
Regulation of Mitochondrial Dynamics in Parkinson’s Disease—Is 2-Methoxyestradiol a Missing Piece?

 ,
,  and
and 
Abstract
:1. Parkinson’s Disease
2. Biomarkers of Oxidative Stress in Physiology and Pathophysiology of Nervous System
3. Mitochondrial Antistress Protective Systems
4. Nitric Oxide as an Ignition Link of Apoptosis
5. 2-Methoxyestradiol (2-ME) a Physiological Compound and an Anticancer Agent
6. Activity of 2-ME in Neurons
7. Mitochondrial Abnormalities as a Mechanism of Neurodegeneration
8. Mitochondrial Biogenesis
9. Fusion/Fission
10. Mitophagy
11. Mitochondrial Biogenesis and Mitochondrial Dynamics as Targets for 2-ME
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathology of Parkinson disease. Park. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Yen, S.H.; McLean, P.J.; Dickson, D.W. Histones facilitate α-synuclein aggregation during neuronal apoptosis. Acta Neuropathol. 2017, 133, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Ammal Kaidery, N.; Thomas, B. Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem. Int. 2018, 117, 91–113. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Dulski, J.; Schinwelski, M.; Konkel, A.; Grabowski, K.; Libionka, W.; Wąż, P.; Sitek, E.J.; Sławek, J. The impact of subthalamic deep brain stimulation on sleep and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord. 2019, 64, 138–144. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Hu, M.T.M. Sleep disturbance as potential risk and progression factor for Parkinson’s disease. J. Park. Dis. 2019, 9, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Leng, Y.; Musiek, E.S.; Hu, K.; Cappuccio, F.P.; Yaffe, K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019, 18, 307–318. [Google Scholar] [CrossRef]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis.Treat. 2008, 4, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Korczyn, A.D. Drug treatment of Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 315–322. [Google Scholar] [CrossRef]
- Zahoor, I.; Shafi, A.; Haq, E. Pharmacological Treatment of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Singapore, 2018; pp. 129–144. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA J. Am. Med. Assoc. 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Wahabi, K.; Perwez, A.; Rizvi, M.A. Parkin in Parkinson’s Disease and Cancer: A Double-Edged Sword. Mol. Neurobiol. 2018, 55, 6788–6800. [Google Scholar] [CrossRef]
- West, A.B.; Dawson, V.L.; Dawson, T.M. To die or grow: Parkinson’s disease and cancer. Trends Neurosci. 2005, 28, 348–352. [Google Scholar] [CrossRef]
- Rojas, N.G.; Cesarini, M.; Etcheverry, J.L.; Prat GADa Arciuch, V.A.; Gatto, E.M. Neurodegenerative diseases and cancer: Sharing common mechanisms in complex interactions. J. Integr. Neurosci. 2020, 19, 187–199. [Google Scholar]
- Brundin, P.; Wyse, R. Cancer enzyme affects Parkinson’s disease. Science 2018, 362, 521–522. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. 2018, 38, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Picchio, M.C.; Martin, E.S.; Cesari, R.; Calin, G.A.; Yendamuri, S.; Kuroki, T.; Pentimalli, F.; Sarti, M.; Yoder, K.; Kaiser, L.R.; et al. Alterations of the tumor suppressor gene parkin in non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 2720–2724. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, Y.; Wu, L.; Dong, Y.; Zhang, J.; Chen, F.; Xie, W.; Huang, J.; Lu, N. Apurinic endonuclease 1 promotes the cisplatin resistance of lung cancer cells by inducing Parkin-mediated mitophagy. Oncol. Rep. 2019, 42, 2245–2254. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Nam, H.J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.Y.; Ham, H.; Evans, D.; Kim, S.H.; et al. Parkin regulates mitosis and genomic stability through Cdc20/Cdh1. Mol. Cell. 2015, 60, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Park, K.R.; Yun, J.S.; Park, M.H.; Jung, Y.Y.; Yeo, I.J.; Nam, K.T.; Kim, H.D.; Song, J.K.; Choi, D.Y.; Park, P.H.; et al. Loss of parkin reduces lung tumor development by blocking p21 degradation. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Denison, S.; Lai, J.P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; et al. Parkin gene alterations in hepatocellular carcinoma. Genes Chromosom. Cancer. 2004, 40, 85–96. [Google Scholar] [CrossRef]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar] [CrossRef] [Green Version]
- Da Silva-Camargo, C.C.V.; Baldin, R.K.S.; Polli, N.L.C.; Agostinho, A.P.; Olandosk, M.; De Noronha, L.; Sotomaior, V.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, N.; Ma, Q.; Chen, Y.; Yao, L.; Zhang, L.; Li, Q.; Shi, M.; Wang, H.; Ying, Z. Somatic and germline mutations in the tumor suppressor gene PARK2 impair PINK1/Parkin-mediated mitophagy in lung cancer cells. Acta Pharmacol. Sin. 2020, 41, 93–100. [Google Scholar] [CrossRef]
