
Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases





Abstract
:1. Introduction
2. Crucial Role of ROS in Physiological and Pathological Mechanisms
2.1. ROS and Mitochondrial Dysfunctions
2.2. Metal Accumulation, ROS Production, and Protein Misfolding
2.3. OS and Protein Misfolding/Accumulation
3. Nature-Based Compounds against Cellular Aging and Neurodegeneration
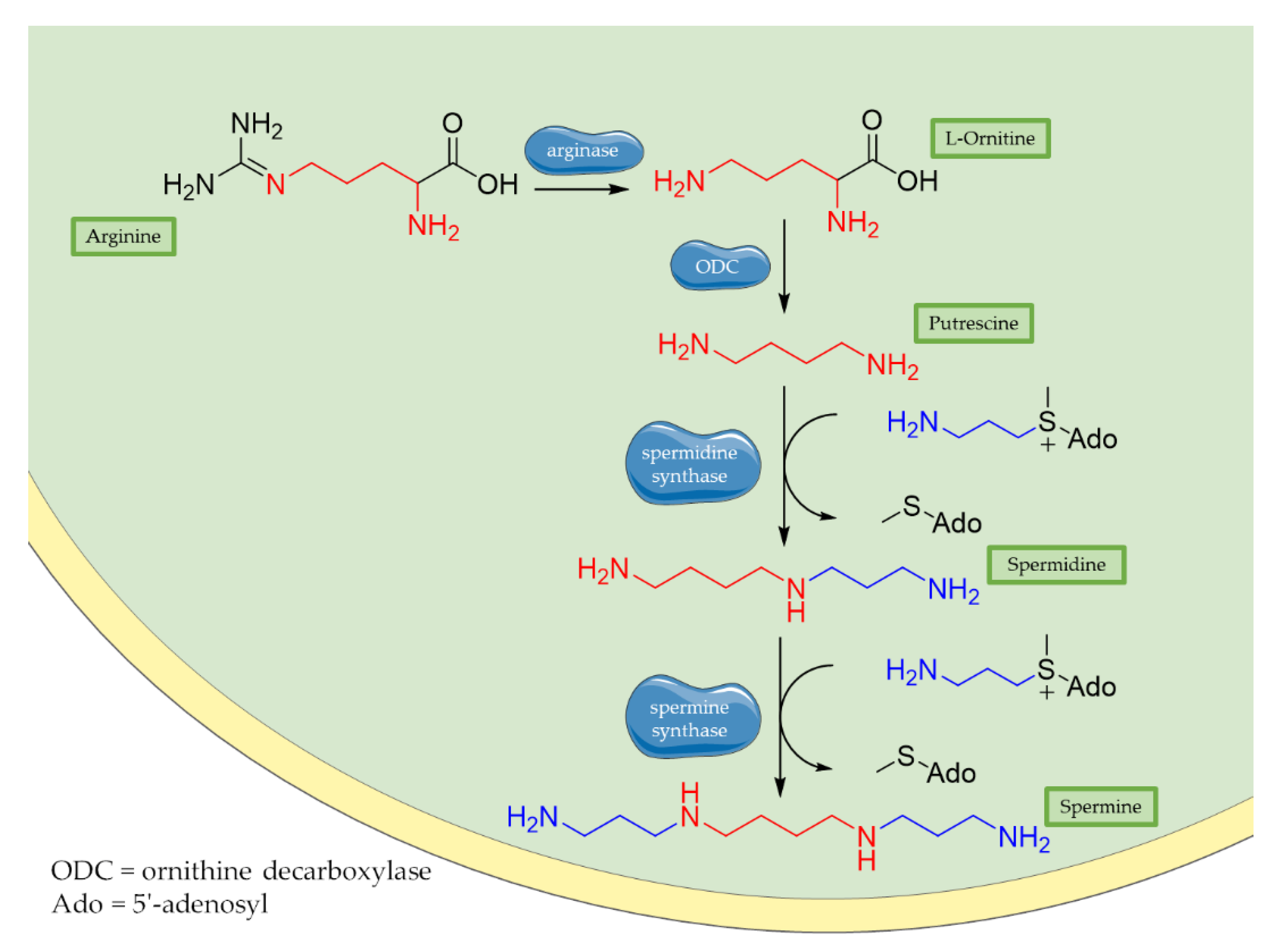
3.1. Polyamines: Spermidine and Spermine
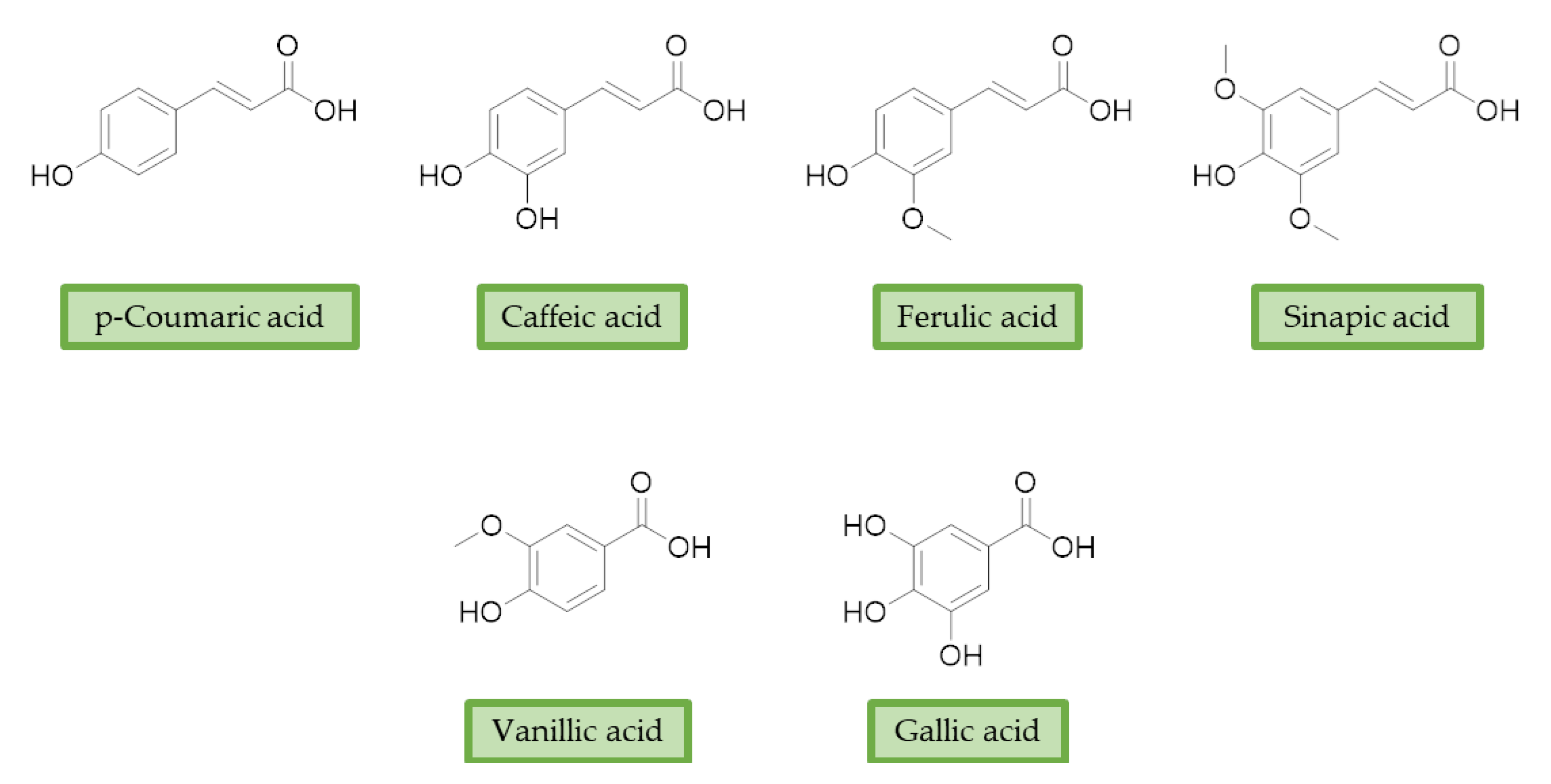
3.2. Phenolic Acids
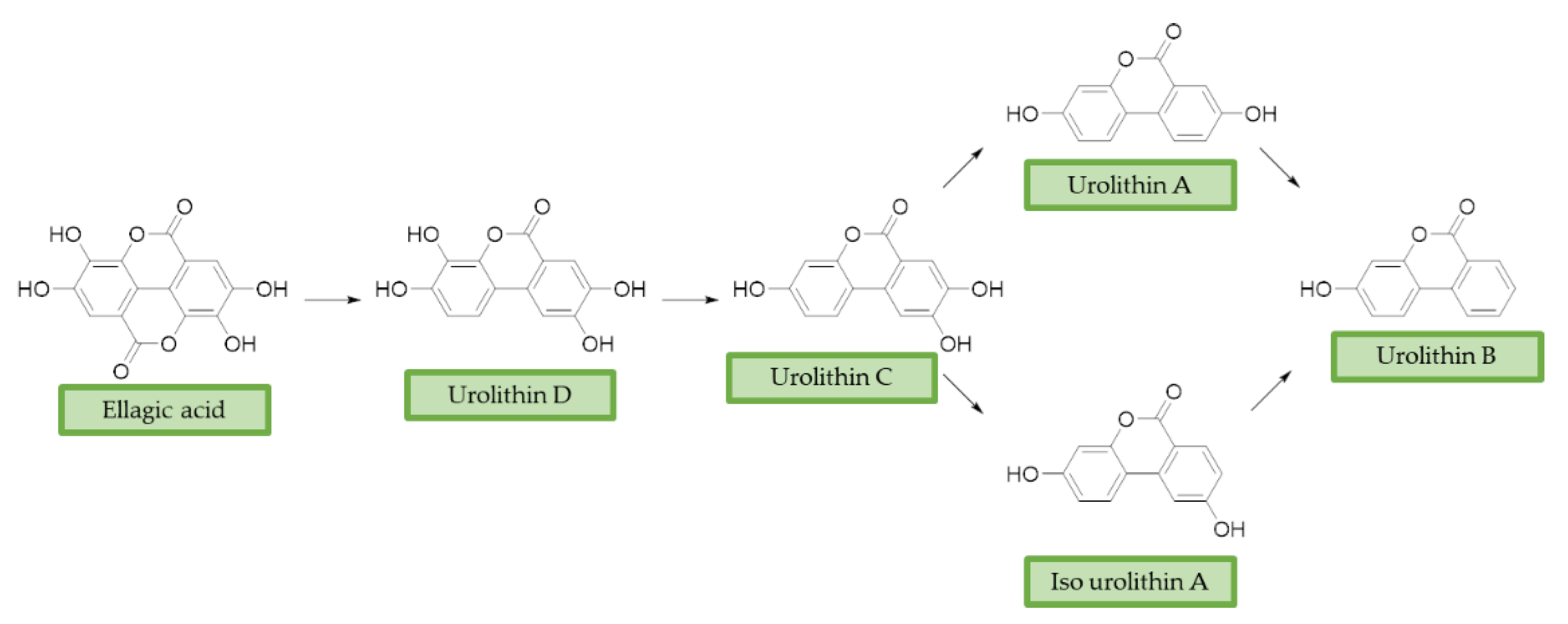
3.3. Urolithins
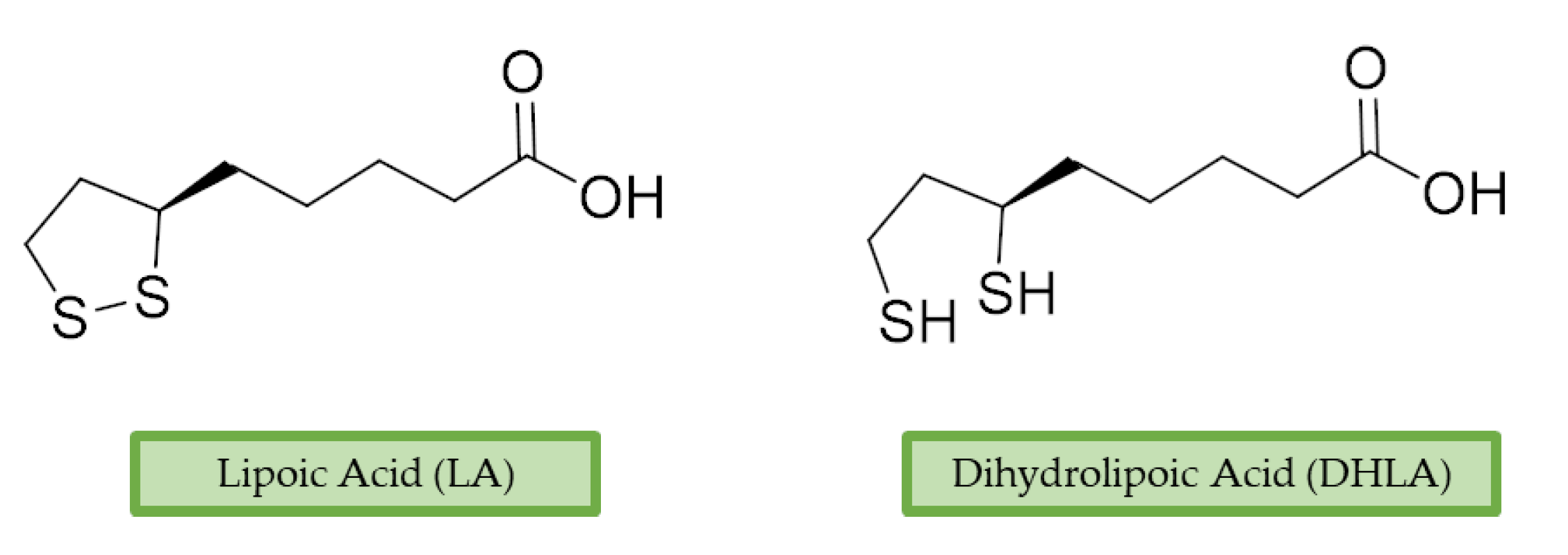
3.4. Lipoic Acid
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, I.P. Age-related neurodegenerative disease research needs aging models. Front. Aging Neurosci. 2015, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, Y.; Domanskyi, A. Detecting Oxidative Stress Biomarkers in Neurodegenerative Disease Models and Patients. Methods Protoc. 2020, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar]
- Nesi, G.; Sestito, S.; Digiacomo, M.; Rapposelli, S. Oxidative Stress, Mitochondrial Abnormalities and Proteins Deposition: Multitarget Approaches in Alzheimer’s Disease. Curr. Top. Med. Chem. 2017, 17, 3062–3079. [Google Scholar] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krisko, A.; Radman, M. Protein damage, ageing and age-related diseases. Open Biol. 2019, 9, 180249. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Schiavone, S.; Trabace, L. Small Molecules: Therapeutic Application in Neuropsychiatric and Neurodegenerative Disorders. Molecules 2018, 23, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flora, S.J.S. Structural, Chemical and Biological Aspects of Antioxidants for Strategies Against Metal and Metalloid Exposure. Oxidative Med. Cell. Longev. 2009, 2, 191–206. [Google Scholar] [CrossRef]
- Zeng, Q.; Siu, W.; Li, L.; Jin, Y.; Liang, S.; Cao, M.; Ma, M.; Wu, Z. Autophagy in Alzheimer’s disease and promising modulatory effects of herbal medicine. Exp. Gerontol. 2019, 119, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-R.; Fu, Y.-S.; Tsai, M.-J.; Cheng, H.; Weng, C.-F. Natural Compounds from Herbs that can Potentially Execute as Autophagy Inducers for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1412. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.J.; Robert, C.; Gough, S.M.; Rassool, F.V.; Aplan, P.D. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk. Res. 2014, 38, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luceri, C.; Bigagli, E.; Femia, A.P.; Caderni, G.; Giovannelli, L.; Lodovici, M. Aging related changes in circulating reactive oxygen species (ROS) and protein carbonyls are indicative of liver oxidative injury. Toxicol. Rep. 2018, 5, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Dhillon, V.S.; Fenech, M. Mutations that affect mitochondrial functions and their association with neurodegenerative diseases. Mutat. Res. Rev. Mutat. Res. 2014, 759, 1–13. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Maremonti, E.; Eide, D.M.; Rossbach, L.M.; Lind, O.C.; Salbu, B.; Brede, D.A. In vivo assessment of reactive oxygen species production and oxidative stress effects induced by chronic exposure to gamma radiation in Caenorhabditis elegans. Free Radic. Biol. Med. 2019, 152, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, J.H.; Stewart, J.B. Mitochondrial DNA: Radically free of free-radical driven mutations. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1354–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; Akar, F.G.; O’Rourke, B. Mitochondrial criticality: A new concept at the turning point of life or death. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 232–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, V.C.; Heales, S.J. Nitric oxide-induced mitochondrial dysfunction: Implications for neurodegeneration. Free. Radic. Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.H.; Lu, C.Y.; Wei, C.Y.; Ma, Y.S.; Lee, H.C. Oxidative stress in human aging and mitochondrial disease-consequences of defective mitochondrial respiration and impaired antioxidant enzyme system. Chin. J. Physiol. 2001, 44, 1–11. [Google Scholar] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Starkov, A.A. Calcium and Mitochondrial Reactive Oxygen Species Generation: How to Read the Facts. J. Alzheimer’s Dis. 2010, 20, S413–S426. [Google Scholar] [CrossRef] [Green Version]
- Tranah, G.J.; Nalls, M.A.; Katzman, S.M.; Yokoyama, J.S.; Lam, E.T.; Zhao, Y.; Mooney, S.; Thomas, F.; Newman, A.B.; Liu, Y.; et al. Mitochondrial DNA Sequence Variation Associated with Dementia and Cognitive Function in the Elderly. J. Alzheimer’s Dis. 2012, 32, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, R.K.; Beal, M.F. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol. Cell. Neurosci. 2013, 55, 101–114. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dexter, D.T.; Wells, F.R.; Lee, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Valko, M.M.H.C.M.; Morris, H.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jiao, Q.; Xu, H.; Du, X.; Shi, L.; Jia, F.; Jiang, H. Biometal Dyshomeostasis and Toxic Metal Accumulations in the Development of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Baros, S.; Valko, M. Redox active metal-induced oxidative stress in biological systems. Transit. Met. Chem. 2012, 37, 127–134. [Google Scholar] [CrossRef]
- Zatta, P.; Kiss, T.; Suwalsky, M.; Berthon, G. Aluminium (III) as a promoter of cellular oxidation. Coord. Chem. Rev. 2002, 228, 271–284. [Google Scholar] [CrossRef]
- Farina, M.; Avila, D.S.; da Rocha, J.B.T.; Aschner, M. Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 2013, 62, 575–594. [Google Scholar] [CrossRef] [Green Version]
- Breydo, L.; Uversky, V.N. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 2011, 3, 1163–1180. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Chen, P.; Martinez-Finley, E.J.; Bornhorst, J.; Chakraborty, S. Metal-induced neurodegeneration in C. elegans. Front. Aging Neurosci. 2013, 5, 18. [Google Scholar]
- Leal, S.S.; Botelho, H.M.; Gomes, C.M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 2012, 256, 2253–2270. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Nunez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognin, S.; Drago, D.; Messori, L.; Zatta, P. Chelation therapy for neurodegenerative diseases. Med. Res. Rev. 2009, 29, 547–570. [Google Scholar] [CrossRef]
- Sales, T.A.; Prandi, I.G.; de Castro, A.A.; Leal, D.H.S.; da Cunha, E.F.F.; Kuca, K.; Ramalho, T.C. Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. Int. J. Mol. Sci. 2019, 20, 1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Bross, P. Protein Misfolding and Cellular Stress: An Overview. Adv. Struct. Saf. Stud. 2010, 648, 3–23. [Google Scholar] [CrossRef]
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and-aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Yue, Z. Neuronal Aggregates: Formation, Clearance, and Spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia–ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, J.R. Oxidative stress impairs autophagy through oxidation of ATG3 and ATG7. Autophagy 2018, 14, 1092–1093. [Google Scholar] [CrossRef] [PubMed]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.-N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural Regen. Res. 2013, 8, 2275–2283. [Google Scholar] [PubMed]
