
Ginsenoside Rg3 Attenuates TNF-α-Induced Damage in Chondrocytes through Regulating SIRT1-Mediated Anti-Apoptotic and Anti-Inflammatory Mechanisms


 and
and 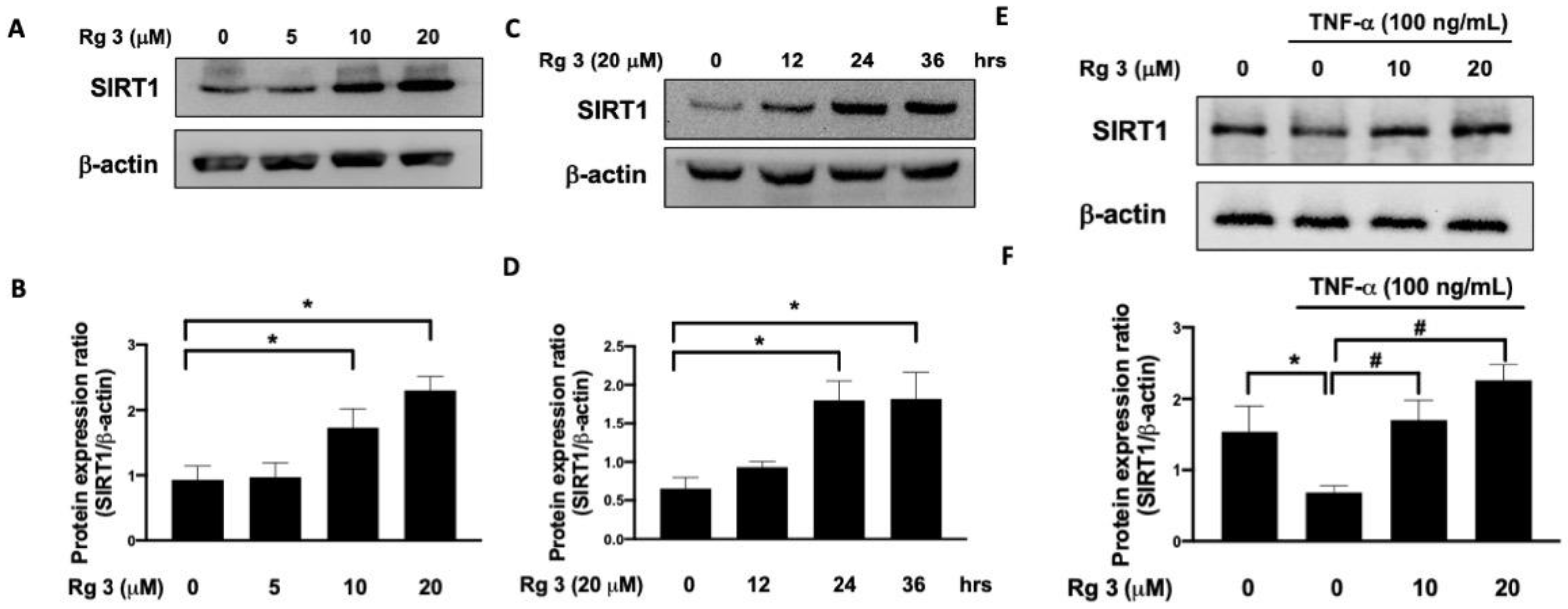{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Immunoblotting
2.3. Investigation of Mitochondrial Biogenesis and Mitochondrial Membrane Potential
2.4. Mitochondrial Superoxide Indicator
2.5. Preparation of Nuclear/Cytosolic Extracts and Mitochondria Isolation
2.6. Co-Immunoprecipitation (Co-IP)
2.7. Investigation of Apoptosis
2.8. Transfection with Small-Interfering RNA
2.9. IL-8 and MMP-9 Release
2.10. Statistical Analyses
3. Result
3.1. Administration of Ginsenoside Rg3 Reverses the TNF-α-Inhibited SIRT1 Expression
3.2. Prevention of TNF-α-Induced Acetylation of CypD Using Ginsenoside Rg3 Is Mediated by SIRT1/PGC-1α/SIRT3 Pathway
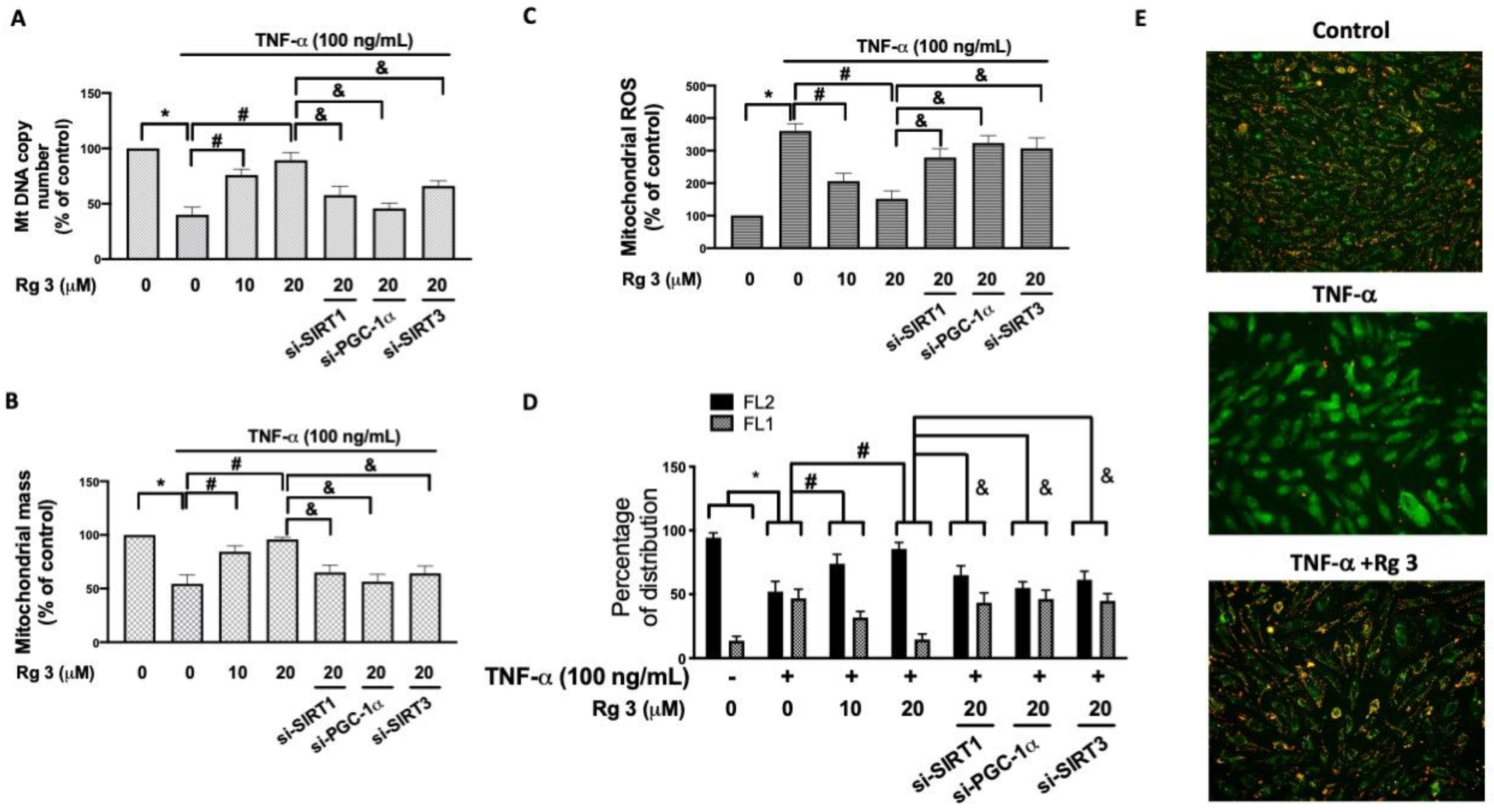
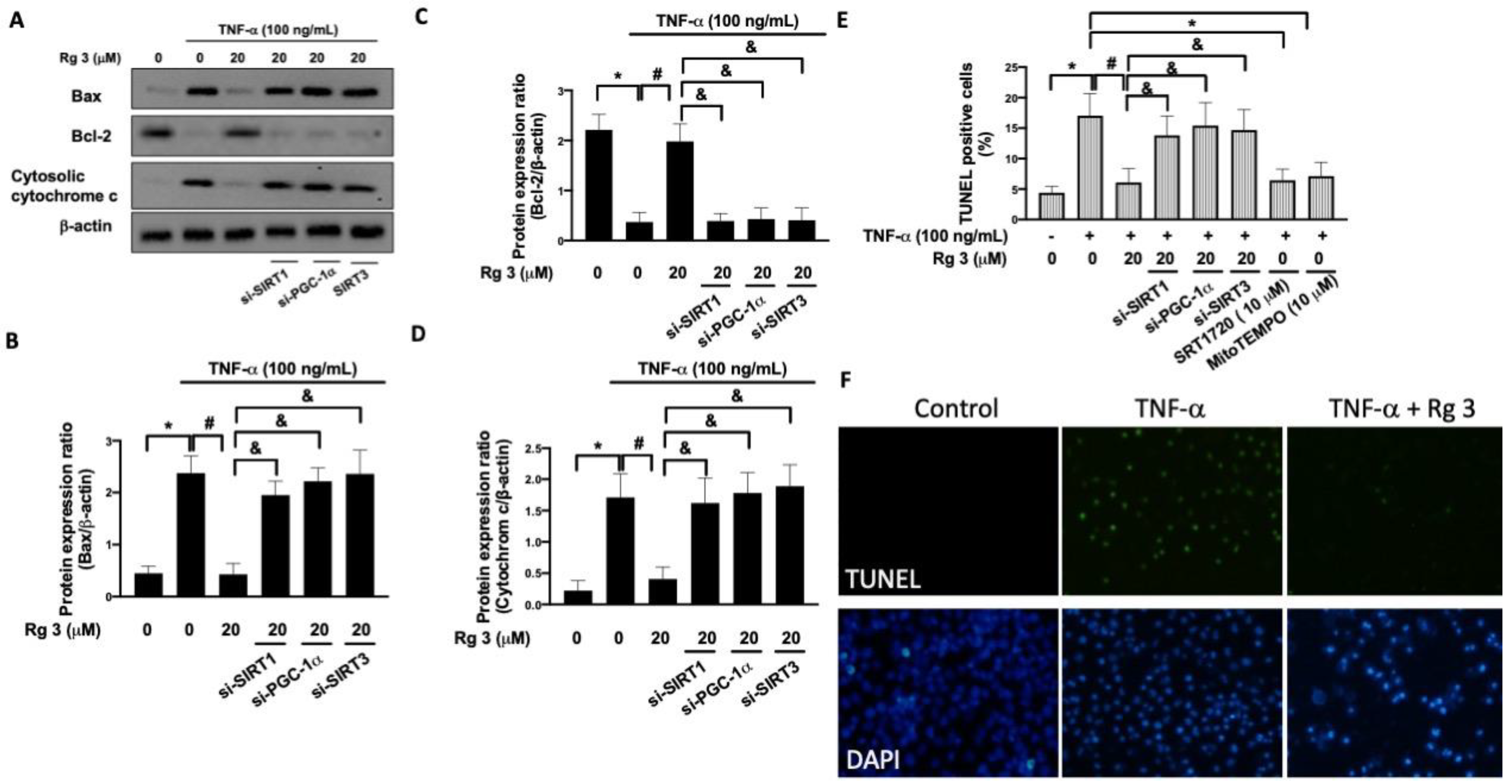
3.3. Ginsenoside Rg3 Ameliorates the TNF-α-Induced Mitochondrial Dysfunction and Apoptosis via SIRT1/PGC-1α/SIRT3 Pathway
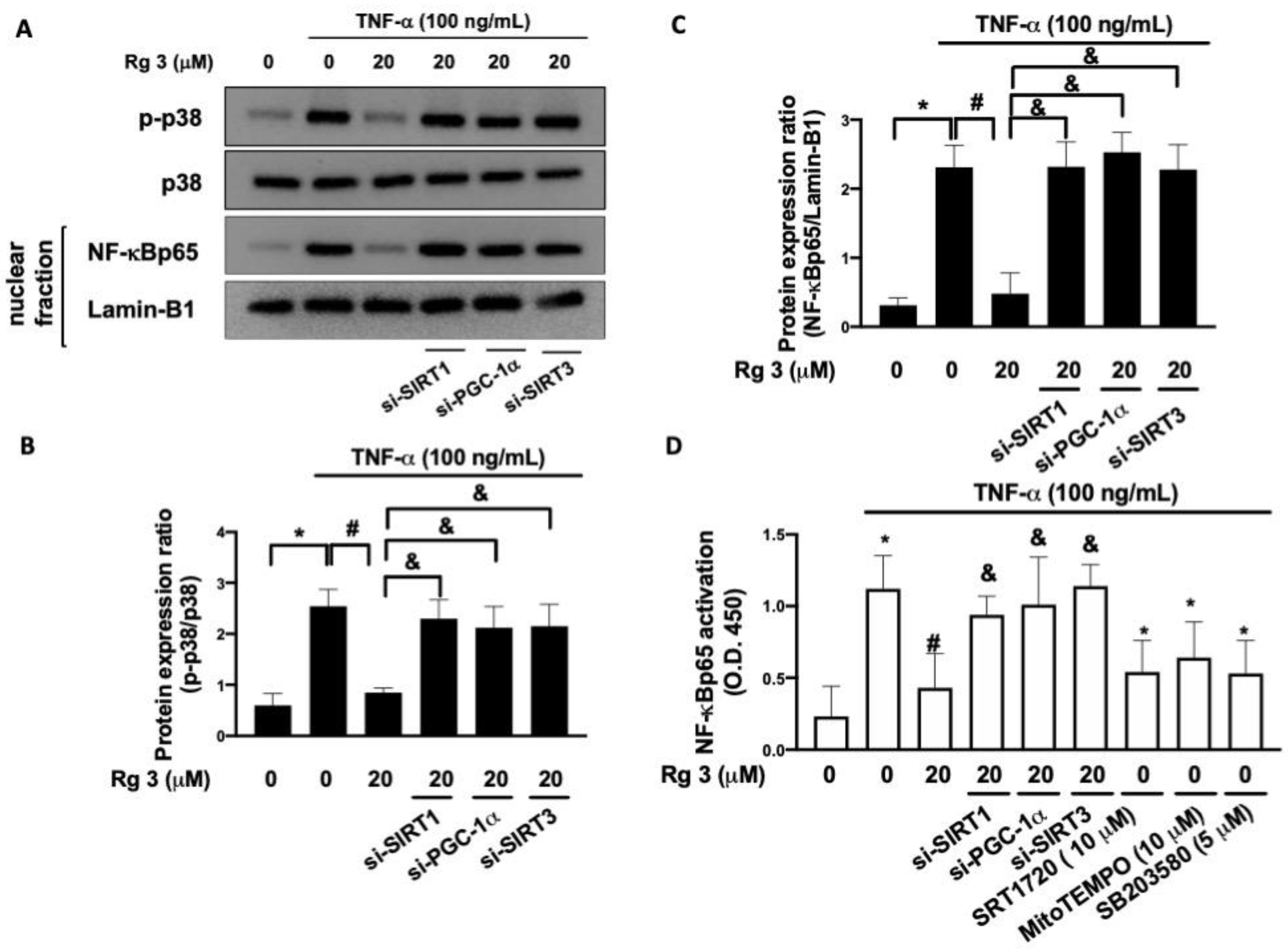
3.4. Ginsenoside Rg3 Suppresses the TNF-α-Stimulated p38 MAPK Phosphorylation and NF-κB Activation through SIRT1/PGC-1α/SIRT3 Signaling
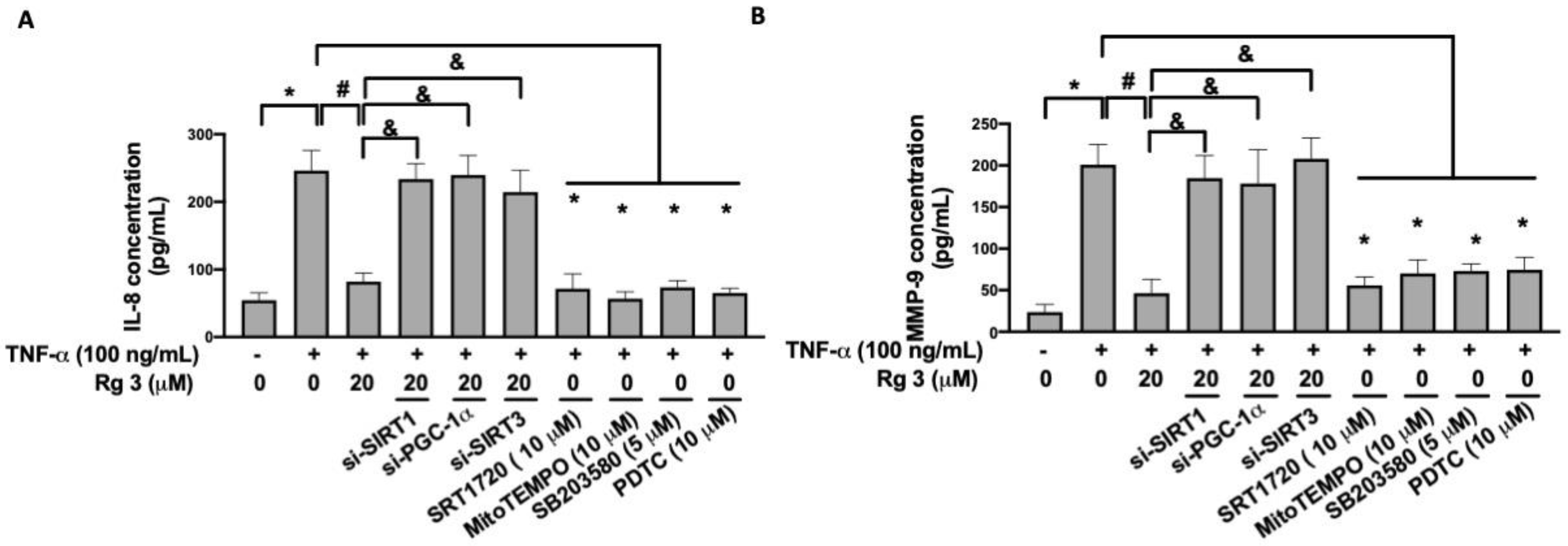
3.5. Ginsenoside Rg3 Inhibits the TNF-α-Increased Production of IL-8 and MMP-9 via SIRT1/PGC-1α/SIRT3/p38 MAPK/NF-κB Pathway
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chu, C.Q.; Field, M.; Feldmann, M.; Maini, R.N. Localization of tumor necrosis factor alpha in synovial tissues and at the cartilage-pannus junction in patients with rheumatoid arthritis. Arthritis Rheum. 1991, 34, 1125–1132. [Google Scholar] [CrossRef]