- Dai, K.; Radin, D.P.; Leonardi, D. PINK1 depletion sensitizes non-small cell lung cancer to glycolytic inhibitor 3-bromopyruvate: Involvement of ROS and mitophagy. Pharmacol. Rep. 2019, 71, 1184–1189. [Google Scholar] [CrossRef]
- Chang, G.; Zhang, W.; Ma, Y.; Wen, Q. PINK1 expression is associated with poor prognosis in lung adenocarcinoma. Tohoku J. Exp. Med. 2018, 245, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.J.; Báez-Becerra, C.; Contreras-Zárate, M.J.; Arboleda, H.; Arboleda, G. PINK1 silencing modifies dendritic spine dynamics of mouse hippocampal neurons. J. Mol. Neurosci. 2019, 69, 570–579. [Google Scholar] [CrossRef]
- Surguchov, A. Intracellular dynamics of synucleins. In International Review of Cell and Molecular Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 103–169. [Google Scholar]
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell responses to extracellular α-Synuclein. Molecules 2019, 24, 305. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.Y. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Rodriguez-Leyva, I.; Chi-Ahumada, E.; Mejía, M.; Castanedo-Cazares, J.P.; Eng, W.; Saikaly, S.K.; Carrizales, J.; Levine, T.D.; Norman, R.A.; Jimenez-Capdeville, M.E. The presence of alpha-synuclein in skin from melanoma and patients with Parkinson’s disease. Mov. Disord. Clin. Pract. 2017, 4, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, M.; Suzuki, S.O.; Doh-Ura, K.; Iwaki, T. α-synuclein is expressed in a variety of brain tumors showing neuronal differentiation. Acta Neuropathol. 2000, 99, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.M.; Rorke, L.B.; Giasson, B.; Lee, V.M.Y.; Trojanowski, J.Q. Expression of α-, β-, and γ-synuclein in glial tumor and medulloblastomas. Acta Neuropathol. 2003, 106, 167–175. [Google Scholar] [CrossRef]
- Raghavan, R.; White, C.L.; Rogers, B.; Coimbra, C.; Rushing, E.J. Alpha-synuclein expression in central nervous system tumors showing neuronal or mixed neuronal/glial differentiation. J. Neuropathol. Exp. Neurol. 2000, 59, 490–494. [Google Scholar] [CrossRef] [Green Version]
- Dean, D.N.; Lee, J.C. Defining an amyloid link between Parkinson’s disease and melanoma. Proc. Natl. Acad. Sci. USA 2020, 117, 22671–22673. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/parkin mediated mitophagy, Ca2+ signalling, and ER–mitochondria contacts in Parkinson’s disease. Int J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [Green Version]
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013, 14, 293–304. [Google Scholar] [CrossRef]
- Ejma, M.; Madetko, N.; Brzecka, A.; Guranski, K.; Alster, P.; Misiuk-Hojło, M.; Somasundaram, S.G.; Kirkland, C.E.; Aliev, G. The links between Parkinson’s disease and cancer. Biomedicines 2020, 8, 416. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain. 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Ward, J.P.T. From physiological redox signalling to oxidant stress. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; pp. 335–342. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, J.; Krause, K. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox mechanisms in neurodegeneration: From disease outcomes to therapeutic opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1599. [Google Scholar] [CrossRef]
- Santos, A.L.; Lindner, A.B. Protein posttranslational modifications: Roles in aging and age-related disease. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Finelli, M.J. Redox post-translational modifications of protein thiols in brain aging and neurodegenerative conditions—Focus on S-nitrosation. Front. Ageing Neurosci. 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of catalase in oxidative stress- And age-Associated degenerative diseases. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Bánhegyi, G.; Bogeski, I.; Davies, K.J.A.; Delaunay-Moisan, A.; Forman, H.J.; Görlach, A.; Kietzmann, T.; Laurindo, F.; Margittai, E.; et al. Transit of H2O2 across the endoplasmic reticulum membrane is not sluggish. Free Radic. Biol. Med. 2016, 94, 157–160. [Google Scholar] [CrossRef] [Green Version]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Van Der Vliet, A.; Janssen-Heininger, Y.M.W. Hydrogen peroxide as a damage signal in tissue injury and inflammation: Murderer, mediator, or messenger? J. Cell Biochem. 2014, 115, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Wang, Q.; Lu, S.; Niu, Y. Hydrogen peroxide: A potential wound therapeutic target? Med. Princ. Pract. 2017, 26, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26. [Google Scholar] [CrossRef]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Wenceslau, C.F.; McCarthy, C.G.; Webb, R.C. To be, or nox to be, endoplasmic reticulum stress in hypertension. Hypertension 2018, 72, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Marschall, R.; Tudzynski, P. Reactive oxygen species in development and infection processes. Semin. Cell Dev. Biol. 2016, 57, 138–146. [Google Scholar] [CrossRef]
