
xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Western Blot Analysis
2.3. Cell Viability Assay
2.4. Conditioned Media Thiol Quantification (Ellman’s Test)
2.5. Total Selenium Measurement by ICP-MS Analysis
2.6. Detection of Lipid Peroxidation
2.7. RNA Expression and Prognostic Value SLC7A11 and SLC3A2 in Normal and Tumor Tissues
2.8. Processing of Human Breast Tissues
2.9. Quantification and Statistical Analysis
3. Results
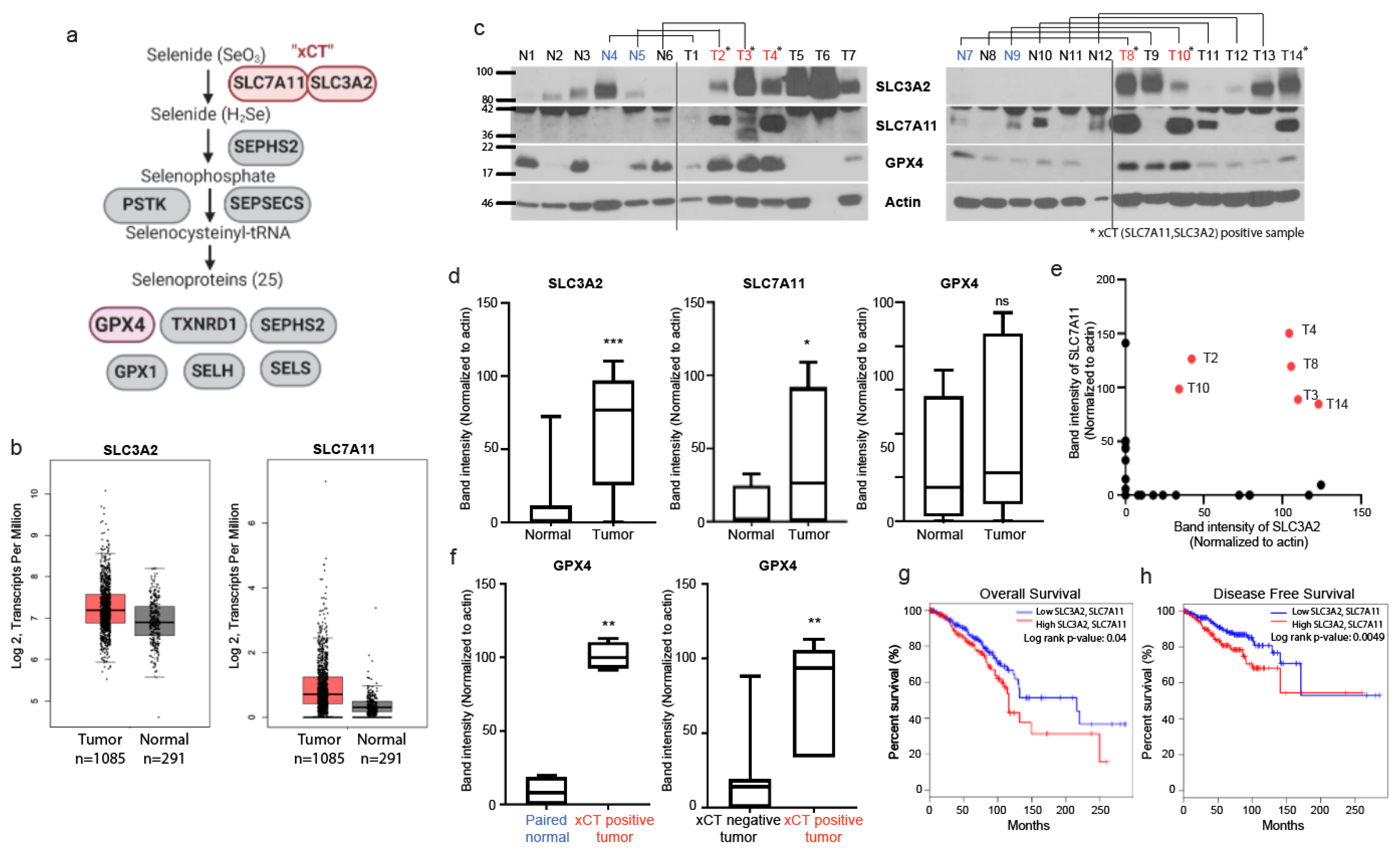
3.1. Expression of xCT, an Initial Step of Selenocysteine Synthesis Pathway, Is Highly Upregulated in Breast Cancer Tissues and Expression of GPX4 Is High in xCT Positivie Tumors
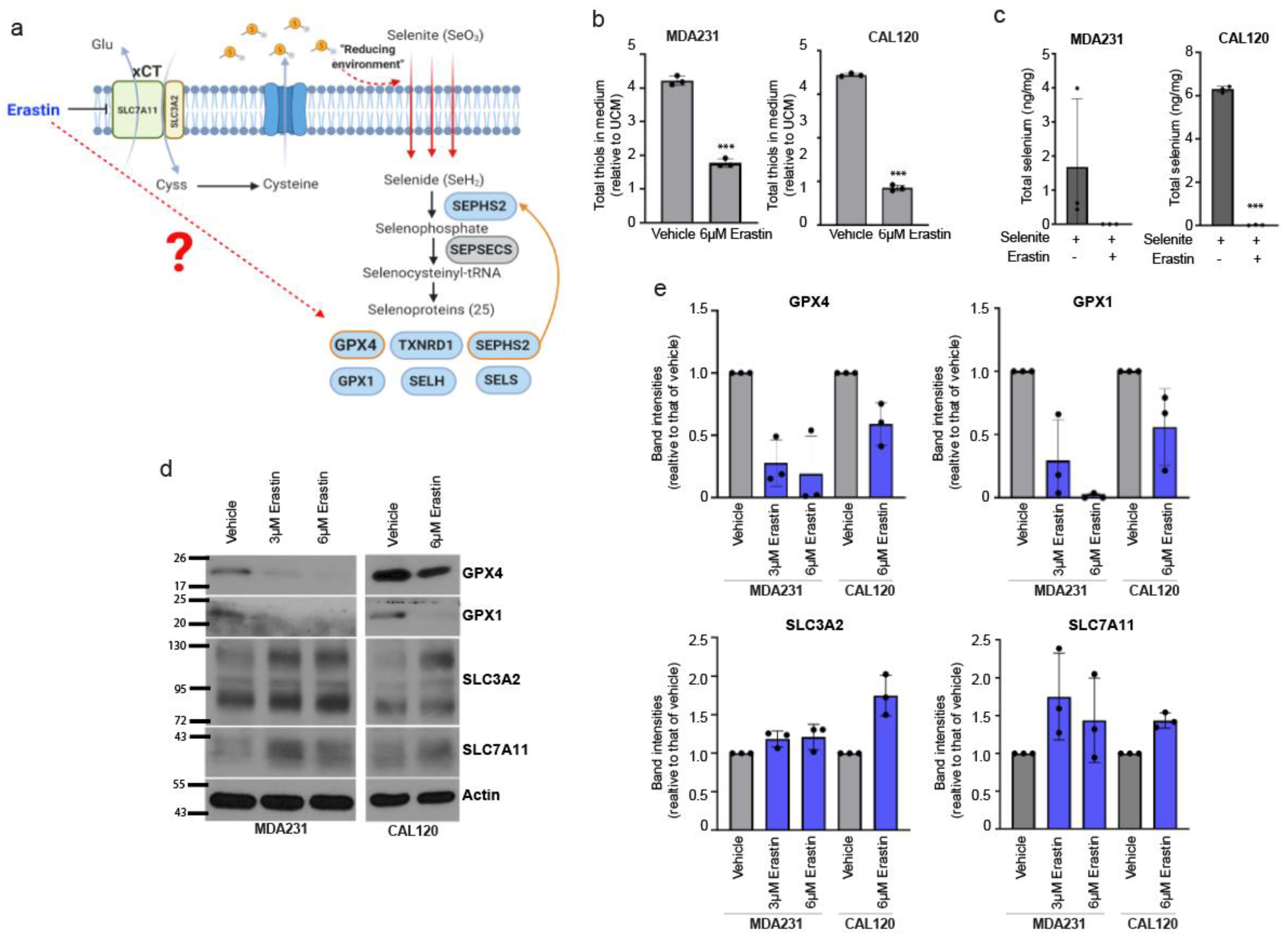
3.2. Erastin Targets the Selenium Uptake and Selenoprotein Expression Promoting Activity of xCT
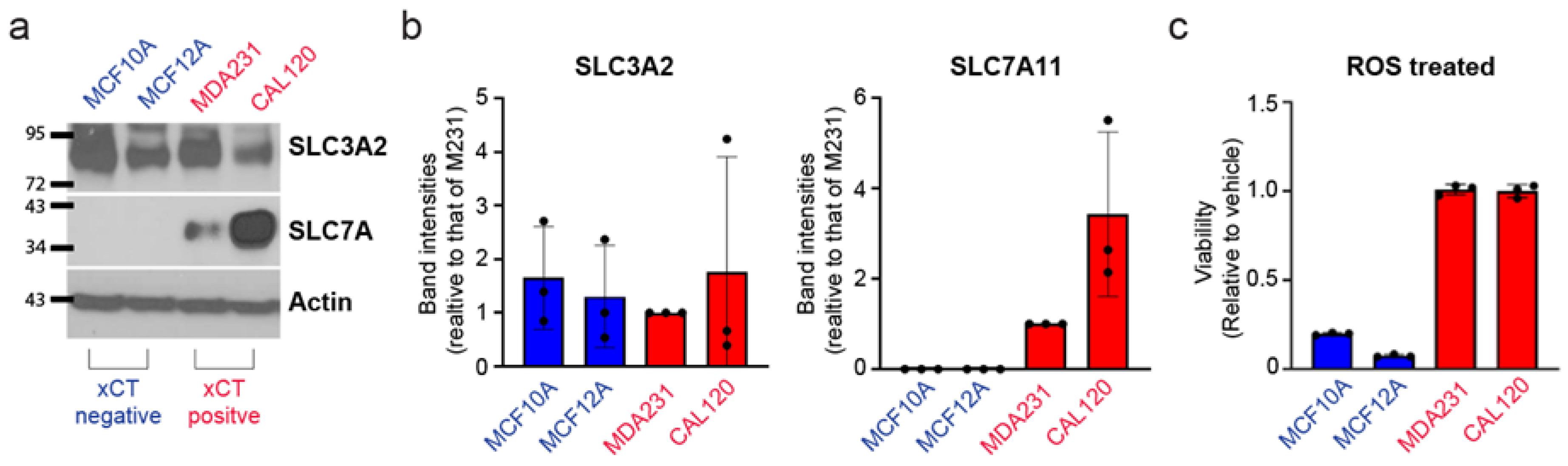
3.3. Breast Cancer Cells Have Increased Resistance against Cell Death Induced by Reactive Oxygen Species Which Correlates with xCT Expression
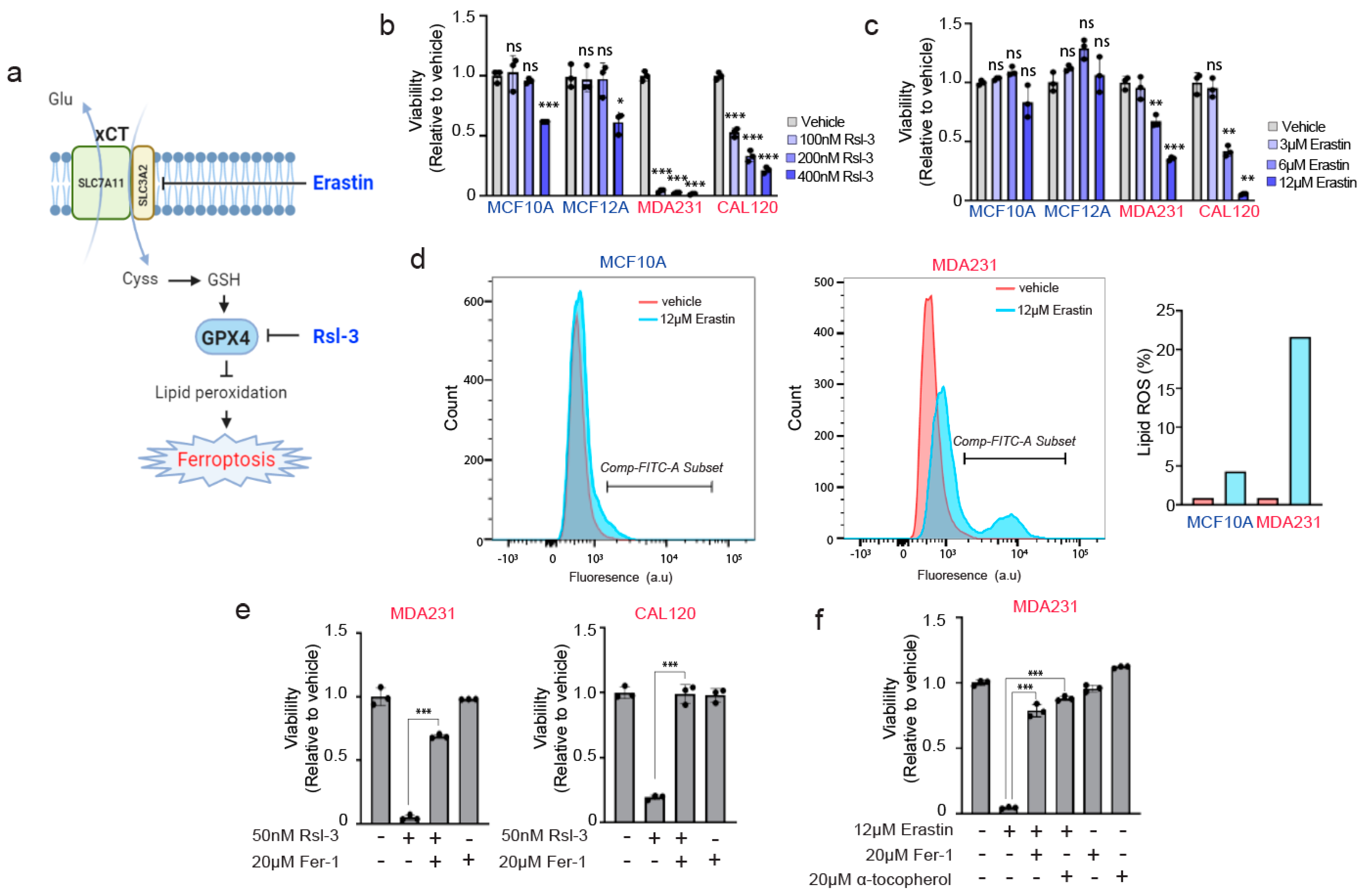
3.4. TNBC Cells Are Paradoxically Hypersensitive to Targeting of Anti-Ferroptotic Machinery by Erastin and Rsl-3
4. Discussion
5. Therapeutic Implications and Concluding Thoughts
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, D.M.; Copeland, P.R. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003, 23, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, A.E.; Lee, N.; Matthew-Onabanjo, A.N.; Spears, M.E.; Park, S.J.; Youkana, D.; Doshi, M.B.; Peppers, A.; Li, R.; Joseph, A.B.; et al. Selenium detoxification is required for cancer-cell survival. Nat. Metab. 2020, 2, 603–611. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem. Biol. 2020, 27, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Vanderplas, A.; Hughes, M.E.; Theriault, R.L.; Edge, S.B.; Wong, Y.N.; Blayney, D.W.; Niland, J.C.; Winer, E.P.; Weeks, J.C. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive Cancer Network. Cancer 2012, 118, 5463–5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, L.; Chen, C.; Zhou, Y.; Hu, D.; Yang, J.; Chen, Y.; Zhuo, W.; Mao, M.; Zhang, X.; et al. Targeting ferroptosis in breast cancer. Biomark. Res. 2020, 8, 58. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Bannai, S.; Christensen, H.N.; Vadgama, J.V.; Ellory, J.C.; Englesberg, E.; Guidotti, G.G.; Gazzola, G.C.; Kilberg, M.S.; Lajtha, A.; Sacktor, B.; et al. Amino acid transport systems. Nature 1984, 311, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olm, E.; Fernandes, A.P.; Hebert, C.; Rundlof, A.K.; Larsen, E.H.; Danielsson, O.; Bjornstedt, M. Extracellular thiol-assisted selenium uptake dependent on the x(c)-cystine transporter explains the cancer-specific cytotoxicity of selenite. Proc. Natl. Acad. Sci. USA 2009, 106, 11400–11405. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar] [PubMed]