- Caramés, B.; López-Armada, M.J.; Cillero-Pastor, B.; Lires-Dean, M.; Vaamonde, C.; Galdo, F.; Blanco, F.J. Differential effects of tumor necrosis factor-alpha and interleukin-1beta on cell death in human articular chondrocytes. Osteoarthr. Cartil. 2008, 16, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Borzi, R.M.; Mazzetti, I.; Macor, S.; Silvestri, T.; Bassi, A.; Cattini, L.; Facchini, A. Flow cytometric analysis of intracellular chemokines in chondrocytes in vivo: Constitutive expression and enhancement in osteoarthritis and rheumatoid arthritis. FEBS Lett. 1999, 455, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Merz, D.; Liu, R.; Johnson, K.; Terkeltaub, R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J. Immunol. 2003, 171, 4406–4415. [Google Scholar] [CrossRef] [Green Version]
- Dreier, R. Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 2010, 12, 216. [Google Scholar] [CrossRef] [Green Version]
- Freemont, A.J.; Hampson, V.; Tilman, R.; Goupille, P.; Taiwo, Y.; Hoyland, J.A. Gene expression of matrix metalloproteinases 1, 3, and 9 by chondrocytes in osteoarthritic human knee articular cartilage is zone and grade specific. Ann. Rheum. Dis. 1997, 56, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Won, H.J.; Kim, H.I.; Park, T.; Kim, H.; Jo, K.; Jeon, H.; Ha, S.J.; Hyun, J.M.; Jeong, A.; Kim, J.S.; et al. Non-clinical pharmacokinetic behavior of ginsenosides. J. Ginseng Res. 2019, 43, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, J.H. A review on the medicinal potentials of ginseng and ginsenosides on