- Bao, L.; Avshalumov, M.V.; Patel, J.C.; Lee, C.R.; Miller, E.W.; Chang, C.J.; Rice, M.E. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 2009, 29, 9002–9010. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chen, G.; Gao, M.; Wang, R.; Liu, Y.; Yu, F. Imaging of endogenous hydrogen peroxide during the process of cell mitosis and mouse brain development with a near-infrared ratiometric fluorescent probe. Anal. Chem. 2019, 91, 1203–1210. [Google Scholar] [CrossRef]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.M.A.; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med. 2002, 32, 1076–1083. [Google Scholar] [CrossRef]
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.P.; Schafer, F.Q.; Goswami, P.C.; Oberley, L.W.; Buettner, G.R. Phospholipid hydroperoxide glutathione peroxidase induces a delay in G1 of the cell cycle. Free Radic. Res. 2003, 37, 621–630. [Google Scholar] [CrossRef]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant properties dedicated to nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Quintana-Cabrera, R.; Bolaños, J.P. Glutathione and γ-glutamylcysteine in hydrogen peroxide detoxification. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2013; pp. 129–144. [Google Scholar] [CrossRef]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Panfili, E.; Sandri, G.; Ernster, L. Distribution of glutathione peroxidases glutathione reductase in rat brain mitochondria. FEBS Lett. 1991, 290, 35–37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Handy, D.E.; Loscalzo, J. Adenosine-dependent induction of glutathione peroxidase 1 in human primary endothelial cells and protection against oxidative stress. Circ. Res. 2005, 96, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: Abnormalities in vascular and cardiac function and structure. Circulation 2002, 106, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Forgione, M.A.; Weiss, N.; Heydrick, S.; Cap, A.; Klings, E.S.; Bierl, C.; Eberhardt, R.T.; Farber, H.W.; Loscalzo, J. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2002, 282, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Taylor, J.M.; Ali, U.; Mansell, A.; Hertzog, P.J. Potential contribution of NF-κB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 2006, 37, 1533–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoehn, B.; Yenari, M.A.; Sapolsky, R.M.; Steinberg, G.K. Glutathione peroxidase overexpression inhibits cytochrome c release and proapoptotic mediators to protect neurons from experimental stroke. Stroke 2003, 34, 2489–2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winternitz, M.C.; Meloy, C.R. On the occurrence of catalase in human tissues and its variations in diseases. J. Exp. Med. 1908, 10, 759–781. [Google Scholar] [CrossRef] [Green Version]
- Ambani, L.M.; Van Woert, M.H.; Murphy, S. Brain peroxidase and catalase in parkinson disease. Arch. Neurol. 1975, 32, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Zivić, S.; Vlaski, J.; Kocić, G.; Pesić, M.; Cirić, V.; Durić, Z. The importance of oxidative stress in pathogenesis of type 1 diabetes--determination of catalase activity in lymphocytes of diabetic patients. Med. Pregl. 2008, 61, 458–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, K.; Tanaka, M.; Iwata, H.; Nishihara, R.; Ishihara, K.; Wang, D.H.; Ogino, K.; Taniuchi, K.; Masuoka, N. Low catalase activity in blood is associated with the diabetes caused by alloxan. Clin. Chim. Acta. 2009, 407, 43–46. [Google Scholar] [CrossRef]
- Sundaram, A.; Siew Keah, L.; Sirajudeen, K.N.S.; Singh, H.J. Upregulation of catalase and downregulation of glutathione peroxidase activity in the kidney precede the development of hypertension in pre-hypertensive SHR. Hypertens. Res. 2013, 36, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikemura, M.; Nishikawa, M.; Hyoudou, K.; Kobayashi, Y.; Yamashita, F.; Hashida, M. Improvement of insulin resistance by removal of systemic hydrogen peroxide by pegylated catalase in obese mice. Mol. Pharm. 2010, 7, 2069–2076. [Google Scholar] [CrossRef]
- Parboosingh, J.S.; Rousseau, M.; Rogan, F.; Amit, Z.; Chertkow, H.; Johnson, W.G.; Manganaro, F.; Schipper, H.N.; Curran, T.J.; Stoessl, J.; et al. Absence of mutations in superoxide dismutase and catalase genes in patients with Parkinson’s Disease. Arch. Neurol. 1995, 52, 1160–1163. [Google Scholar] [CrossRef]