- Liu, J.; Xia, X.; Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- El Ansari, R.; Craze, M.L.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; Green, A.R. The multifunctional solute carrier 3A2 (SLC3A2) confers a poor prognosis in the highly proliferative breast cancer subtypes. Br. J. Cancer 2018, 118, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Cheng, D.; Hou, L.; Zhou, S.; Ying, T.; Yang, Q. SLC3A2 is upregulated in human osteosarcoma and promotes tumor growth through the PI3K/Akt signaling pathway. Oncol. Rep. 2017, 37, 2575–2582. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Wada, K.; Toyooka, T.; Shinomiya, N.; Shimazaki, H.; Nakanishi, K.; Nagatani, K.; Otani, N.; Osada, H.; Uozumi, Y.; et al. Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery 2013, 72, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Pointer, K.B.; Kuo, J.S. Excitotoxic SLC7A11 Expression Is a Marker of Poor Glioblastoma Survival and a Potential Therapeutic Target. Neurosurgery 2015, 77, N16–N17. [Google Scholar] [CrossRef] [Green Version]
- Shiozaki, A.; Iitaka, D.; Ichikawa, D.; Nakashima, S.; Fujiwara, H.; Okamoto, K.; Kubota, T.; Komatsu, S.; Kosuga, T.; Takeshita, H.; et al. xCT, component of cysteine/glutamate transporter, as an independent prognostic factor in human esophageal squamous cell carcinoma. J. Gastroenterol. 2014, 49, 853–863. [Google Scholar] [CrossRef]
