
Preventing Oxidative Stress in the Liver: An Opportunity for GLP-1 and/or PASK

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Liver Metabolic Functions and Detoxification
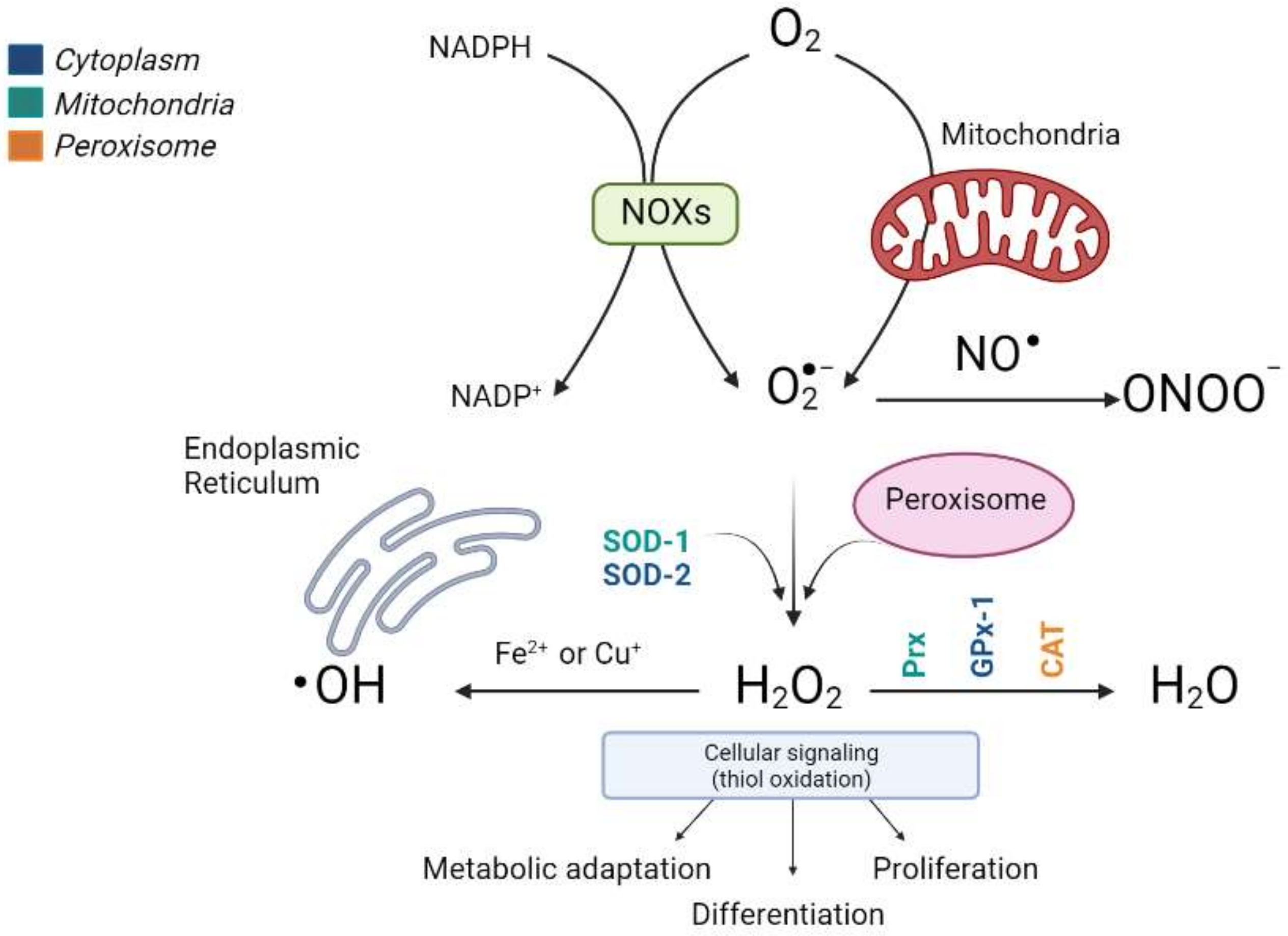
2.1. ROS and Antioxidant Defense
2.2. Hepatic Oxidative Stress and Nutritional Status
3. Nutrient Sensors and Oxidative Stress
3.1. AMPK and mTOR
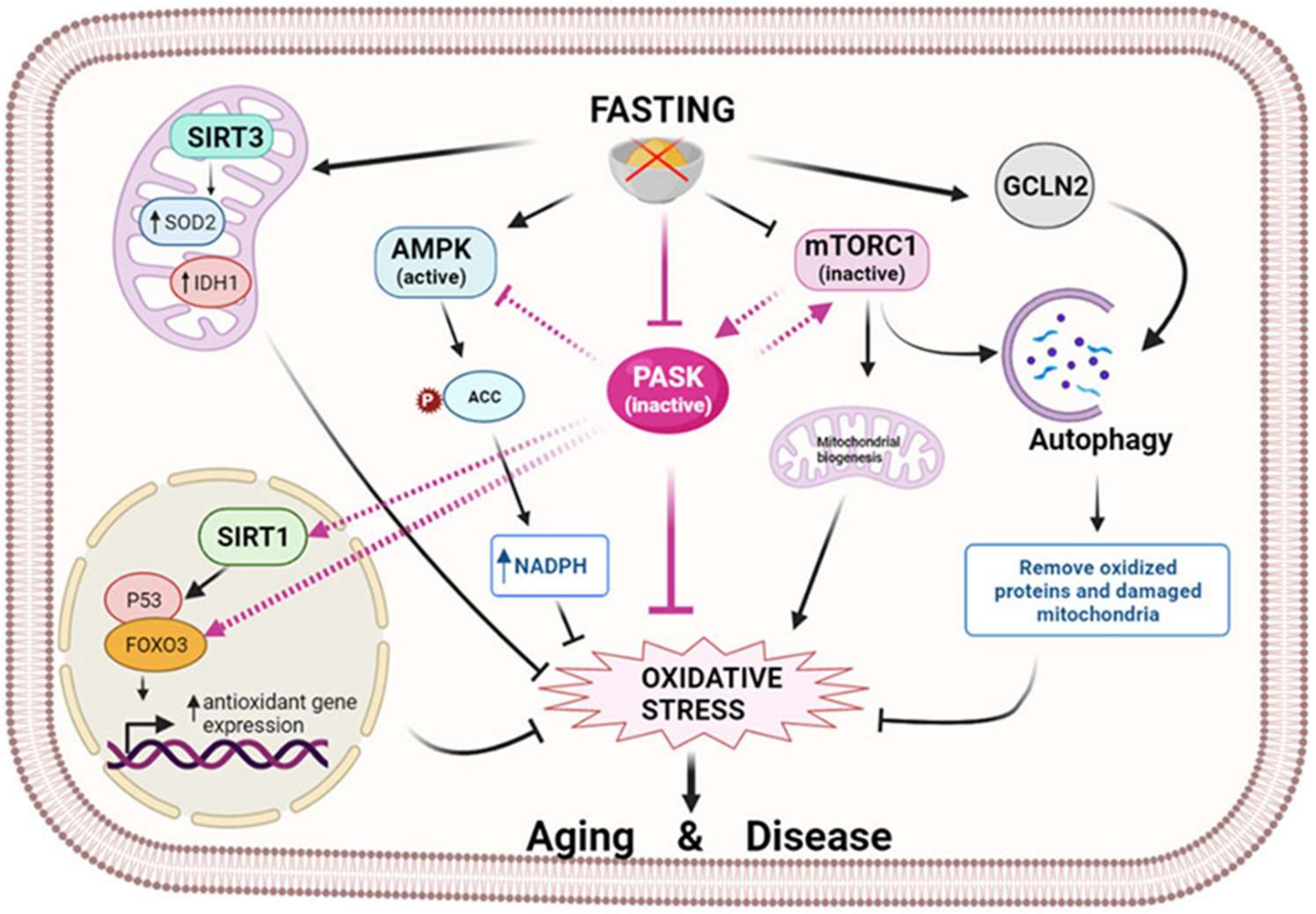
3.2. PASK
3.3. Sirtuin Family
4. Potential Role of PASK and Exendin-4/GLP-1 in Therapy
4.1. PASK Deficiency Reduces Hepatic Oxidative Stress
4.2. GLP-1 Role in Oxidative Stress
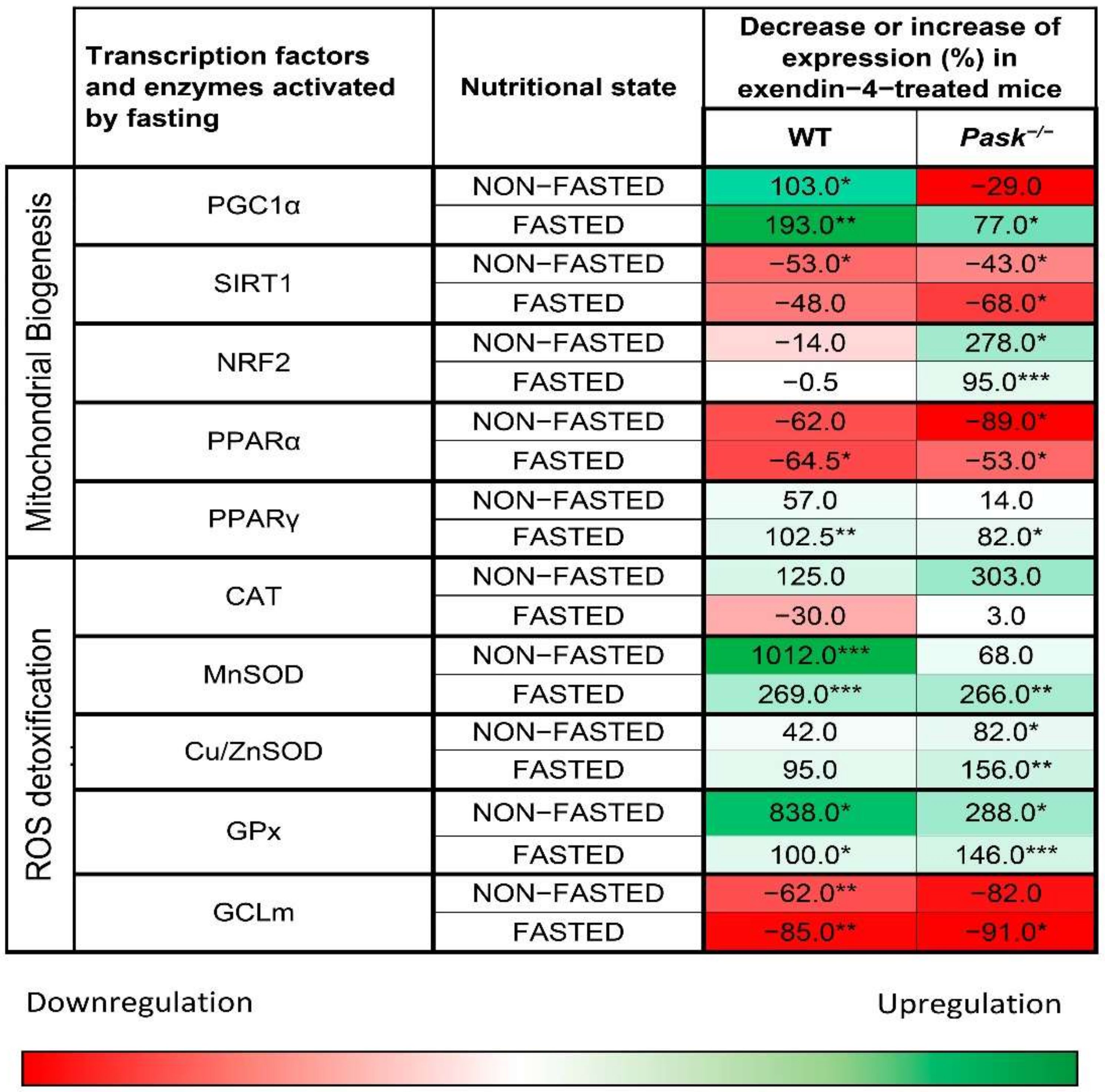
4.3. Evidence for Exendin-4/GLP-1 and PASK Interplay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [Green Version]
- Chiu, S.; Mulligan, K.; Schwarz, J.-M. Dietary carbohydrates and fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 277–282. [Google Scholar] [CrossRef]
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [Green Version]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Stud. Surf. Sci. Catal. 2015, 74, 35–84. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Fukami, T.; Yokoi, T.; Nakajima, M. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab. Pharmacokinet. 2015, 30, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Edeas, M.; Attaf, D.; Mailfert, A.-S.; Nasu, M.; Joubet, R. Maillard Reaction, mitochondria and oxidative stress: Potential role of antioxidants. Pathol. Biol. 2010, 58, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Ramachandran, A. Antioxidant Defense Mechanisms. Compr. Toxicol. 2018, 2, 277–295. [Google Scholar]
- Zhu, L.; Lu, Y.; Zhang, J.; Hu, Q. Subcellular Redox Signaling. Adv. Exp. Med. Biol. 2017, 967, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ramachandran, A. Reactive oxygen species in the normal and acutely injured liver. J. Hepatol. 2011, 55, 227–228. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Murray, F.J.; Monnot, A.D.; Jacobson-Kram, D.; Cohen, S.M.; Hardisty, J.F.; Atillasoy, E.; Hermanowski-Vosatka, A.; Kuffner, E.; Wikoff, D.; et al. Assessment of the biochemical pathways for acetaminophen toxicity: Implications for its carcinogenic hazard potential. Regul. Toxicol. Pharmacol. 2021, 120, 104859. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Valle, V.; Chavez-Tapia, N.; Uribe, M.; Mendez-Sanchez, N. Role of Oxidative Stress and Molecular Changes in Liver Fibrosis: A Review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Sorensen, M.; Sanz, A.; Gomez, J.; Pamplona, R.; Portero-Otín, M.; Gredilla, R.; Barja, G. Effects of fasting on oxidative stress in rat liver mitochondria. Free. Radic. Res. 2006, 40, 339–347. [Google Scholar] [CrossRef]
- Walsh, M.E.; Shi, Y.; van Remmen, H. The effects of dietary restriction on oxidative stress in rodents. Free. Radic. Biol. Med. 2014, 66, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Hamm, A.C.; Bonaus, M.; Jacob, A.; Jaekel, J.; Schorle, H.; Pankratz, M.J.; Katzenberger, J.D. Starvation response in mouse liver shows strong correlation with life-span-prolonging processes. Physiol. Genom. 2004, 17, 230–244. [Google Scholar] [CrossRef] [Green Version]