- Almeida, S.; Alves, M.G.; Sousa, M.; Oliveira, P.F.; Silva, B.M. Are Polyphenols Strong Dietary Agents Against Neurotoxicity and Neurodegeneration? Neurotox. Res. 2016, 30, 345–366. [Google Scholar] [CrossRef]
- Pohl, F.; Lin, P.K.T. The Potential Use of Plant Natural Products and Plant Extracts with Antioxidant Properties for the Prevention/Treatment of Neurodegenerative Diseases: In Vitro, In Vivo and Clinical Trials. Molecules 2018, 23, 3283. [Google Scholar] [CrossRef] [Green Version]
- Pallauf, K.; Rimbach, G. Autophagy, polyphenols and healthy ageing. Ageing Res. Rev. 2013, 12, 237–252. [Google Scholar] [CrossRef]
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.F.M.; Pogačnik, L. Polyphenols from Food and Natural Products: Neuroprotection and Safety. Antioxidants 2020, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Koudoufio, M.; Desjardins, Y.; Feldman, F.; Spahis, S.; Delvin, E.; Levy, E. Insight into Polyphenol and Gut Microbiota Crosstalk: Are Their Metabolites the Key to Understand Protective Effects against Metabolic Disorders? Antioxidants 2020, 9, 982. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, U. The early history of polyamine research. Plant Physiol. Biochem. 2010, 48, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Esparza, N.C.; Latorre-Moratalla, M.L.; Comas-Basté, O.; Toro-Funes, N.; Veciana-Nogués, M.T.; Vidal-Carou, M.C. Polyamines in Food. Front. Nutr. 2019, 6, 108. [Google Scholar] [CrossRef]
- Wallace, H.M. The polyamines: Past, present and future. Essays Biochem. 2009, 46, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucciarelli, S.; Moreschini, B.; Micozzi, D.; de Fronzo, G.S.; Carpi, F.M.; Polzonetti, V.; Vincenzetti, S.; Mignini, F.; Napolioni, V. Spermidine and Spermine Are Enriched in Whole Blood of Nona/Centenarians. Rejuvenation Res. 2012, 15, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.K.; Tabor, C.W.; Tabor, H. Polyamine deficiency leads to accumulation of reactive oxygen species in a spe2Delta mutant of Saccharomyces cerevisiae. Yeast 2006, 23, 751–761. [Google Scholar] [CrossRef]
- Sava, I.G.; Battaglia, V.; Rossi, C.A.; Salvi, M.; Toninello, A. Free radical scavenging action of the natural polyamine spermine in rat liver mitochondria. Free. Radic. Biol. Med. 2006, 41, 1272–1281. [Google Scholar] [CrossRef]
- Jeong, J.-W.; Cha, H.-J.; Han, M.H.; Hwang, S.J.; Lee, D.-S.; Yoo, J.S.; Choi, I.-W.; Kim, S.; Kim, H.-S.; Kim, G.-Y.; et al. Spermidine Protects against Oxidative Stress in Inflammation Models Using Macrophages and Zebrafish. Biomol. Ther. 2018, 26, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, I.; Sankhe, R.; Mudgal, J.; Arora, D.; Nampoothiri, M. Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 2020, 83, 102083. [Google Scholar] [CrossRef] [PubMed]
- Molnar, M.M.; Liddell, S.C.; Wadkins, R.M. Effects of Polyamine Binding on the Stability of DNA i-Motif Structures. ACS Omega 2019, 4, 8967–8973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacomino, G.; Picariello, G.; d’Agostino, L. DNA and nuclear aggregates of polyamines. Biochim. Biophys. Acta Bioenerg. 2012, 1823, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Gong, H.; Sun, Q.; Zhao, R.; Jia, Y. Spermidine-Activated Satellite Cells Are Associated with Hypoacetylation in ACVR2B and Smad3 Binding to Myogenic Genes in Mice. J. Agric. Food Chem. 2018, 66, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Nisosantano, M.; Pedro, J.M.B.-S.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Marino, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2014, 22, 509–516. [Google Scholar] [CrossRef]