cardiovascular diseases. J. Ginseng Res. 2014, 38, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.A.; Kim, S.; Chang, S.H.; Hwang, H.J.; Choi, Y.N. Anti-arthritic effect of ginsenoside Rb1 on collagen induced arthritis in mice. Int. Immunopharmacol. 2007, 7, 1286–1291. [Google Scholar] [CrossRef]
- Kim, S.; Na, J.Y.; Song, K.B.; Choi, D.S.; Kim, J.H.; Kwon, Y.B.; Kwon, J. Protective Effect of Ginsenoside Rb1 on Hydrogen Peroxide-induced Oxidative Stress in Rat Articular Chondrocytes. J. Ginseng Res. 2012, 36, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Na, J.Y.; Kim, S.; Song, K.; Lim, K.H.; Shin, G.W.; Kim, J.H.; Kim, B.; Kwon, Y.B.; Kwon, J. Anti-apoptotic Activity of Ginsenoside Rb1 in Hydrogen Peroxide-treated Chondrocytes: Stabilization of Mitochondria and the Inhibition of Caspase-3. J. Ginseng Res. 2012, 36, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lim, H.; Shehzad, O.; Kim, Y.S.; Kim, H.P. Ginsenosides from Korean red ginseng inhibit matrix metalloproteinase-13 expression in articular chondrocytes and prevent cartilage degradation. Eur. J. Pharmacol. 2014, 724, 145–151. [Google Scholar] [CrossRef] [PubMed]
- So, M.W.; Lee, E.J.; Lee, H.S.; Koo, B.S.; Kim, Y.G.; Lee, C.K.; Yoo, B. Protective effects of ginsenoside Rg3 on human osteoarthritic chondrocytes. Mod. Rheumatol. 2013, 23, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Dvir-Ginzberg, M.; Mobasheri, A.; Kumar, A. The Role of Sirtuins in Cartilage Homeostasis and Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 43. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Lai, X.D.; Ouyang, J.; Yang, J.D. Effects of Ginsenoside Rg3 on fatigue resistance and SIRT1 in aged rats. Toxicology 2018, 409, 144–151. [Google Scholar] [CrossRef]