- Stockman, M.-C.; Thomas, D.; Burke, J.; Apovian, C.M. Intermittent Fasting: Is the Wait Worth the Weight? Curr. Obes. Rep. 2018, 7, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’Yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815.e4. [Google Scholar] [CrossRef] [Green Version]
- Hatchwell, L.; Harney, D.J.; Cielesh, M.; Young, K.; Koay, Y.C.; O’Sullivan, J.F.; Larance, M. Multi-omics Analysis of the Intermittent Fasting Response in Mice Identifies an Unexpected Role for HNF4α. Cell Rep. 2020, 30, 3566–3582.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, S.S.; Vieira, C.P.; McFarland, D.; Sandler, M.; Levitsky, Y.; Dorweiler, T.F.; Lydic, T.A.; Asare-Bediako, B.; Adu-Agyeiwaah, Y.; Sielski, M.S.; et al. Fasting and fasting-mimicking treatment activate SIRT1/LXRα and alleviate diabetes-induced systemic and microvascular dysfunction. Diabetologia 2021, 64, 1674–1689. [Google Scholar] [CrossRef]
- Yan, L.-J. Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Benavides, G.A.; Vassilopoulos, A.; Gius, D.; Darley-Usmar, V.; Zhang, J. Bioenergetic and autophagic control by Sirt3 in response to nutrient deprivation in mouse embryonic fibroblasts. Biochem. J. 2013, 454, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Bruss, M.D.; Khambatta, C.F.; Ruby, M.A.; Aggarwal, I.; Hellerstein, M.K. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am. J. Physiol. Metab. 2010, 298, E108–E116. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, Y.; Tan, S.; Liu, L.; Hu, S.; Huo, H.; Li, M.; Cui, Q.; Yu, M. Nutrient deprivation-related OXPHOS/glycolysis interconversion via HIF-1α/C-MYC pathway in U251 cells. Tumor Biol. 2015, 37, 6661–6671. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie Restriction Promotes Mitochondrial Biogenesis by Inducing the Expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Korac, B.; Kalezic, A.; Pekovic-Vaughan, V.; Korac, A.; Jankovic, A. Redox changes in obesity, metabolic syndrome, and diabetes. Redox Biol. 2021, 42, 101887. [Google Scholar] [CrossRef]
- Li, Y.-L.; Xie, H.; Musha, H.; Xing, Y.; Mei, C.-X.; Wang, H.-J.; Wulasihan, M. The Risk Factor Analysis for Type 2 Diabetes Mellitus Patients with Nonalcoholic Fatty Liver Disease and Positive Correlation with Serum Uric Acid. Cell Biophys. 2015, 72, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, A.; Bonelli, P.; Tuccillo, F.M.; Goldfine, I.D.; Evans, J.L.; Buonaguro, F.M.; Mancini, A. Role of gut microbiota and oxidative stress in the progression of non-alcoholic fatty liver disease to hepatocarcinoma: Current and innovative therapeutic approaches. Redox Biol. 2018, 15, 467–479. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free. Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Nair, J.; Srivatanakul, P.; Haas, C.; Jedpiyawongse, A.; Khuhaprema, T.; Seitz, H.K.; Bartsch, H. High urinary excretion of lipid peroxidation-derived DNA damage in patients with cancer-prone liver diseases. Mutat. Res. Mol. Mech. Mutagen. 