- Gsell, W.; Conrad, R.; Hickethier, M.; Sofic, E.; Frölich, L.; Wichart, I.; Jellinger, K.; Moll, G.; Ransmayr, G.; Beckmann, H.; et al. Decreased catalase activity but unchanged superoxide dismutase activity in brains of patients with dementia of alzheimer type. J. Neurochem. 1995, 64, 1216–1223. [Google Scholar] [CrossRef]
- Goulas, A.; Fidani, L.; Kotsis, A.; Mirtsou, V.; Petersen, R.C.; Tangalos, E.; Hardy, J. An association study of a functional catalase gene polymorphism, -262C→T, and patients with Alzheimer’s disease. Neurosci. Lett. 2002, 330, 210–212. [Google Scholar] [CrossRef]
- De Sousa, R.T.; Zarate, C.A.; Zanetti, M.V.; Costa, A.C.; Talib, L.L.; Gattaz, W.F.; Machado-Vieira, R. Oxidative stress in early stage bipolar disorder and the association with response to lithium. J. Psychiatr. Res. 2014, 50, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Selek, S.; Altindag, A.; Saracoglu, G.; Aksoy, N. Oxidative markers of myeloperoxidase and catalase and their diagnostic performance in bipolar disorder. J. Affect. Disord. 2015, 181, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Rukmini, M.S.; D’Souza, B.; D’Souza, V. Superoxide dismutase and catalase activities and their correlation with malondialdehyde in schizophrenic patients. Indian J. Clin. Biochem. 2004, 19, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Rad. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.T.; Khazaei, M.; Khazaei, M. The effect of catalase C262T gene polymorphism in susceptibility to ovarian cancer in Kermanshah province, Western Iran. J. Obstet. Gynaecol. 2018, 38, 562–566. [Google Scholar] [CrossRef]
- Song, X.; Xu, J.; Liang, C.; Chao, Y.; Jin, Q.; Wang, C.; Chen, M.; Liu, Z. Self-supplied tumor oxygenation through separated liposomal delivery of H2O2 and catalase for enhanced radio-immunotherapy of cancer. Nano Lett. 2018, 18, 6360–6368. [Google Scholar] [CrossRef]
- Lee, K.T.; Lu, Y.J.; Mi, F.L.; Burnouf, T.; Wei, Y.T.; Chiu, S.C.; Chuang, E.Y.; Lu, S.Y. Catalase-modulated heterogeneous fenton reaction for selective cancer cell eradication: SnFe2O4 nanocrystals as an effective reagent for treating lung cancer cells. ACS Appl. Mater. Interfaces 2017, 9, 1273–1279. [Google Scholar] [CrossRef]
- Glorieux, C.; Sandoval, J.M.; Dejeans, N.; Nonckreman, S.; Bahloula, K.; Poirel, H.A.; Calderon, P.B. Evaluation of potential mechanisms controlling the catalase expression in breast cancer cells. Oxid. Med. Cell Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Altobelli, G.G.; Van Noorden, S.; Balato, A.; Cimini, V. Copper/zinc superoxide dismutase in human skin: Current knowledge. Front. Med. 2020, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.L.; Willassen, N.P.; Leiros, I. The first structure of a cold-adapted superoxide dismutase (SOD): Biochemical and structural characterization of iron SOD from Aliivibrio salmonicida. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.C.; Johnson, O.E.; Cabelli, D.E.; Brunold, T.C.; Maroney, M.J. Nickel superoxide dismutase: Structural and functional roles of Cys2 and Cys6. J. Biol. Inorg. Chem. 2010, 15, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver. Cu,Zn-SOD in mitochondria. J. Biol Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattan, Z.; Minig, V.; Leroy, P.; Dauça, M.; Becuwe, P. Role of manganese superoxide dismutase on growth and invasive properties of human estrogen-independent breast cancer cells. Breast Cancer Res. Treat. 2008, 108, 203–215. [Google Scholar] [CrossRef]
- Chuang, T.C.; Liu, J.Y.; Lin, C.T.; Tang, Y.T.; Yeh, M.H.; Chang, S.C.; Li, J.W.; Kao, M.C. Human manganese superoxide dismutase suppresses HER2/neu-mediated breast cancer malignancy. FEBS Lett. 2007, 581, 4443–4449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soini, Y.; Vakkala, M.; Kahlos, K.; Pääkkö, P.; Kinnula, V. MnSOD expression is less frequent in tumour cells of invasive breast carcinomas than in in situ carcinomas or non-neoplastic breast epithelial cells. J. Pathol. 2001, 195, 156–162. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren-Sandberg, A.; Domellof, L. Pancreatitis and the risk of pancreatic cancer. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef]
- Hu, Y.; Rosen, D.G.; Yang, G.; Zhou, Y.; Liu, J.; Huang, P. Expression of manganese superoxide dismutase (MnSOD) in human ovarian carcinoma and its role in cancer cell proliferation. Cancer Res. 2005, 65, 1127. [Google Scholar]