- Oppenheimer, H.; Gabay, O.; Meir, H.; Haze, A.; Kandel, L.; Liebergall, M.; Gagarina, V.; Lee, E.J.; Dvir-Ginzberg, M. 75-kd sirtuin 1 blocks tumor necrosis factor α-mediated apoptosis in human osteoarthritic chondrocytes. Arthritis Rheum. 2012, 64, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany N. Y.) 2010, 2, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef]
- Rego, A.C.; Vesce, S.; Nicholls, D.G. The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT1-7 neural cells. Cell Death Differ. 2001, 8, 995–1003. [Google Scholar] [CrossRef]
- Zwerina, J.; Hayer, S.; Redlich, K.; Bobacz, K.; Kollias, G.; Smolen, J.S.; Schett, G. Activation of p38 MAPK is a key step in tumor necrosis factor-mediated inflammatory bone destruction. Arthritis Rheum. 2006, 54, 463–472. [Google Scholar] [CrossRef]
- Ulivi, V.; Giannoni, P.; Gentili, C.; Cancedda, R.; Descalzi, F. p38/NF-kB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J. Cell Biochem. 2008, 104, 1393–1406. [Google Scholar] [CrossRef]
- Pulai, J.I.; Chen, H.; Im, H.J.; Kumar, S.; Hanning, C.; Hegde, P.S.; Loeser, R.F. NF-kappa B mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J. Immunol. 2005, 174, 5781–5788. [Google Scholar] [CrossRef] [Green Version]
- Shakibaei, M.; John, T.; Schulze-Tanzil, G.; Lehmann, I.; Mobasheri, A. Suppression of NF-kappaB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: Implications for the treatment of osteoarthritis. Biochem. Pharmacol. 2007, 73, 1434–1445. [Google Scholar] [CrossRef]