2010, 683, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Mihaylova, M.M.; Shaw, R.J. AMPK. In The Liver; Wiley Online Library: Hoboken, NJ, USA, 2020; pp. 472–484. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976, Erratum in Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Chiang, H.-H.; Louw, M.; Susanto, A.; Chen, D. Nutrient Sensing and the Oxidative Stress Response. Trends Endocrinol. Metab. 2017, 28, 449–460. [Google Scholar] [CrossRef]
- Lee, E.H.; Baek, S.Y.; Park, J.Y.; Kim, Y.W. Rifampicin activates AMPK and alleviates oxidative stress in the liver as mediated with Nrf2 signaling. Chem. Interact. 2020, 315, 108889. [Google Scholar] [CrossRef]
- Ren, Y.; Shen, H.-M. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019, 25, 101154. [Google Scholar] [CrossRef]
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428. [Google Scholar] [CrossRef] [Green Version]
- Sid, B.; Verrax, J.; Calderon, P.B. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem. Pharmacol. 2013, 86, 200–209. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-activated Protein Kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, S.L.; Moncada, S. AMPKα1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.-L.; Liu, Y.; Zheng, P. TSC–mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nacarelli, T.; Azar, A.; Sell, C. Inhibition of mTOR Prevents ROS Production Initiated by Ethidium Bromide-Induced Mitochondrial DNA Depletion. Front. Endocrinol. 2014, 5, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravindran, R.; Loebbermann, J.; Nakaya, H.I.; Khan, N.; Ma, H.; Gama, L.; Machiah, D.K.; Lawson, B.; Hakimpour, P.; Wang, Y.-C.; et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 2016, 531, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikani, C.K.; Antonysamy, S.A.; Bonanno, J.B.; Romero, R.; Zhang, F.F.; Russell, M.; Gheyi, T.; Iizuka, M.; Emtage, S.; Sauder, J.M.; et al. Structural Bases of PAS Domain-regulated Kinase (PASK) Activation in the Absence of Activation Loop Phosphorylation. J. Biol. Chem. 2010, 285, 41034–41043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möglich, A.; Ayers, R.A.; Moffat, K. Structure and Signaling Mechanism of Per-ARNT-Sim Domains. Structure 2009, 17, 1282–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, J.T.; Crosson, S. Ligand-Binding PAS Domains in a Genomic, Cellular, and Structural Context. Annu. Rev. Microbiol. 2011, 65, 261–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amezcua, C.A.; Harper, S.M.; Rutter, J.; Gardner, K.H. Structure and Interactions of PAS Kinase N-Terminal PAS Domain: Model for Intramolecular Kinase Regulation. Structure 2002, 10, 1349–1361. [Google Scholar] [CrossRef] [Green Version]
- Schläfli, P.; Tröger, J.; Eckhardt, K.; Borter, E.; Spielmann, P.; Wenger, R.H. Substrate preference and phosphatidylinositol monophosphate inhibition of the catalytic domain of the Per-Arnt-Sim domain kinase PASKIN. FEBS J. 2011, 278, 1757–1768. [Google Scholar] [CrossRef]