- Nishida, T.; Sugiyama, T.; Kataoka, A.; Tashiro, M.; Yakushiji, M.; Ishikawa, M. Serum manganese superoxide dismutase (MnSOD) and histological virulence of ovarian cancer. Asia Oceania J. Obstet. Gynaecol. 1993, 19, 427–431. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Nelson, K.K.; Rodriguez, A.M.; Kim, K.H.; Tower, G.B.; Rutter, J.L.; Brinckerhoff, C.E.; Huang, T.T.; Epstein, C.J.; Jeffrey, J.J.; et al. Manganese superoxide dismutase signals matrix metalloproteinase expression via H2O2-dependent ERK1/2 activation. J. Biol. Chem. 2001, 276, 14264–14270. [Google Scholar] [CrossRef] [Green Version]
- Ough, M.; Lewis, A.; Zhang, Y.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Inhibition of cell growth by overexpression of manganese superoxide dismutase (MnSOD) in human pancreatic carcinoma. Free Radic. Res. 2004, 38, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Behrend, L.; Mohr, A.; Dick, T.; Zwacka, R.M. Manganese superoxide dismutase induces p53-dependent senescence in colorectal cancer Cells. Mol. Cell Biol. 2005, 25, 7758–7769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uudsemaa, M.; Tamm, T. Density-functional theory calculations of aqueous redox potentials of fourth-period transition metals. J. Phys. Chem. A 2003, 107, 9997–10003. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mungrue, I.N.; Bredt, D.S. nNOS at a glance: Implications for brain and brawn. J. Cell Sci. 2004, 117, 2627–2629. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on immune cells and its role in diseases. Int. J. Mol. Sci. 2018, 12, 3805. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Natal, C.; Modol, T.; Osés-Prieto, J.A.; López-Moratalla, N.; Iraburu, M.J.; López-Zabalza, M.J. Specific protein nitration in nitric oxide-induced apoptosis of human monocytes. Apoptosis 2008, 13, 1356–1367. [Google Scholar] [CrossRef]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide Biol. Chem. 2019, 93, 102–114. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat Rev Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Parada-Bustamante, A.; Valencia, C.; Reuquen, P.; Diaz, P.; Rincion-Rodriguez, R.; Orihuela, P. Role of 2-methoxyestradiol, an endogenous estrogen metabolite, in health and disease. Mini Rev. Med. Chem. 2015, 15, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooberry, S.L. New insights into 2-methoxyestradiol, a promising antiangiogenic and antitumor agent. Curr. Opin. Oncol. 2003, 15, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.R.; Hahn, N.M.; Pili, R.; Oh, W.K.; Hammers, H.; Sweeney, C.; Kim, K.; Perlman, S.; Arnott, J.; Sidor, C.; et al. A phase II study of 2-methoxyestradiol (2ME2) NanoCrystal® dispersion (NCD) in patients with taxane-refractory, metastatic castrate-resistant prostate cancer (CRPC). Investig. New Drugs 2011, 29, 1465–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tevaarwerk, A.J.; Holen, K.D.; Alberti, D.B.; Sidor, C.; Arnott, J.; Quon, C.; Wilding, G.; Liu, G. Phase i trial of 2-methoxyestradioI NanoCrystal dispersion in advanced solid malignancies. Clin. Cancer Res. 2009, 15, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.S.; Raghuvanshi, D.S.; Hasanain, M.; Alam, S.; Sarkar, J.; Mitra, K.; Khan, F.; Negi, A.S. Recent advances in chemistry and pharmacology of 2-methoxyestradiol: An anticancer investigational drug. Steroids 2016, 110, 9–34. [Google Scholar] [CrossRef] [PubMed]
- LaVallee, T.M.; Burke, P.A.; Swartz, G.M.; Hamel, E.; Agoston, G.E.; Shah, J.; Suwandi, L.; Hanson, A.D.; Fogler, W.E.; Sidor, C.F.; et al. Significant antitumor activity in vivo following treatment with the microtubule agent ENMD-1198. Mol. Cancer Ther. 2008, 7, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Dahut, W.L.; Lakhani, N.J.; Gulley, J.L.; Arlen, P.M.; Kohn, E.C.; Kotz, H.; McNally, D.; Pair, A.; Nguyen, D.; Yang, S.X.; et al. Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol. Ther. 2006, 5, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.P.; Medina, R.A. Owen GI. 2-methoxyestradiol and disorders of female reproductive tissues. Horm. Cancer 2014, 5, 274–283. [Google Scholar] [CrossRef]
- Zhang, N.; Xu, Y.; Xin, X.; Huo, P.; Zhang, Y.; Chen, H.; Feng, N.; Feng, Q.; Zhang, Z. Dual-modal imaging-guided theranostic nanocarriers based on 2-methoxyestradiol and indocyanine green. Int. J. Pharm. 2021, 592, 120098. [Google Scholar] [CrossRef]
- Al-Kazaale, N.; Tran, P.T.; Haidari, F.; Solum, E.J.; Liekens, S.; Vervaeke, P.; Sylte, I.; Cheng, J.-J.; Vik, A.; Hansen, T.V. Synthesis, molecular modeling and biological evaluation of potent analogs of 2-methoxyestradiol. Steroids 2018, 136, 47–55. [Google Scholar] [CrossRef]