- Zhou, Y.D.; Hou, J.G.; Liu, W.; Ren, S.; Wang, Y.P.; Zhang, R.; Chen, C.; Wang, Z.; Li, W. 20(R)-ginsenoside Rg3, a rare saponin from red ginseng, ameliorates acetaminophen-induced hepatotoxicity by suppressing PI3K/AKT pathway-mediated inflammation and apoptosis. Int. Immunopharmacol. 2018, 59, 21–30. [Google Scholar] [CrossRef]
- Kee, J.Y.; Hong, S.H. Ginsenoside Rg3 suppresses mast cell-mediated allergic inflammation via mitogen-activated protein kinase signaling pathway. J. Ginseng Res. 2019, 43, 282–290. [Google Scholar] [CrossRef]
- Lee, I.S.; Uh, I.; Kim, K.S.; Kim, K.H.; Park, J.; Kim, Y.; Jung, J.H.; Jung, H.J.; Jang, H.J. Anti-Inflammatory Effects of Ginsenoside Rg3 via NF-κB Pathway in A549 Cells and Human Asthmatic Lung Tissue. J. Immunol. Res. 2016, 2016, 7521601. [Google Scholar] [CrossRef] [Green Version]
- Tu, C.; Wan, B.; Zeng, Y. Ginsenoside Rg3 alleviates inflammation in a rat model of myocardial infarction via the SIRT1/NF-κB pathway. Exp. Ther. Med. 2020, 20, 238. [Google Scholar] [CrossRef]
- Ren, B.; Feng, J.; Yang, N.; Guo, Y.; Chen, C.; Qin, Q. Ginsenoside Rg3 attenuates angiotensin II-induced myocardial hypertrophy through repressing NLRP3 inflammasome and oxidative stress via modulating SIRT1/NF-κB pathway. Int. Immunopharmacol. 2021, 98, 107841. [Google Scholar] [CrossRef]
- Takayama, K.; Ishida, K.; Matsushita, T.; Fujita, N.; Hayashi, S.; Sasaki, K.; Tei, K.; Kubo, S.; Matsumoto, T.; Fujioka, H.; et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 2009, 60, 2731–2740. [Google Scholar] [CrossRef]