- Grose, J.H.; Rutter, J. The Role of PAS Kinase in PASsing the Glucose Signal. Sensors 2010, 10, 5668–5682. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Carneiro, V.; Pérez-García, A.; Alvarez, E.; Sanz, C. Role of Nutrient and Energy Sensors in the Development of Type 2 Diabetes. In Type 2 Diabetes; Stoian, A.P., Ed.; InTech—Open Access Publisher: London, UK, 2021; pp. 343–369. ISBN 978-1-83881-904-0. Available online: https://www.intechopen.com/chapters/74657 (accessed on 19 October 2021).
- Hao, H.-X.; Rutter, J. The role of PAS kinase in regulating energy metabolism. IUBMB Life 2008, 60, 204–209. [Google Scholar] [CrossRef]
- DeMille, D.; Grose, J.H. PAS kinase: A nutrient sensing regulator of glucose homeostasis. IUBMB Life 2013, 65, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.-D.; Zhang, J.-G.; Wang, Y.-Z.; Liu, Y.; Liu, G.-L.; Li, X.-Y. Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism. Nutrients 2015, 7, 7437–7450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado-Carneiro, V.; Pérez-García, A.; Alvarez, E.; Sanz, C. PAS Kinase: A Nutrient and Energy Sensor “Master Key” in the Response to Fasting/Feeding Conditions. Front. Endocrinol. 2020, 11, 594053. [Google Scholar] [CrossRef] [PubMed]
- Xavier, G.D.S.; Farhan, H.; Kim, H.; Caxaria, S.; Johnson, P.; Hughes, S.; Bugliani, M.; Marselli, L.; Marchetti, P.; Birzele, F.; et al. Per-arnt-sim (PAS) domain-containing protein kinase is downregulated in human islets in type 2 diabetes and regulates glucagon secretion. Diabetologia 2010, 54, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Semplici, F.; Mondragon, A.; MacIntyre, B.; Madeyski-Bengston, K.; Persson-Kry, A.; Barr, S.; Ramne, A.; Marley, A.; McGinty, J.; French, P.; et al. Cell type-specific deletion in mice reveals roles for PAS kinase in insulin and glucagon production. Diabetologia 2016, 59, 1938–1947. [Google Scholar] [CrossRef] [Green Version]
- Karakkat, J.V.; Kaimala, S.; Sreedharan, S.P.; Jayaprakash, P.; Adeghate, E.A.; Ansari, S.A.; Guccione, E.; Mensah-Brown, E.P.K.; Emerald, B.S. The metabolic sensor PASK is a histone 3 kinase that also regulates H3K4 methylation by associating with H3K4 MLL2 methyltransferase complex. Nucleic Acids Res. 2019, 47, 10086–10103. [Google Scholar] [CrossRef]
- Kikani, C.K.; Wu, X.; Paul, L.; Sabic, H.; Shen, Z.; Shakya, A.; Keefe, A.; Villanueva, C.; Kardon, G.; Graves, B.; et al. Pask integrates hormonal signaling with histone modification via Wdr5 phosphorylation to drive myogenesis. eLife 2016, 5, 791. [Google Scholar] [CrossRef] [Green Version]