- Borahay, M.A.; Vincent, K.L.; Motamedi, M.; Tekedereli, I.; Salama, S.A.; Ozpolat, B.; Kilic, G.S. Liposomal 2-methoxyestradiol nanoparticles for treatment of uterine leiomyoma in a patient-derived xenograft mouse model. Reprod. Sci. 2021, 28, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Fotsis, T.; Zhang, Y.; Pepper, M.S.; Adlercreutz, H.; Montesano, R.; Nawroth, P.P.; Schweigerer, L. The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature 1994, 368, 237–239. [Google Scholar] [CrossRef]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17β-estradiol and estrone formed by 15 selectively expressed human cytochrome P450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef]
- Matsumoto, M.; Weickert, C.S.; Akil, M.; Lipska, B.K.; Hyde, T.M.; Herman, M.M.; Kleinman, J.E.; Weinberger, D.R. Catechol O-methyltransferase mRNA expression in human and rat brain: Evidence for a role in cortical neuronal function. Neuroscience 2003, 116, 127–137. [Google Scholar] [CrossRef]
- Gorska, M.; Kuban-Jankowska, A.; Slawek, J.; Wozniak, M. New insight into 2-methoxyestradiol- a possible physiological link between neurodegeneration and cancer cell death. Curr. Med. Chem. 2016, 23, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Thaler, F.; Kuss, E. Concentrations of 2-hydroxyoestrogens in human sera measured by a heterologous immunoassay with an 125I-labelled ligand. Acta Endocrinol. 1982, 100, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Mueck, A.O.; Seeger, H. 2-methoxyestradiol—Biology and mechanism of action. Steroids 2010, 75, 625–631. [Google Scholar] [CrossRef]
- Sweeney, C.; Liu, G.; Yiannoutsos, C.; Kolesar, J.; Horvath, D.; Staab, M.J.; Fife, K.; Armstrong, V.; Treston, A.; Sidor, C.; et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin. Cancer Res. 2005, 11, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Gorska, M.; Kuban-Jankowska, A.; Zmijewski, M.; Gorzynik, M.; Szkatula, M.; Wozniak, M. Neuronal nitric oxide synthase induction in the antitumorigenic and neurotoxic effects of 2-methoxyestradiol. Molecules 2014, 19, 13267–13281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorska, M.; Kuban-Jankowska, A.; Zmijewski, M.; Gammazza, A.M.; Cappello, F.; Wnuk, M.; Gorzynik, M.; Rzeszutek, I.; Daca, A.; Lewinska, A.; et al. DNA strand breaks induced by nuclear hijacking of neuronal NOS as an anti-cancer effect of 2-methoxyestradiol. Oncotarget 2015, 6, 15449–15463. [Google Scholar] [CrossRef] [Green Version]
- Gorska-Ponikowska, M.; Ploska, A.; Jacewicz, D.; Szkatula, M.; Barone, G.; Lo Bosco, G.; Lo Celso, F.; Dabrowska, A.M.; Kuban-Jankowska, A.; Gorzynik-Debicka, M.; et al. Modification of DNA structure by reactive nitrogen species as a result of 2-methoxyestradiol–induced neuronal nitric oxide synthase uncoupling in metastatic osteosarcoma cells. Redox Biol. 2020, 32, 101522. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Lin, C.M.; Flynn, E.; Folkman, J.; Hamel, E. 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc. Natl. Acad. Sci. USA 1994, 91, 3964–3968. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [Green Version]
- Attalla, H.; Westberg, J.A.; Andersson, L.C.; Adlercreutz, H.; Mäkelä, T.P. 2-methoxyestradiol-induced phosphorylation of Bcl-2: Uncoupling from JNK/SAPK activation. Biochem. Biophys. Res. Commun. 1998, 247, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Haldar, S. Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 2003, 538, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Jee, S.B.; Park, W.Y.; Choi, Y.J.; Kim, B.; Kim, Y.H.; Jun, D.Y.; Kim, Y.H. Tumor suppressor protein p53 promotes 2-methoxyestradiol-induced activation of bak and bax, leading to mitochondria-dependent apoptosis in human colon cancer HCT116 Cells. J. Microbiol. Biotechnol. 2014, 24, 1654–1663. [Google Scholar] [CrossRef] [Green Version]
- Gorska, M.; Zmijewski, M.A.; Kuban-Jankowska, A.; Wnuk, M.; Rzeszutek, I.; Wozniak, M. Neuronal Nitric oxide synthase-mediated genotoxicity of 2-methoxyestradiol in hippocampal HT22 cell line. Mol. Neurobiol. 2016, 53, 5030–5040. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Kuban-Jankowska, A.; Milczarek, R.; Wozniak, M. Nitro-oxidative stress is involved in anticancer activity of 17beta-estradiol derivative in neuroblastoma cells. Anticancer Res. 2016, 36, 1693–1698. [Google Scholar]
- Gorska-Ponikowska, M.; Kuban-Jankowska, A.; Eisler, S.A.; Perricone, U.; Lo Bosco, G.; Barone, G.; Nussberger, S. 2-Methoxyestradiol affects mitochondrial biogenesis pathway and succinate dehydrogenase complex flavoprotein subunit A in osteosarcoma cancer cells. Cancer Genom. Proteom. 2018, 15, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Lis, A.; Ciesielski, M.J.; Barone, T.A.; Scott, B.E.; Fenstermaker, R.A.; Plunkett, R.J. 2-Methoxyestradiol inhibits proliferation of normal and neoplastic glial cells, and induces cell death, in vitro. Cancer Lett. 2004, 213, 57–65. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, A.S.; Zhang, C.-J.; Katsumoto, A.; Teixeira, A.L. Hippocampal adult neurogenesis: Does the immune system matter? J. Neurol. Sci. 2017, 372, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Tsikas, D. Analytical methods for 3-nitrotyrosine quantification in biological samples: The unique role of tandem mass spectrometry. Amino Acids 2012, 45–63. [Google Scholar] [CrossRef]