- Burgio, G.; Corona, D.F.V.; Nicotra, C.M.A.; Carruba, G.; Taibi, G. P/CAF-mediated spermidine acetylation regulates histone acetyltransferase activity. J. Enzym. Inhib. Med. Chem. 2016, 31, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Yue, F.; Li, W.; Zou, J.; Jiang, X.; Xu, G.; Huang, H.; Liu, L. Spermidine Prolongs Lifespan and Prevents Liver Fibrosis and Hepatocellular Carcinoma by Activating MAP1S-Mediated Autophagy. Cancer Res. 2017, 77, 2938–2951. [Google Scholar] [CrossRef] [Green Version]
- Phadwal, K.; Kurian, D.; Salamat, M.K.F.; Macrae, V.E.; Diack, A.B.; Manson, J.C. Spermine increases acetylation of tubulins and facilitates autophagic degradation of prion aggregates. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Soda, K. Spermine and gene methylation: A mechanism of lifespan extension induced by polyamine-rich diet. Amino Acids 2019, 52, 213–224. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, H.; Zhang, J.; Yu, H.; Lin, Z.; Cai, Y. Spermidine Exhibits Protective Effects Against Traumatic Brain Injury. Cell. Mol. Neurobiol. 2020, 40, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Signor, C.; Mello, C.F.; Porto, G.P.; Ribeiro, D.A.; Rubin, M.A. Spermidine improves fear memory persistence. Eur. J. Pharmacol. 2014, 730, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilad, G.M.; Gilad, V.H. Novel polyamine derivatives as neuroprotective agents. J. Pharmacol. Exp. Ther. 1999, 291, 39–43. [Google Scholar] [PubMed]
- Melchiorre, C.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V. Polyamines in Drug Discovery: From the Universal Template Approach to the Multitarget-Directed Ligand Design Strategy. J. Med. Chem. 2010, 53, 5906–5914. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A.; Melchiorre, C. Memoquin: A multi-target-directed ligand as an innovative therapeutic opportunity for Alzheimer’s disease. Neurotherapeutics 2009, 6, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Hu, K.; Bai, P.; Yu, L.; Ma, Q.; Li, T.; Zhang, X.; Chen, C.; Peng, K.; Liu, W.; et al. Design, synthesis and evaluation of novel ferulic acid-memoquin hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2016, 26, 2539–2543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Hong, C.; Luo, W.; Wang, C. Design, synthesis and evaluation of genistein-polyamine conjugates as multi-functional anti-Alzheimer agents. Acta Pharm. Sin. B 2015, 5, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Simoni, E.; Caporaso, R.; Bergamini, C.; Fiori, J.; Fato, R.; Miszta, P.; Filipek, S.; Caraci, F.; Giuffrida, M.L.; Andrisano, V.; et al. Polyamine Conjugation as a Promising Strategy to Target Amyloid Aggregation in the Framework of Alzheimer’s Disease. ACS Med. Chem. Lett. 2016, 7, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-Q.; Fan, H.-X.; He, R.-R.; Xiao, J.; Tsoi, B.; Lan, K.-H.; Kurihara, H.; So, K.-F.; Yao, X.-S.; Gao, H. Lycibarbarspermidines A–O, New Dicaffeoylspermidine Derivatives from Wolfberry, with Activities against Alzheimer’s Disease and Oxidation. J. Agric. Food Chem. 2016, 64, 2223–2237. [Google Scholar] [CrossRef]
- Gao, H.; Yao, X.; He, R.; Chen, G.; Zhou, Z.; Wang, C.; Hu, D.; Fan, H. Dicaffeoyl spermidine cyclized derivatives and use thereof. U.S. Patent No 10,457,702, 29 October 2019. [Google Scholar]