- Kikani, C.K.; Wu, X.; Fogarty, S.; Kang, S.A.W.; Dephoure, N.; Gygi, S.P.; Sabatini, D.M.; Rutter, J. Activation of PASK by mTORC1 is required for the onset of the terminal differentiation program. Proc. Natl. Acad. Sci. USA 2019, 116, 10382–10391. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.-X.; Cardon, C.M.; Swiatek, W.; Cooksey, R.C.; Smith, T.L.; Wilde, J.; Boudina, S.; Abel, E.D.; McClain, D.; Rutter, J. PAS kinase is required for normal cellular energy balance. Proc. Natl. Acad. Sci. USA 2007, 104, 15466–15471. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Carneiro, V.; Roncero, I.; Blazquez, E.; Alvarez, E.; Sanz, C. PAS Kinase as a Nutrient Sensor in Neuroblastoma and Hypothalamic Cells Required for the Normal Expression and Activity of Other Cellular Nutrient and Energy Sensors. Mol. Neurobiol. 2013, 48, 904–920. [Google Scholar] [CrossRef]
- Hurtado-Carneiro, V.; Roncero, I.; Egger, S.S.; Wenger, R.H.; Blazquez, E.; Sanz, C.; Alvarez, E. PAS Kinase Is a Nutrient and Energy Sensor in Hypothalamic Areas Required for the Normal Function of AMPK and mTOR/S6K1. Mol. Neurobiol. 2014, 50, 314–326. [Google Scholar] [CrossRef]
- Dongil, P.; García, A.P.; Hurtado-Carneiro, V.; Herrero, C.; Blazquez, E.; Alvarez, E.; Sanz, C. Pas Kinase Deficiency Triggers Antioxidant Mechanisms in the Liver. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Dongil, P.; García, A.P.; Hurtado-Carneiro, V.; Herrero-De-Dios, C.; Álvarez, E.; Sanz, C. PAS kinase deficiency reduces aging effects in mice. Aging 2020, 12, 2275–2301. [Google Scholar] [CrossRef]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Strycharz, J.; Rygielska, Z.; Swiderska, E.; Drzewoski, J.; Szemraj, J.; Szmigiero, L.; Sliwinska, A. SIRT1 as a Therapeutic Target in Diabetic Complications. Curr. Med. Chem. 2018, 25, 1002–1035. [Google Scholar] [CrossRef]
- Merksamer, P.I.; Liu, Y.; He, W.; Hirschey, M.D.; Chen, D.; Verdin, E. The sirtuins, oxidative stress and aging: An emerging link. Aging 2013, 5, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Pignatti, C.; D’Adamo, S.; Stefanelli, C.; Flamigni, F.; Cetrullo, S. Nutrients and Pathways that Regulate Health Span and Life Span. Geriatrics 2020, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.; Verdin, E.; Chen, D. Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 Mediates Reduction of Oxidative Damage and Prevention of Age-Related Hearing Loss under Caloric Restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Iwahara, T.; Bonasio, R.; Narendra, V.; Reinberg, D. SIRT3 Functions in the Nucleus in the Control of Stress-Related Gene Expression. Mol. Cell. Biol. 2012, 32, 5022–5034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Deng, C.-X.; Finley, L.W.; Kim, H.-S.; Gius, D. SIRT3 Is a Mitochondrial Tumor Suppressor: A Scientific Tale That Connects Aberrant Cellular ROS, the Warburg Effect, and Carcinogenesis: Figure 1. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef] [Green Version]
- Mercken, E.M.; Mitchell, S.J.; Martin-Montalvo, A.; Minor, R.K.; Almeida, M.; Gomes, A.P.; Scheibye-Knudsen, M.; Palacios, H.H.; Licata, J.J.; Zhang, Y.; et al. SRT 2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 2014, 13, 787–796. [Google Scholar] [CrossRef]