- Niederer, F.; Ospelt, C.; Brentano, F.; Hottiger, M.O.; Gay, R.E.; Gay, S.; Detmar, M.; Kyburz, D. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann. Rheum. Dis 2011, 70, 1866–1873. [Google Scholar] [CrossRef]
- Dvir-Ginzberg, M.; Gagarina, V.; Lee, E.J.; Booth, R.; Gabay, O.; Hall, D.J. Tumor necrosis factor α-mediated cleavage and inactivation of SirT1 in human osteoarthritic chondrocytes. Arthritis Rheum. 2011, 63, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xiao, W.; Wu, P.; Deng, Z.; Zeng, C.; Li, H.; Yang, T.; Lei, G. The expression of SIRT1 in articular cartilage of patients with knee osteoarthritis and its correlation with disease severity. J. Orthop. Surg. Res. 2016, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Takayama, K.; Kurosaka, M.; Kuroda, R. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J. Orthop. Res. 2011, 29, 511–515. [Google Scholar] [CrossRef]
- Sacitharan, P.K.; Bou-Gharios, G.; Edwards, J.R. SIRT1 directly activates autophagy in human chondrocytes. Cell Death Discov. 2020, 6, 41. [Google Scholar] [CrossRef]
- Gagarina, V.; Gabay, O.; Dvir-Ginzberg, M.; Lee, E.J.; Brady, J.K.; Quon, M.J.; Hall, D.J. SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010, 62, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Jackson, C.J.; Park, S.Y. SIRT1, a class III histone deacetylase, regulates TNF-α-induced inflammation in human chondrocytes. Osteoarthr. Cartil. 2013, 21, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Sasaki, H.; Takayama, K.; Ishida, K.; Matsumoto, T.; Kubo, S.; Matsuzaki, T.; Nishida, K.; Kurosaka, M.; Kuroda, R. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1β in human chondrocytes. J. Orthop. Res. 2013, 31, 531–537. [Google Scholar] [CrossRef]