- Grosso, G. Effects of Polyphenol-Rich Foods on Human Health. Nutrients 2018, 10, 1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Mello, A.J.M.; Fasolo, D. Chapter 20—Polyphenol Antioxidants from Natural Sources and Contribution to Health Promotion. In Polyphenols in Human Health and Disease; Watson, R.R., Preedy, R.V., Zibadi, S., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 253–265. [Google Scholar]
- Handique, J.; Baruah, J. Polyphenolic compounds: An overview. React. Funct. Polym. 2002, 52, 163–188. [Google Scholar] [CrossRef]
- Haque, M.E.; Javed, H.; Azimullah, S.; Khair, S.B.A.; Ojha, S. Neuroprotective potential of ferulic acid in the rotenone model of Parkinson’s disease. Drug Des. Dev. Ther. 2015, 9, 5499–5510. [Google Scholar] [CrossRef] [Green Version]
- Magnani, C.; Chiari, B.G.; Isaac, V.L.B.; Correa, M.A.; Salgado, H.R.N. In Vitro Safety Evaluation of Caffeic Acid. Athens J. Health 2014, 1, 181–188. [Google Scholar] [CrossRef]
- Zanwar, A.A.; Badole, S.L.; Shende, P.S.; Hegde, M.V.; Bodhankar, S.L. Role of Gallic Acid in Cardiovascular Disorders. In Polyphenols in Human Health and Disease; Elsevier BV: Amsterdam, The Netherlands, 2014; pp. 1045–1047. [Google Scholar]
- Shahidi, F.; Janitha, P.K.; Wanasundara, P.D. Phenolic antioxidants. Crit. Rev. Food Sci. Nutr. 1992, 32, 67–103. [Google Scholar] [CrossRef]
- Ren, Z.; Zhang, R.; Li, Y.; Li, Y.; Yang, Z.; Yang, H. Ferulic acid exerts neuroprotective effects against cerebral ischemia/reperfusion-induced injury via antioxidant and anti-apoptotic mechanisms in vitro and in vivo. Int. J. Mol. Med. 2017, 40, 1444–1456. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekhar, Y.; Kumar, G.P.; Ramya, E.M.; Anilakumar, K.R. Gallic Acid Protects 6-OHDA Induced Neurotoxicity by Attenuating Oxidative Stress in Human Dopaminergic Cell Line. Neurochem. Res. 2018, 43, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Kantham, S.; Rao, V.M.; Palanivelu, M.K.; Pham, H.L.; Shaw, P.N.; McGeary, R.P.; Ross, B.P. Metal chelation, radical scavenging and inhibition of Aβ42 fibrillation by food constituents in relation to Alzheimer’s disease. Food Chem. 2016, 199, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andjelkovic, M.; Vancamp, J.; Demeulenaer, B.; Depaemelaere, G.; Socaciu, C.; Verloo, M.; Verhe, R. Iron-chelation properties of phenolic acids bearing catechol and galloyl groups. Food Chem. 2006, 98, 23–31. [Google Scholar] [CrossRef]
- Yu, M.; Chen, X.; Liu, J.; Ma, Q.; Zhuo, Z.; Chen, H.; Zhou, L.; Yang, S.; Zheng, L.; Hou, S.T.; et al. Gallic acid disruption of Abeta1-42 aggregation rescues cognitive decline of APP/PS1 double transgenic mouse. Neurobiol. Dis. 2019, 124, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Paleologou, K.E.; Elv, G.; Khair, S.B.A.; Kazim, A.S.; Minhas, S.T.; Al-Tel, T.H.; Al-Hayani, A.A.; Haque, M.E.; Eeliezer, D.; et al. Structure activity relationship of phenolic acid inhibitors of α-synuclein fibril formation and toxicity. Front. Aging Neurosci. 2014, 6, 197. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Chang, J.-C.; Lin, W.-Y.; Li, C.-C.; Hsieh, M.; Chen, H.-W.; Wang, T.-S.; Wu, W.-T.; Liu, C.-S.; Liu, K.-L. Caffeic acid and resveratrol ameliorate cellular damage in cell and Drosophila models of spinocerebellar ataxia type 3 through upregulation of Nrf2 pathway. Free. Radic. Biol. Med. 2018, 115, 309–317. [Google Scholar] [CrossRef]
- Askar, M.H.; Hussein, A.M.; Al-Basiony, S.F.; Meseha, R.K.; Metias, E.F.; Salama, M.M.; Antar, A.; El-Sayed, A. Effects of Exercise and Ferulic Acid on Alpha Synuclein and Neuroprotective Heat Shock Protein 70 in An Experimental Model of Parkinsonism Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 156–169. [Google Scholar] [CrossRef]