- Alam, F.; Syed, H.; Amjad, S.; Baig, M.; Khan, T.A.; Rehman, R. Interplay between oxidative stress, SIRT1, reproductive and metabolic functions. Curr. Res. Physiol. 2021, 4, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.-I.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Swiatek, W.; Parnell, K.M.; Nickols, G.A.; Scharschmidt, B.F.; Rutter, J. Validation of PAS Kinase, a Regulator of Hepatic Fatty Acid and Triglyceride Synthesis, as a Therapeutic Target for Nonalcoholic Steatohepatitis. Hepatol. Commun. 2020, 4, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; A Nauck, M. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Cuthbertson, D.J.; Irwin, A.; Gardner, C.J.; Daousi, C.; Purewal, T.; Furlong, N.; Goenka, N.; Thomas, E.L.; Adams, V.L.; Pushpakom, S.P.; et al. Improved Glycaemia Correlates with Liver Fat Reduction in Obese, Type 2 Diabetes, Patients Given Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists. PLoS ONE 2012, 7, e50117. [Google Scholar] [CrossRef] [PubMed]
- Parlevliet, E.T.; Wang, Y.; Geerling, J.J.; Der Elst, J.P.S.-V.; Picha, K.; O’Neil, K.; Stojanovic-Susulic, V.; Ort, T.; Havekes, L.M.; Romijn, J.A.; et al. GLP-1 Receptor Activation Inhibits VLDL Production and Reverses Hepatic Steatosis by Decreasing Hepatic Lipogenesis in High-Fat-Fed APOE*3-Leiden Mice. PLoS ONE 2012, 7, e49152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.A.; Kolachala, V.L.; Jiang, R.; Abramowsky, C.; Romero, R.; Fifadara, N.; Anania, F.; Knechtle, S.; Kirk, A. The Glucagon-Like Peptide-1 Receptor Agonist Exendin 4 Has a Protective Role in Ischemic Injury of Lean and Steatotic Liver by Inhibiting Cell Death and Stimulating Lipolysis. Am. J. Pathol. 2012, 181, 1693–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.S.; Jun, H.-S. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int. J. Mol. Sci. 2017, 19, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Romero, D.; Swiatek, W.I.; Dorweiler, I.; Kikani, C.; Sabic, H.; Zweifel, B.S.; McKearn, J.; Blitzer, J.T.; Nickols, G.A.; et al. PAS Kinase Drives Lipogenesis through SREBP-1 Maturation. Cell Rep. 2014, 8, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Pérez-García, A.; Dongil, P.; Hurtado-Carneiro, V.; Blázquez, E.; Sanz, C.; Álvarez, E. High-fat diet alters PAS kinase regulation by fasting and feeding in liver. J. Nutr. Biochem. 2018, 57, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; DeMarco, V.G. Oxidative Stress and Obesity: The Chicken or the Egg? Diabetes 2014, 63, 2216–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Soyal, S.; Krempler, F.; Oberkofler, H.; Patsch, W. PGC-1α: A potent transcriptional cofactor involved in the pathogenesis of type 2 diabetes. Diabetology 2006, 49, 1477–1488. [Google Scholar] [CrossRef]
- Cheng, C.-F.; Ku, H.-C.; Lin, H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef] [Green Version]
