from Clinical and Pre-Clinical Findings. Curr. Mol. Pharmacol. 2021, 14, 263–280. [Google Scholar] [CrossRef]
- Enogieru, A.B.; Haylett, W.; Hiss, D.C.; Bardien, S.; Ekpo, O.E. Rutin as a Potent Antioxidant: Implications for Neurodegenerative Disorders. Oxidative Med. Cell. Longev. 2018, 2018, 6241017. [Google Scholar] [CrossRef] [PubMed]
- Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of Sulforaphane in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8637. [Google Scholar] [CrossRef]
- Denzer, I.; Münch, G.; Friedland, K. Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacol. Res. 2015, 103, 80–94. [Google Scholar] [CrossRef]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Merino, C.; Lopez-Sanchez, C.; Lagoa, R.; Samhan-Arias, A.K.; Bueno, C.; Garcia-Martinez, V. Neuroprotective actions of flavonoids. Curr. Med. Chem. 2011, 18, 1195–1212. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Wen, L.L.; Huang, Y.-N.; Chen, Y.-T.; Ku, M.-C. Dual Effects of Antioxidants in Neurodegeneration: Direct Neuroprotection against Oxidative Stress and Indirect Protection via Suppression of Gliamediated Inflammation. Curr. Pharm. Des. 2006, 12, 3521–3533. [Google Scholar] [CrossRef]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef]
- Salmina, A.B.; Komleva, Y.K.; Szijártó, I.A.; Gorina, Y.V.; Lopatina, O.L.; Gertsog, G.E.; Filipovic, M.R.; Gollasch, M. H2S- and NO-Signaling Pathways in Alzheimer’s Amyloid Vasculopathy: Synergism or Antagonism? Front. Physiol. 2015, 6, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, T.; Saha, P.; Jiang, T.; Sen, N. Sulfhydration of AKT triggers Tau-phosphorylation by activating glycogen synthase kinase 3β in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 4418–4427. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid β formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Tusi, S.K.; Nouhi, F.; Abdi, A.; Khodagholi, F. Dietary supplementation with tBHQ, an Nrf2 stabilizer molecule, confers neuroprotection against apoptosis in amyloid beta-injected rat. J. Biotechnol. 2010, 150, 455. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, C.; Guo, Y.; Han, Y.; Wang, C.; Chu, H.; Kong, L.; Ma, H. TBHQ Attenuates Neurotoxicity Induced by Methamphetamine in the VTA through the Nrf2/HO-1 and PI3K/AKT Signaling Pathways. Oxidative Med. Cell. Longev. 2020, 2020, 8787156. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.; Pehar, M.; Cassina, P.; Beckman, J.S.; Barbeito, L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J. Neurochem. 2006, 97, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-Inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef]
- Yamazaki, H.; Tanji, K.; Wakabayashi, K.; Matsuura, S.; Itoh, K. Role of the Keap1/Nrf2 pathway in neurodegenerative diseases. Pathol. Int. 2015, 65, 210–219. [Google Scholar] [CrossRef]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson’s Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-κB/Nuclear Transcription Factor Related to NF-E2. Antioxidants Redox Signal. 2017, 27, 453–471. [Google Scholar] [CrossRef] [Green Version]
- Vucic, S.; Ryder, J.; Mekhael, L.; Rd, H.; Mathers, S.; Needham, M.; Dw, S.; MC, K. Phase 2 randomized placebo controlled double blind study to assess the efficacy and safety of tecfidera in patients with amyotrophic lateral sclerosis (TEALS Study). Medicine 2020, 99, e18904. [Google Scholar] [CrossRef]
- Ettcheto, M.; Cano, A.; Manzine, P.R.; Busquets, O.; Verdaguer, E.; Castro-Torres, R.D.; García, M.L.; Beas-Zarate, C.; Olloquequi, J.; Auladell, C.; et al. Epigallocatechin-3-Gallate (EGCG) Improves Cognitive Deficits Aggravated by an Obesogenic Diet Through Modulation of Unfolded Protein Response in APPswe/PS1dE9 Mice. Mol. Neurobiol. 2019, 57, 1814–1827. [Google Scholar] [CrossRef]