- Lianxu, C.; Hongti, J.; Changlong, Y. NF-kappaBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and TNF-alpha-induced chondrocytes. Osteoarthr. Cartil. 2006, 14, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.H.; Wu, C.H.; Jou, I.M.; Tu, Y.K.; Hung, C.H.; Hsieh, P.L.; Tsai, K.L. PKR activation causes inflammation and MMP-13 secretion in human degenerated articular chondrocytes. Redox Biol. 2018, 14, 72–81. [Google Scholar] [CrossRef]
- Ma, C.H.; Wu, C.H.; Jou, I.M.; Tu, Y.K.; Hung, C.H.; Chou, W.C.; Chang, Y.C.; Hsieh, P.L.; Tsai, K.L. PKR Promotes Oxidative Stress and Apoptosis of Human Articular Chondrocytes by Causing Mitochondrial Dysfunction through p38 MAPK Activation-PKR Activation Causes Apoptosis in Human Chondrocytes. Antioxidants 2019, 8, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Wang, Y.; Terkeltaub, R.; Liu-Bryan, R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr. Cartil. 2018, 26, 1539–1550. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, K.; Huang, C.; Lin, D.; Zhou, Y.; Wu, Y.; Tian, N.; Fan, P.; Pan, X.; Xu, D.; et al. SIRT3 Activation by Dihydromyricetin Suppresses Chondrocytes Degeneration via Maintaining Mitochondrial Homeostasis. Int. J. Biol. Sci. 2018, 14, 1873–1882. [Google Scholar] [CrossRef] [Green Version]
- Baeza, J.; Smallegan, M.J.; Denu, J.M. Mechanisms and Dynamics of Protein Acetylation in Mitochondria. Trends Biochem. Sci. 2016, 41, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Zhou, Y.; Dai, Q.; Qiao, C.; Zhao, L.; Hui, H.; Lu, N.; Guo, Q.L. Oroxylin A induces dissociation of hexokinase II from the