- Gurd, B.J.; Yoshida, Y.; McFarlan, J.T.; Holloway, G.P.; Moyes, C.D.; Heigenhauser, G.J.F.; Spriet, L.; Bonen, A. Nuclear SIRT1 activity, but not protein content, regulates mitochondrial biogenesis in rat and human skeletal muscle. Am. J. Physiol. Integr. Comp. Physiol. 2011, 301, R67–R75. [Google Scholar] [CrossRef] [PubMed]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.-X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-Associated Reductions in AMP-Activated Protein Kinase Activity and Mitochondrial Biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Madden, S.; Itzhaki, L.S. Structural and mechanistic insights into the Keap1-Nrf2 system as a route to drug discovery. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2020, 1868, 140405. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Yamamoto, M. Molecular Mechanisms Activating the Nrf2-Keap1 Pathway of Antioxidant Gene Regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Zhang, Y.; Luo, M.; Wu, Q.; Yu, L.; Chu, H. Transcriptional upregulation centra of HO-1 by EGB via the MAPKs/Nrf2 pathway in mouse C2C12 myoblasts. Toxicol. Vitr. 2015, 29, 380–388. [Google Scholar] [CrossRef]
- Barančík, M.; Grešová, L.; Bartekova, M.; Dovinova, I. Nrf2 as a Key Player of Redox Regulation in Cardiovascular Diseases. Physiol. Res. 2016, 65 (Suppl. S1), S1–S10. [Google Scholar] [CrossRef] [PubMed]
- García, A.P.; Dongil, P.; Hurtado-Carneiro, V.; Blazquez, E.; Sanz, C.; Alvarez, E. PAS Kinase deficiency alters the glucokinase function and hepatic metabolism. Sci. Rep. 2018, 8, 11091. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/ -TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme Oxygenase-1 Regulates Cardiac Mitochondrial Biogenesis via Nrf2-Mediated Transcriptional Control of Nuclear Respiratory Factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The Energy Sensor AMP-activated Protein Kinase Directly Regulates the Mammalian FOXO3 Transcription Factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsalve, M.; Borniquel, S.; Valle, I.; Lamas, S. Mitochondrial dysfunction in human pathologies. Front. Biosci. 2007, 12, 1131–1153. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, I.; Álvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Mei, Y.; Zhang, Y.; Yamamoto, K.; Xie, W.; Mak, T.W.; You, H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 5153–5158. [Google Scholar] [CrossRef] [Green Version]
- Mojsov, S.; Heinrich, G.; Wilson, I.; Ravazzola, M.; Orci, L.; Habener, J.F. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J. Biol. Chem. 1986, 261, 11880–11889. [Google Scholar] [CrossRef]
- Blázquez, E.; Alvarez, E.; Navarro, M.; Roncero, I.; Rodríguez-Fonseca, F.; Chowen, J.A.; Zueco, J.A. Glucagon-like peptide-1 (7–36) amide as a novel neuropeptide. Mol. Neurobiol. 1998, 18, 157–173. [Google Scholar] [CrossRef]
- Hurtado-Carneiro, V.; Roncero, I.; Blazquez, E.; Alvarez, E.; Sanz, C. Glucagon-Like Peptide-1 and Its Implications in Obesity. In Hot Topics in Endocrine and Endocrine-Related Diseases; Fedele, M., Ed.; Intech Open Access Publisher: Rijeka, Croatia, 2013; pp. 165–195. ISBN 978-953-51-1080-4. Available online: https://www.intechopen.com/chapters/44429 (accessed on 19 October 2021).