- Kinoshita, H.; Okabe, H.; Beppu, T.; Chikamoto, A.; Hayashi, H.; Imai, K.; Mima, K.; Nakagawa, S.; Ishimoto, T.; Miyake, K.; et al. Cystine/glutamic acid transporter is a novel marker for predicting poor survival in patients with hepatocellular carcinoma. Oncol. Rep. 2013, 29, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.S.; Jeong, B.S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugano, K.; Maeda, K.; Ohtani, H.; Nagahara, H.; Shibutani, M.; Hirakawa, K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015, 35, 677–682. [Google Scholar] [PubMed]
- Lu, X. The Role of Large Neutral Amino Acid Transporter (LAT1) in Cancer. Curr. Cancer Drug Targets 2019, 19, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Walko, C.M.; Grande, C. Management of common adverse events in patients treated with sorafenib: Nurse and pharmacist perspective. Semin. Oncol. 2014, 41 (Suppl. 2), S17–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e629. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting xCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anticancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef]
- Sugiyama, A.; Ohta, T.; Obata, M.; Takahashi, K.; Seino, M.; Nagase, S. xCT inhibitor sulfasalazine depletes paclitaxel-resistant tumor cells through ferroptosis in uterine serous carcinoma. Oncol. Lett. 2020, 20, 2689–2700. [Google Scholar] [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef] [Green Version]