mitochondria and inhibits glycolysis by SIRT3-mediated deacetylation of cyclophilin D in breast carcinoma. Cell Death Dis. 2013, 4, e601. [Google Scholar] [CrossRef] [Green Version]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Díaz-Delfín, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem. 2011, 286, 16958–16966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, C.-H.; Chou, W.-C.; Wu, C.-H.; Jou, I.-M.; Tu, Y.-K.; Hsieh, P.-L.; Tsai, K.-L. Ginsenoside Rg3 Attenuates TNF-α-Induced Damage in Chondrocytes through Regulating SIRT1-Mediated Anti-Apoptotic and Anti-Inflammatory Mechanisms. Antioxidants 2021, 10, 1972. https://doi.org/10.3390/antiox10121972
Ma C-H, Chou W-C, Wu C-H, Jou I-M, Tu Y-K, Hsieh P-L, Tsai K-L. Ginsenoside Rg3 Attenuates TNF-α-Induced Damage in Chondrocytes through Regulating SIRT1-Mediated Anti-Apoptotic and Anti-Inflammatory Mechanisms. Antioxidants. 2021; 10(12):1972. https://doi.org/10.3390/antiox10121972
Chicago/Turabian StyleMa, Ching-Hou, Wan-Ching Chou, Chin-Hsien Wu, I-Ming Jou, Yuan-Kun Tu, Pei-Ling Hsieh, and Kun-Ling Tsai. 2021. "Ginsenoside Rg3 Attenuates TNF-α-Induced Damage in Chondrocytes through Regulating SIRT1-Mediated Anti-Apoptotic and Anti-Inflammatory Mechanisms" Antioxidants 10, no. 12: 1972. https://doi.org/10.3390/antiox10121972
APA StyleMa, C. -H., Chou, W. -C., Wu, C. -H., Jou, I. -M., Tu, Y. -K., Hsieh, P. -L., & Tsai, K. -L. (2021). Ginsenoside Rg3 Attenuates TNF-α-Induced Damage in Chondrocytes through Regulating SIRT1-Mediated Anti-Apoptotic and Anti-Inflammatory Mechanisms. Antioxidants, 10(12), 1972. https://doi.org/10.3390/antiox10121972