- Malik, J.; Roohi, N. GLP-1, a powerful physiological incretin: An update. J. Biol. Regul. Homeost Agents 2018, 32, 1171–1176. [Google Scholar] [PubMed]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsbøll, T. Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef]
- Drucker, D.J. Glucagon-Like Peptides: Regulators of Cell Proliferation, Differentiation, and Apoptosis. Mol. Endocrinol. 2003, 17, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, E.; Roncero, I.; Chowen, J.A.; Thorens, B.; Blázquez, E. Expression of the Glucagon-Like Peptide-1 Receptor Gene in Rat Brain. J. Neurochem. 2002, 66, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Martinez, M.D.; Roncero, I.; Chowen, J.A.; Garcia-Cuartero, B.; Gispert, J.D.; Sanz, C.; Vazquez, P.; Maldonado, A.; de Caceres, J.; et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J. Neurochem. 2005, 92, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; de Fonseca, F.R.; Alvarez, E.; Chowen, J.A.; Zueco, J.A.; Gomez, R.; Eng, J.; Blázquez, E. Colocalization of Glucagon-Like Peptide-1 (GLP-1) Receptors, Glucose Transporter GLUT-2, and Glucokinase mRNAs in Rat Hypothalamic Cells: Evidence for a Role of GLP-1 Receptor Agonists as an Inhibitory Signal for Food and Water Intake. J. Neurochem. 2002, 67, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Bain, S.C.; Mosenzon, O.; Arechavaleta, R.; Bogdanski, P.; Comlekci, A.; Consoli, A.; Deerochanawong, C.; Dungan, K.; Faingold, M.C.; Farkouh, M.E.; et al. Cardiovascular safety of oral semaglutide in patients with type 2 diabetes: Rationale, design and patient baseline characteristics for the PIONEER 6 trial. Diabetes Obes. Metab. 2018, 21, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef]
- Seghieri, M.; Christensen, A.S.; Andersen, A.; Solini, A.; Knop, F.K.; Vilsbøll, T. Future Perspectives on GLP-1 Receptor Agonists and GLP-1/glucagon Receptor Co-agonists in the Treatment of NAFLD. Front. Endocrinol. 2018, 9, 649. [Google Scholar] [CrossRef]
- Zhou, R.; Lin, C.; Cheng, Y.; Zhuo, X.; Li, Q.; Xu, W.; Zhao, L.; Yang, L. Liraglutide Alleviates Hepatic Steatosis and Liver Injury in T2MD Rats via a GLP-1R Dependent AMPK Pathway. Front. Pharmacol. 2021, 11, 600175. [Google Scholar] [CrossRef]
- Pérez-García, A.; Hurtado-Carneiro, V.; Herrero-De-Dios, C.; Dongil, P.; García-Mauriño, J.; Sánchez, M.; Sanz, C.; Álvarez, E. Storage and Utilization of Glycogen by Mouse Liver during Adaptation to Nutritional Changes Are GLP-1 and PASK Dependent. Nutrients 2021, 13, 2552. [Google Scholar] [CrossRef]
- Lu, M.; Wang, P.; Qiao, Y.; Jiang, C.; Ge, Y.; Flickinger, B.; Malhotra, D.K.; Dworkin, L.D.; Liu, Z.; Gong, R. GSK3β-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019, 26, 101275. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Joharapurkar, A.; Dhanesha, N.; Kshirsagar, S.; Detroja, J.; Patel, K.; Gandhi, T.; Patel, K.; Bahekar, R.; Jain, M. Combination of omeprazole with GLP-1 agonist therapy improves insulin sensitivity and antioxidant activity in liver in type 1 diabetic mice. Pharmacol. Rep. 2013, 65, 927–936. [Google Scholar] [CrossRef]
- Niu, S.; Wang, L.; He, M.; Peng, Y.; Li, S. Exendin-4 regulates redox homeostasis in rats fed with high-fat diet. Acta Biochim. Biophys. Sin. 2015, 47, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis inob/ob mice. Hepatology 2005, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Liu, D. Controlling Obesity and Metabolic Diseases by Hydrodynamic Delivery of a Fusion Gene of Exendin-4 and α1 Antitrypsin. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurtado-Carneiro, V.; Dongil, P.; Pérez-García, A.; Álvarez, E.; Sanz, C. Preventing Oxidative Stress in the Liver: An Opportunity for GLP-1 and/or PASK. Antioxidants 2021, 10, 2028. https://doi.org/10.3390/antiox10122028
Hurtado-Carneiro V, Dongil P, Pérez-García A, Álvarez E, Sanz C. Preventing Oxidative Stress in the Liver: An Opportunity for GLP-1 and/or PASK. Antioxidants. 2021; 10(12):2028. https://doi.org/10.3390/antiox10122028
Chicago/Turabian StyleHurtado-Carneiro, Verónica, Pilar Dongil, Ana Pérez-García, Elvira Álvarez, and Carmen Sanz. 2021. "Preventing Oxidative Stress in the Liver: An Opportunity for GLP-1 and/or PASK" Antioxidants 10, no. 12: 2028. https://doi.org/10.3390/antiox10122028
APA StyleHurtado-Carneiro, V., Dongil, P., Pérez-García, A., Álvarez, E., & Sanz, C. (2021). Preventing Oxidative Stress in the Liver: An Opportunity for GLP-1 and/or PASK. Antioxidants, 10(12), 2028. https://doi.org/10.3390/antiox10122028