- Chen, J.-L.; Duan, W.-J.; Luo, S.; Li, S.; Ma, X.-H.; Hou, B.-N.; Cheng, S.-Y.; Fang, S.-H.; Wang, Q.; Huang, S.-Q.; et al. Ferulic acid attenuates brain microvascular endothelial cells damage caused by oxygen-glucose deprivation via punctate-mitochondria-dependent mitophagy. Brain Res. 2017, 1666, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Osseni, R.A.; Debbasch, C.; Christen, M.O.; Rat, P.; Warnet, J.M. Tacrine-induced Reactive Oxygen Species in a Human Liver Cell Line: The Role of Anethole Dithiolethione as a Scavenger. Toxicol. Vitr. 1999, 13, 683–688. [Google Scholar] [CrossRef]
- Pi, R.; Mao, X.; Chao, X.; Cheng, Z.; Liu, M.; Duan, X.; Ye, M.; Chen, X.; Mei, Z.; Han, Y.; et al. Tacrine-6-ferulic acid, a novel multifunctional dimer, inhibits amyloid-beta-mediated Alzheimer’s disease-associated pathogenesis in vitro and in vivo. PLoS ONE 2012, 7, e31921. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yang, H.; Chen, Y.; Lin, H.; Li, Q.; Mo, J.; Bian, Y.; Pei, Y.; Sun, H. Synthesis, pharmacology and molecular docking on multifunctional tacrine-ferulic acid hybrids as cholinesterase inhibitors against Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2018, 33, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Miyazawa, M. Serotonin derivatives as inhibitors of beta-secretase (BACE 1). Pharmacie 2011, 66, 301–305. [Google Scholar]
- Dhiman, P.; Malik, N.; Khatkar, A. Hybrid caffeic acid derivatives as monoamine oxidases inhibitors: Synthesis, radical scavenging activity, molecular docking studies and in silico ADMET analysis. Chem. Central J. 2018, 12, 1–17. [Google Scholar] [CrossRef]
- Estrada, M.; Herrera-Arozamena, C.; Pérez, C.; Viña, D.; Romero, A.; Morales-García, J.A.; Pérez-Castillo, A.; Rodríguez-Franco, M.I. New cinnamic—N-benzylpiperidine and cinnamic—N,N-dibenzyl(N-methyl)amine hybrids as Alzheimer-directed multitarget drugs with antioxidant, cholinergic, neuroprotective and neurogenic properties. Eur. J. Med. Chem. 2016, 121, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benchekroun, M.; Pachón-Angona, I.; Luzet, V.; Martin, H.; Oset-Gasque, M.J.; Marco-Contelles, J.; Ismaili, L. Synthesis, antioxidant and Aβ anti-aggregation properties of new ferulic, caffeic and lipoic acid derivatives obtained by the Ugi four-component reaction. Bioorg. Chem. 2019, 85, 221–228. [Google Scholar] [CrossRef]
- Nesi, G.; Chen, Q.; Sestito, S.; Digiacomo, M.; Yang, X.; Wang, S.; Pi, R.; Rapposelli, S. Nature-based molecules combined with rivastigmine: A symbiotic approach for the synthesis of new agents against Alzheimer’s disease. Eur. J. Med. Chem. 2017, 141, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Simoni, E.; Serafini, M.M.; Bartolini, M.; Caporaso, R.; Pinto, A.; Necchi, D.; Fiori, J.; Andrisano, V.; Minarini, A.; Rosini, M.; et al. Nature-Inspired Multifunctional Ligands: Focusing on Amyloid-Based Molecular Mechanisms of Alzheimer’s Disease. ChemMedChem 2016, 11, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Wang, K.; Han, X.; Cao, M.; Tan, Z.; Liu, W. Design, Synthesis, and Evaluation of Novel Ferulic Acid Derivatives as Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 1008–1024. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Choubey, P.K.; Sharma, P.; Seth, A.; Saraf, P.; Shrivastava, S.K. Design, synthesis, and biological evaluation of ferulic acid based 1,3,4-oxadiazole hybrids as multifunctional therapeutics for the treatment of Alzheimer’s disease. Bioorg. Chem. 2020, 95, 103506. [Google Scholar] [CrossRef]
- Landete, J. Ellagitannins, ellagic acid and their derived metabolites: A review about source, metabolism, functions and health. Food Res. Int. 2011, 44, 1150–1160. [Google Scholar] [CrossRef]