- Tinggi, U. Selenium: Its role as antioxidant in human health. Environ. Health Prev. Med. 2008, 13, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279 e1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, N.; Khanahmad, H.; Hakimian, S.M.; Sheikhi, A.; Rashidi, B.; Pourfarzam, M. Evaluation of long-chain acyl-coenzyme A synthetase 4 (ACSL4) expression in human breast cancer. Res. Pharm. Sci. 2020, 15, 48–56. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, N.; Carlisle, A.E.; Peppers, A.; Park, S.J.; Doshi, M.B.; Spears, M.E.; Kim, D. xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers. Antioxidants 2021, 10, 317. https://doi.org/10.3390/antiox10020317
Lee N, Carlisle AE, Peppers A, Park SJ, Doshi MB, Spears ME, Kim D. xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers. Antioxidants. 2021; 10(2):317. https://doi.org/10.3390/antiox10020317
Chicago/Turabian StyleLee, Namgyu, Anne E. Carlisle, Austin Peppers, Sung Jin Park, Mihir B. Doshi, Meghan E. Spears, and Dohoon Kim. 2021. "xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers" Antioxidants 10, no. 2: 317. https://doi.org/10.3390/antiox10020317
APA StyleLee, N., Carlisle, A. E., Peppers, A., Park, S. J., Doshi, M. B., Spears, M. E., & Kim, D. (2021). xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers. Antioxidants, 10(2), 317. https://doi.org/10.3390/antiox10020317