- Ahsan, H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, D.; Fernandes, R.; Prudêncio, C.; Vieira, M. 3-Nitrotyrosine quantification methods: Current concepts and future challenges. Biochimie 2016, 125, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarlane, D.; Dybdal, N.; Donaldson, M.T.; Miller, L.; Cribb, A.E. Nitration and increased?-Synuclein expression associated with dopaminergic neurodegeneration in equine pituitary pars intermedia dysfunction. J. Neuroendocrinol. 2005, 17, 73–80. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Warren Olanow, C. Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef]
- Adamczyk, A.; Kaźmierczak, A.; Czapski, G.A.; Strosznajder, J.B. α-Synuclein induced cell death in mouse hippocampal (HT22) cells is mediated by nitric oxide-dependent activation of caspase-3. FEBS Lett. 2010, 584, 3504–3508. [Google Scholar] [CrossRef] [Green Version]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aygun, N. Biological and genetic features of neuroblastoma and their clinical importance. Curr. Pediatr. Rev. 2018, 14, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.R.; Hu LSen Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.N.; Pathak, S.S. Correlation between plasma neurotransmitters and memory loss in pregnancy. J. Reprod Med. 2002, 47, 494–496. [Google Scholar] [CrossRef]
- Ong, E.L.H.; Goldacre, R.; Goldacre, M. Differential risks of cancer types in people with Parkinson’s disease: A national record-linkage study. Eur J. Cancer 2014, 50, 2456–2462. [Google Scholar] [CrossRef]
- Kuzumaki, N.; Suda, Y.; Iwasawa, C.; Narita, M.; Sone, T.; Watanabe, M.; Maekawa, A.; Matsumoto, T.; Akamatsu, W.; Igarashi, K.; et al. Cell-specific overexpression of COMT in dopaminergic neurons of Parkinson’s disease. Brain 2019, 142, 1675–1689. [Google Scholar] [CrossRef]
- Kanasaki, K.; Palmsten, K.; Sugimoto, H.; Ahmad, S.; Hamano, Y.; Xie, L.; Parry, S.; Augustin, H.G.; Gattone, V.H.; Folkman, J.; et al. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 2008, 453, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Frontiers in Neuroanatomy. Front. Res. Found. 2015, 9, 91. [Google Scholar]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia Nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Rane, A.; Rajagopalan, S.; Chinta, S.J.; Andersen, J.K. Detrimental effects of oxidative losses in parkin activity in a model of sporadic Parkinson’s disease are attenuated by restoration of PGC1alpha. Neurobiol. Dis. 2016, 93, 115–120. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca 2+ -induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [Green Version]
- Yavich, L.; Tanila, H.; Vepsäläinen, S.; Jäkälä, P. Role of α-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.H.; Alaimo, A.; Gorojod, R.M.; Porte Alcon, S.; Fuentes, F.; Coluccio Leskow, F.; Kotler, M.L. Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol. Cell Neurosci. 2018, 88, 107–117. [Google Scholar] [CrossRef]
- Audano, M.; Schneider, A.; Mitro, N. Mitochondria, lysosomes and dysfunction: Their meaning in neurodegeneration. J. Neurochem. 2018, 291–309. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Hirabayashi, Y.; Kwon, S.-K.; Lewis, T.L.; Polleux, F. Emerging roles of mitochondria in synaptic transmission and neurodegeneration. Curr. Opin. Physiol. 2018, 3, 82–93. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid—Different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Mullin, S.; Schapira, A. α-Synuclein and mitochondrial dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Brown GC, Murphy MP, editors. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Langston, J.W. The MPTP story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinsons disease. Cell Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Guerra De Souza, A.C.; Prediger, R.D.; Cimarosti, H. SUMO-regulated mitochondrial function in Parkinson’s disease. J. Neurochem. 2016, 137, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to cancer development and therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug. Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.-T.; Wang, Z.-Z.; Yuan, Y.-H.; Wang, X.-L.; Sun, H.-M.; Chen, N.-H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Singh, S.; Tiwari, V.; Bano, S.; Shukla, S. Dopamine D1 receptor agonism induces dynamin related protein-1 inhibition to improve mitochondrial biogenesis and dopaminergic neurogenesis in rat model of Parkinson’s disease. Behav. Brain Res. 2020, 378, 112304. [Google Scholar] [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Senft, D.; Ronai, Z.A. Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 2016, 39, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Gorska-Ponikowska, M.; Bastian, P.; Zauszkiewicz-Pawlak, A.; Ploska, A.; Zubrzycki, A.; Kuban-Jankowska, A.; Nussberger, S.; Kalinowski, L.; Kmiec, Z. Regulation of mitochondrial dynamics in 2-methoxyestradiol-mediated osteosarcoma cell death. Sci. Rep. 2021, 11, 1616. [Google Scholar] [CrossRef]