- Llorach, R.; Cerdá, B.; Cerón, J.J.; Espín, J.C.; Tomás-Barberán, F.A. Evaluation of the bioavailability and metabolism in the rat of punicalagin, an antioxidant polyphenol from pomegranate juice. Eur. J. Nutr. 2003, 42, 18–28. [Google Scholar] [CrossRef]
- Jurgoński, A.; Juśkiewicz, J.; Fotschki, B.; Kołodziejczyk, K.; Milala, J.; Kosmala, M.; Grzelak-Błaszczyk, K.; Markiewicz, L. Metabolism of strawberry mono- and dimeric ellagitannins in rats fed a diet containing fructo-oligosaccharides. Eur. J. Nutr. 2017, 56, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Tomás-Barberán, F.A.; García-Villalba, R.; González-Sarrías, A.; Selma, M.V.; Espín, J.C. Ellagic Acid Metabolism by Human Gut Microbiota: Consistent Observation of Three Urolithin Phenotypes in Intervention Trials, Independent of Food Source, Age, and Health Status. J. Agric. Food Chem. 2014, 62, 6535–6538. [Google Scholar] [CrossRef]
- Cerdá, B.; Periago, P.M.; Espín, A.J.C.; Tomás-Barberán, F.A. Identification of Urolithin A as a Metabolite Produced by Human Colon Microflora from Ellagic Acid and Related Compounds. J. Agric. Food Chem. 2005, 53, 5571–5576. [Google Scholar] [CrossRef] [PubMed]
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem. Toxicol. 2017, 108, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Sasso, G.L.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Zhang, Y.; McKeever, R.; Henning, S.M.; Lee, R.-P.; Suchard, M.A.; Li, Z.; Chen, S.; Thames, G.; Zerlin, A.; et al. Pomegranate Juice and Extracts Provide Similar Levels of Plasma and Urinary Ellagitannin Metabolites in Human Subjects. J. Med. Food 2008, 11, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Bialonska, D.; Kasimsetty, S.G.; Khan, S.I.; Ferreira, D. Urolithins, Intestinal Microbial Metabolites of Pomegranate Ellagitannins, Exhibit Potent Antioxidant Activity in a Cell-Based Assay. J. Agric. Food Chem. 2009, 57, 10181–10186. [Google Scholar] [CrossRef]
- Cásedas, G.; Les, F.; Choya-Foces, C.; Hugo, M.; López, V. The Metabolite Urolithin-A Ameliorates Oxidative Stress in Neuro-2a Cells, Becoming a Potential Neuroprotective Agent. Antioxidants 2020, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Yeoh, B.S.; Singh, R.; Chandrasekar, B.; Vemula, P.K.; Haribabu, B.; Vijay-Kumar, M.; Jala, V.R. Gut Microbiota Conversion of Dietary Ellagic Acid into Bioactive Phytoceutical Urolithin a Inhibits Heme Perox-idases. PLoS ONE 2016, 11, e0156811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boakye, Y.D.; Groyer, L.; Heiss, E.H. An increased autophagic flux contributes to the anti-inflammatory potential of urolithin A in macrophages. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 61–70. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, F.; Guo, Z.; Zhao, J.; Song, X.; Yang, H. Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells. Mol. Carcinog. 2018, 57, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Velagapudi, R.; Lepiarz, I.; El-Bakoush, A.; Katola, F.O.; Bhatia, H.; Fiebich, B.L.; Olajide, O.A. Induction of Autophagy and Activation of SIRT-1 Deacetylation Mechanisms Mediate Neuroprotection by the Pomegranate Metabolite Urolithin A in BV2 Microglia and Differentiated 3D Human Neural Progenitor Cells. Mol. Nutr. Food Res. 2019, 63, e1801237. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, A.; Zheng, Y.; Wu, X.; Tang, W.; Liu, M.; Ma, S.; Jiang, L.; Hu, W.; Zhang, X.; Chen, Z. Urolithin A-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo. CNS Neurosci. Ther. 2019, 25, 976–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liu, Z.; Zhou, Y.; Hou, N.; Yan, W.; Qin, Y.; Ye, Q.; Cheng, X.-Y.; Xiao, Q.; Wu, X.; et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 2020, 153, 104655. [Google Scholar] [CrossRef]