- Cano, A.; Ettcheto, M.; Chang, J.-H.; Barroso, E.; Espina, M.; Kühne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J. Control. Release 2019, 301, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, S.; Arsic, A.; Ristic-Medic, D.; Cvetkovic, Z.; Vucic, V. Lipid Peroxidation and Antioxidant Supplementation in Neurodegenerative Diseases: A Review of Human Studies. Antioxidants 2020, 9, 1128. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liu, M.; Yao, W.; Du, K.; He, M.; Jin, X.; Jiao, L.; Ma, G.; Wei, B.; Wei, M. Epigallocatechin-3-Gallate Attenuates Microglial Inflammation and Neurotoxicity by Suppressing the Activation of Canonical and Noncanonical Inflammasome via TLR4/NF-κB Pathway. Mol. Nutr. Food Res. 2019, 63, 1801230. [Google Scholar] [CrossRef]
- Koh, S.-H.; Lee, S.M.; Kim, H.Y.; Lee, K.-Y.; Lee, Y.J.; Kim, H.-T.; Kim, J.; Kim, M.-H.; Hwang, M.S.; Song, C.; et al. The effect of epigallocatechin gallate on suppressing disease progression of ALS model mice. Neurosci. Lett. 2006, 395, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Bradley-Whitman, M.A.; Timmons, M.D.; Beckett, T.L.; Murphy, M.P.; Lynn, B.C.; Lovell, M.A. Nucleic acid oxidation: An early feature of Alzheimer’s disease. J. Neurochem. 2013, 128, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Yong, H.Y.F.; Rawji, K.S.; Ghorbani, S.; Xue, M.; Yong, V.W. The benefits of neuroinflammation for the repair of the injured central nervous system. Cell. Mol. Immunol. 2019, 16, 540–546. [Google Scholar] [CrossRef]
- Bargiela, D.; Chinnery, P.F. Mitochondria in neuroinflammation—Multiple sclerosis (MS), leber hereditary optic neuropathy (LHON) and LHON-MS. Neurosci. Lett. 2019, 710, 132932. [Google Scholar] [CrossRef]
- Pinti, M.; Ferraro, D.; Nasi, M. Microglia activation: A role for mitochondrial DNA? Neural. Regen. Res. 2021, 16, 2393–2394. [Google Scholar] [CrossRef]
- Zeng, F.; Wu, Y.; Li, X.; Ge, X.; Guo, Q.; Lou, X.; Cao, Z.; Hu, B.; Long, N.J.; Mao, Y.; et al. Custom-Made Ceria Nanoparticles Show a Neuroprotective Effect by Modulating Phenotypic Polarization of the Microglia. Angew. Chem. Int. Ed. 2018, 57, 5808–5812. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Yu, K.; Youshani, A.S.; Wilkinson, F.L.; O’Leary, C.; Cook, P.; Laaniste, L.; Liao, A.; Mosses, D.; Waugh, C.; Shorrock, H.; et al. A nonmyeloablative chimeric mouse model accurately defines microglia and macrophage contribution in glioma. Neuropathol. Appl. Neurobiol. 2018, 45, 119–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Krysko, D.; Agostinis, P.; Krysko, O.; Garg, A.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Cheng, A.N.; Cheng, L.-C.; Kuo, C.-L.; Lo, Y.K.; Chou, H.-Y.; Chen, C.-H.; Wang, Y.-H.; Chuang, T.-H.; Cheng, S.-J.; Lee, A.Y.-L. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1–mediated immunoescape via STING-IFN signaling and extracellular vesicles. J. Immunother. Cancer 2020, 8, e001372. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e5. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.; Zhao, J.; Zhou, X.; Yang, Q.; Chen, Y.; Zhu, J.; Yuan, P.; Yang, J.; Qin, T.; Wan, S.; et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene 2019, 38, 5007–5020. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Tan, X.; Li, S.; Al-Nusaif, M.; Le, W. Role of Glia-Derived Extracellular Vesicles in Neurodegenerative Diseases. Front. Aging Neurosci. 2021, 13, 765395. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernández-Delgado, I.; Latorre-Pellicer, A.; Acín-Pérez, R.; Martín-Cófreces, N.B.; Jaso-Tamame, Á.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through anti-gen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Cheng, J.; Kong, X.; Li, S.; Li, X.; Zhang, M.; Zhang, H.; Yang, T.; Dong, Y.; Li, J.; et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 2020, 10, 9644–9662. [Google Scholar] [CrossRef] [PubMed]