- Karbowski, M.; Spodnik, J.H.; Teranishi, M.A.; Wozniak, M.; Nishizawa, Y.; Usukura, J.; Wakabayashi, T. Opposite effects of microtubule-stabilizing and microtubule-destabilizing drugs on biogenesis of mitochondria in mammalian cells. J. Cell Sci. 2001, 114, 281–291. [Google Scholar]
- Jiang, J.X.; Riquelme, M.A.; Zhou, J.Z. ATP, a double-edged sword in cancer. Oncoscience 2015, 2, 673–674. [Google Scholar] [CrossRef]
- Beijer, S.; Hupperets, P.S.; Van Den Borne, B.E.; Eussen, S.R.; Van Henten, A.M.; Van Den Beuken-Van Everdingen, M.; De Graeff, A.; Ambergen, T.A.; Van Den Brandt, P.A.; Dagnelie, P.C. Effect of adenosine 5′-triphosphate infusions on the nutritional status and survival of preterminal cancer patients. Anticancer Drugs 2009, 20, 625–633. [Google Scholar] [CrossRef] [PubMed]


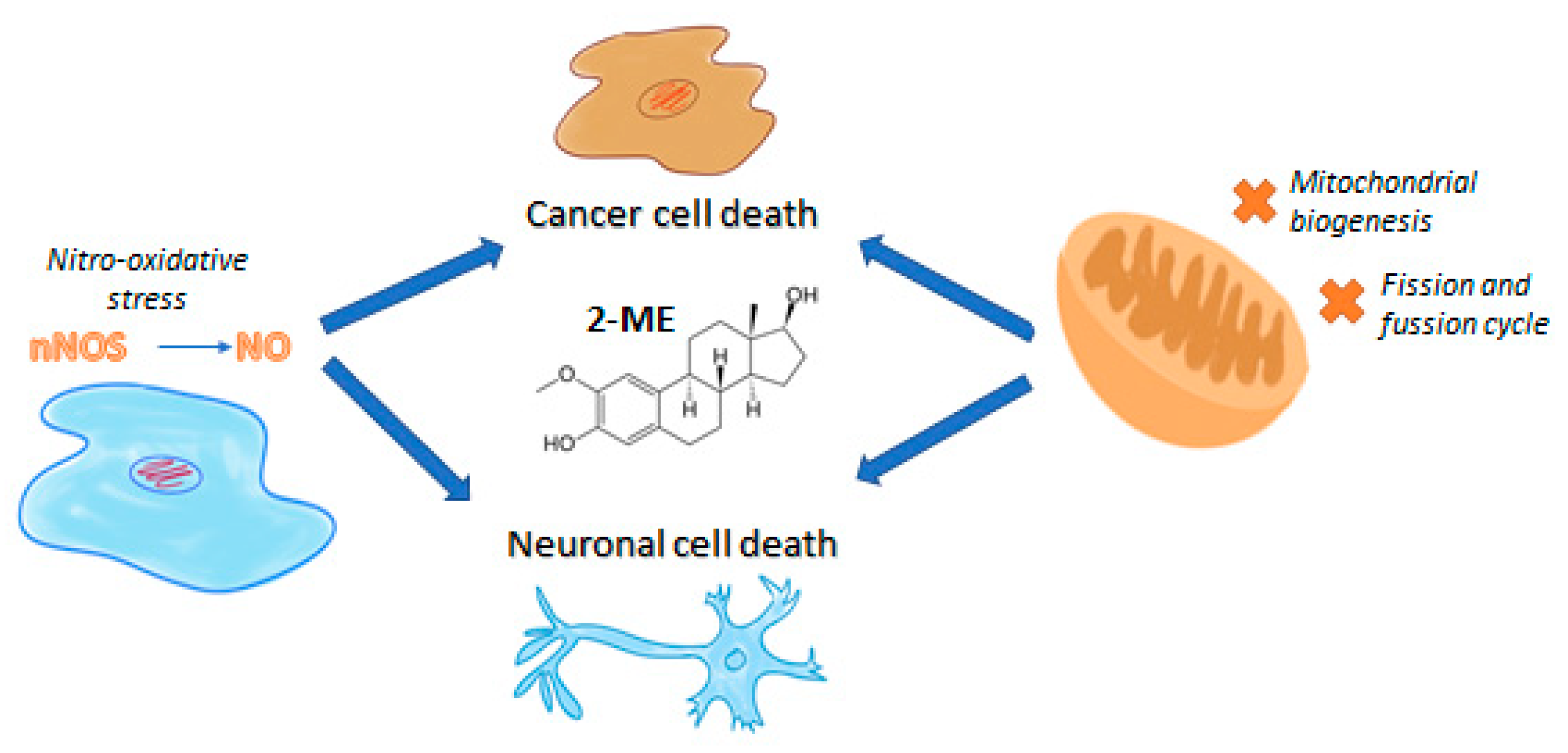{kind=link}
| Mutated Genes and Pathogenetic Functions | Involvement in PD | Involvement in Cancer | Reference |
|---|---|---|---|
| α-synuclein | Crucial component of Lewy bodies | Accumulation and aggregation e.g., in melanoma, brain and glial tumors | [33,34,35,36,37] |
| Parkin | Loss of function; crucial for accurate mitophagy initiation | Loss of function; increased sensitiveness to some cancers; initiate a tumor formation process; mutations present on e.g., lung, liver, intestine, and brain cancers | [19,20,21,22,23,24,25] |
| PINK1 | Loss of function; stabilize the mitochondrial membrane potential; deficiency impairs the plasticity of stratium and hippocampus | High expression in lung cancer; probable factor of chemo-resistance | [26,27,28,29] |
| Nitro-oxidative stress, mitochondrial dysfunction | Progression of neurodegeneration; damage DNA, lipid, and proteins; inducing apoptosis | Progression of cancer cells proliferation; damage DNA, lipid, and proteins; inducing apoptosis | [42,43,44,45] |
| Mutated Gene | Description of Gene | Influence of Mutations on Mitochondrial Function | References |
|---|---|---|---|
| α-synuclein | Crucial component of Lewy bodies; regulate synaptic vesicle transportation and endocytosis | Disturbed mitochondrial trafficking; fragmented mitochondria; inhibition of respiratory complex I; misfolding into oligomeric which are toxic to the mitochondria; induces the mitochondrial fragmentation | [2,171,172,173,174] |
| PINK1 | Kinase localized in mitochondria; crucial for accurate mitophagy initiation | Accumulates on the OMM of damaged mitochondria, and recruits Parkin to the dysfunctional mitochondrion | [175,176,177,178] |
| Parkin | Cytosolic E3 ubiquitin ligase located in mitochondria; crucial for accurate mitophagy initiation | Ubiquitinates outer mitochondrial membrane proteins and leads to their degradation by the proteasome | [175,176,177,178] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastian, P.; Dulski, J.; Roszmann, A.; Jacewicz, D.; Kuban-Jankowska, A.; Slawek, J.; Wozniak, M.; Gorska-Ponikowska, M. Regulation of Mitochondrial Dynamics in Parkinson’s Disease—Is 2-Methoxyestradiol a Missing Piece? Antioxidants 2021, 10, 248. https://doi.org/10.3390/antiox10020248
Bastian P, Dulski J, Roszmann A, Jacewicz D, Kuban-Jankowska A, Slawek J, Wozniak M, Gorska-Ponikowska M. Regulation of Mitochondrial Dynamics in Parkinson’s Disease—Is 2-Methoxyestradiol a Missing Piece? Antioxidants. 2021; 10(2):248. https://doi.org/10.3390/antiox10020248
Chicago/Turabian StyleBastian, Paulina, Jaroslaw Dulski, Anna Roszmann, Dagmara Jacewicz, Alicja Kuban-Jankowska, Jaroslaw Slawek, Michal Wozniak, and Magdalena Gorska-Ponikowska. 2021. "Regulation of Mitochondrial Dynamics in Parkinson’s Disease—Is 2-Methoxyestradiol a Missing Piece?" Antioxidants 10, no. 2: 248. https://doi.org/10.3390/antiox10020248
APA StyleBastian, P., Dulski, J., Roszmann, A., Jacewicz, D., Kuban-Jankowska, A., Slawek, J., Wozniak, M., & Gorska-Ponikowska, M. (2021). Regulation of Mitochondrial Dynamics in Parkinson’s Disease—Is 2-Methoxyestradiol a Missing Piece? Antioxidants, 10(2), 248. https://doi.org/10.3390/antiox10020248