- Mathur, V.; Burai, R.; Vest, R.T.; Bonanno, L.; Lehallier, B.; Zardeneta, M.E.; Mistry, K.N.; Do, D.; Marsh, S.; Abud, E.M.; et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron 2017, 96, 1290–1302.e6. [Google Scholar] [CrossRef] [Green Version]
- Tsilioni, I.; Theoharides, T.C. Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1β. J. Neuroinflamm. 2018, 15, 1–8. [Google Scholar] [CrossRef]
- Nakanishi, H.; Hayashi, Y.; Wu, Z. The role of microglial mtDNA damage in age-dependent prolonged LPS-induced sickness behavior. Neuron Glia Biol. 2011, 7, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Nasi, M.; De Gaetano, A.; Bianchini, E.; De Biasi, S.; Gibellini, L.; Neroni, A.; Mattioli, M.; Pinti, M.; Tartaro, D.L.; Borella, R.; et al. Mitochondrial damage-associated molecular patterns stimulate reactive oxygen species production in human microglia. Mol. Cell. Neurosci. 2020, 108, 103538. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.; Kang, H.Y.; Lim, H.R.; Kwon, Y.; Jo, M.; Jeon, Y.M.; Kim, S.R.; Kim, K.; Ha, C.M.; et al. The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory re-sponse. J. Neuroinflamm. 2020, 17, 299. [Google Scholar] [CrossRef]
- Hu, J.; Bibli, S.I.; Wittig, J.; Zukunft, S.; Lin, J.; Hammes, H.-P.; Popp, R.; Fleming, I. Soluble epoxide hydrolase promotes astrocyte survival in retinopathy of prematurity. J. Clin. Investig. 2019, 129, 5204–5218. [Google Scholar] [CrossRef]
- Ignatenko, O.; Chilov, D.; Paetau, I.; De Miguel, E.; Jackson, C.B.; Capin, G.; Paetau, A.; Terzioglu, M.; Euro, L.; Suomalainen, A. Loss of mtDNA activates astrocytes and leads to spongiotic encephalopathy. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Liu, Q.; Li, X.; Li, S.; Chen, J.; Hong, Z.; Wu, X.; Zhao, Y.; Ren, J. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis. 2020, 11, 1050. [Google Scholar] [CrossRef] [PubMed]
- E Leurs, C.; Podlesniy, P.; Trullas, R.; Balk, L.; Steenwijk, M.D.; Malekzadeh, A.; Piehl, F.; Uitdehaag, B.M.; Killestein, J.; Van Horssen, J.; et al. Cerebrospinal fluid mtDNA concentration is elevated in multiple sclerosis disease and responds to treatment. Mult. Scler. J. 2017, 24, 472–480. [Google Scholar] [CrossRef]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Cervera-Carles, L.; Alcolea, D.; Estanga, A.; Ecay-Torres, M.; Izagirre, A.; Clerigué, M.; Garcia-Sebastian, M.; Villanúa, J.; Escalas, C.; Blesa, R.; et al. Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. Neurobiol. Aging 2017, 53, 192.e1–192.e4. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, G.; Gallotti, C. [Aneurysms of the hepatic artery. Clinico-radiologic study]. Radiol. Med. 1987, 74, 49–58. [Google Scholar]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA–induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.; Gale, M. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef]
- Han, Y.; Chen, L.; Liu, H.; Jin, Z.; Wu, Y.; Wu, Y.; Li, W.; Ying, S.; Chen, Z.; Shen, H.; et al. Airway Epithelial cGAS Is Critical for Induction of Experimental Allergic Airway Inflammation. J. Immunol. 2020, 204, 1437–1447. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Y.; Liu, H.; Liu, Z.; Zhao, J. Mitochondrial DNA from hepatocytes induces upregulation of interleukin-33 expression of macrophages in nonalcoholic steatohepatitis. Dig. Liver Dis. 2020, 52, 637–643. [Google Scholar] [CrossRef]
- Ozasa, K.; Temizoz, B.; Kusakabe, T.; Kobari, S.; Momota, M.; Coban, C.; Ito, S.; Kobiyama, K.; Kuroda, E.; Ishii, K.J. Cyclic GMP-AMP Triggers Asthma in an IL-33-Dependent Manner That Is Blocked by Amlexanox, a TBK1 Inhibitor. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Xu, H.; Sun, L.; He, Y.; Yuan, X.; Niu, J.; Su, J.; Li, D. Deficiency in IL-33/ST2 Axis Reshapes Mitochondrial Metabolism in Lipopolysaccharide-Stimulated Macrophages. Front. Immunol. 2019, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Li, D.; Ma, J.; Zhao, Y.; Xu, L.; Tian, R.; Liu, Y.; Sun, L.; Su, J. The IL-33/ST2 axis affects tumor growth by regulating mitophagy in macrophages and reprogramming their polarization. Cancer Biol. Med. 2021, 18, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Hu, M.-M.; Bian, L.-J.; Liu, Y.; Chen, Q.; Shu, H.-B. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.; Martinez, L.A. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021, 39, 494–508.e5. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Antioxidants | Research Progress | Antioxidant Mechanism | Relative Pathway | Diseases | References |
|---|---|---|---|---|---|
| Selegiline/Rasagiline | Clinical applications | Monoamine oxidase type B (MAO-B) inhibitor | PD | [13] | |
| GBE | In Vivo and in vitro models; clinical trials | Free radical–scavenging action | JNK, ERK1/2, Akt | AD | [14,15] |
| Anthocyanins extracted | In Vivo models | Free radical–scavenging action | PI3K/Akt/Nrf2 | AD | [16,17] |
| Resveratrol | In Vivo and in vitro models | Maintaining the levels of antioxidant enzymes; free radical–scavenging action | AMPK, PI3K/Akt/GSK-3β | AD, PD, ALS | [18,19] |
| Coenzyme Q10 | In Vivo and in vitro models; clinical trials | Antioxidant in mitochondria and lipid membranes | PD, ALS | [20,21,22] | |
| Rutin | In vitro and in vivo models | Directly scavenge ROS | JNK, p38 MAPK, | AD, PD | [23] |
| SFN | In vitro and in vivo models | Free radical–scavenging action | Nrf2/ARE | AD, PD, ALS | [24,25] |
| Flavonoids | In Vivo models | Free radical–scavenging action | NF-κB/iNOS | AD, PD, ALS | [26,27,28] |
| H2S | In vitro and in vivo models | Mediating the activities of glutathione peroxidase, SOD and catalase | Akt/Nrf2/GSK-3β,NO pathway | AD, PD, ALS | [29,30,31,32,33] |
| tBHQ | In Vivo models | An Nrf2 Stabilizer | NF-κB/HSP70, PI3K/Akt | AD, ALS | [34,35,36,37] |
| DMF | In vitro and in vivo models | An Nrf2 activator | p62/Keap1/Nrf2/ARE, NF-κB | AD, PD, ALS | [38,39,40,41] |
| EGCG | In vitro and in vivo models | Free radical–scavenging action | ATF4/PTP1B, TLR4/NF-κB | AD, ALS | [42,43,44,45,46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917. https://doi.org/10.3390/antiox10121917
Zhao Y, Liu B, Xu L, Yu S, Fu J, Wang J, Yan X, Su J. ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants. 2021; 10(12):1917. https://doi.org/10.3390/antiox10121917
Chicago/Turabian StyleZhao, Yuanxin, Buhan Liu, Long Xu, Sihang Yu, Jiaying Fu, Jian Wang, Xiaoyu Yan, and Jing Su. 2021. "ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression" Antioxidants 10, no. 12: 1917. https://doi.org/10.3390/antiox10121917
APA StyleZhao, Y., Liu, B., Xu, L., Yu, S., Fu, J., Wang, J., Yan, X., & Su, J. (2021). ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants, 10(12), 1917. https://doi.org/10.3390/antiox10121917