- Gulcan, H.O.; Unlu, S.; Esiringu, I.; Ercetin, T.; Sahin, Y.; Oz, D.; Sahin, M.F. Design, synthesis and biological evaluation of novel 6H-benzo[c]chromen-6-one, and 7,8,9,10-tetrahydro-benzo[c]chromen-6-one derivatives as potential cholinesterase inhibitors. Bioorg. Med. Chem. 2014, 22, 5141–5154. [Google Scholar] [CrossRef]
- Norouzbahari, M.; Burgaz, E.V.; Erçetin, T.; Fallah, A.; Foroumadi, A.; Firoozpour, L.; Sahin, M.F.; Gazi, M.; Gulcan, H.O. Design, Synthesis and Characterization of Novel Urolithin Derivatives as Cholinesterase Inhibitor Agents. Lett. Drug Des. Discov. 2018, 15, 1131–1140. [Google Scholar] [CrossRef]
- Blanquet, P. Casein kinase 2 as a potentially important enzyme in the nervous system. Prog. Neurobiol. 2000, 60, 211–246. [Google Scholar] [CrossRef]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2010, 31, 924–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozza, G.; Gianoncelli, A.; Bonvini, P.; Zorzi, E.; Pasquale, R.; Rosolen, A.; Pinna, L.A.; Meggio, F.; Zagotto, G.; Moro, S. Urolithin as a converging scaffold linking ellagic acid and coumarin analogues: Design of potent protein kinase CK2 inhibitors. ChemMedChem 2011, 6, 2273–2286. [Google Scholar] [CrossRef]
- Xie, S.-S.; Lan, J.-S.; Wang, X.; Wang, Z.-M.; Jiang, N.; Li, F.; Wu, J.-J.; Wang, J.; Kong, L.-Y. Design, synthesis and biological evaluation of novel donepezil–coumarin hybrids as multi-target agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2016, 24, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Walgren, J.L.; Amani, Z.; McMillan, J.M.; Locher, M.; Buse, M.G. Effect of R(+)alpha-lipoic acid on pyruvate metabolism and fatty acid oxidation in rat hepatocytes. Metabolism 2004, 53, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-lipoic acid as a biological antioxidant. Free. Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Y.; Yu, W.; Jiang, S. Scavenging ability on ROS of alpha-lipoic acid (ALA). Food Chem. 2004, 84, 563–567. [Google Scholar] [CrossRef]
- Moraes, T.B.; Zanin, F.; da Rosa, A.; de Oliveira, A.; Coelho, J.; Petrillo, F.; Wajner, M.; Dutra-Filho, C.S. Lipoic acid prevents oxidative stress in vitro and in vivo by an acute hyperphenylalaninemia chemically-induced in rat brain. J. Neurol. Sci. 2010, 292, 89–95. [Google Scholar] [CrossRef]
- Suh, J.H.; Wang, H.; Liu, R.-M.; Liu, J.; Hagen, T.M. (R)-alpha-lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: Evidence for increased cysteine requirement for GSH synthesis. Arch. Biochem. Biophys. 2004, 423, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ersahin, M.; Toklu, H.Z.; Çetinel, Ş.; Yüksel, M.; Erzik, C.; Berkman, M.Z.; Yeğen, B.Ç.; Sener, G.; Yeǧen, B.Ç. Alpha Lipoic Acid Alleviates Oxidative Stress and Preserves Blood Brain Permeability in Rats with Subarachnoid Hemorrhage. Neurochem. Res. 2009, 35, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Camiolo, G.; Tibullo, D.; Giallongo, C.; Romano, A.; Parrinello, N.L.; Musumeci, G.; di Rosa, M.; Vicario, N.; Brundo, M.V.; Amenta, F.; et al. α-Lipoic Acid Reduces Iron-induced Toxicity and Oxidative Stress in a Model of Iron Overload. Int. J. Mol. Sci. 2019, 20, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, F.; Nomier, M.A.; Sabik, L.M.E.; Shaheen, M.A. Manganese-induced neurotoxicity and the potential protective effects of lipoic acid and Spirulina platensis. Toxicol. Mech. Methods 2020, 30, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Al-Otaibi, S.S.; Arafah, M.M.; Sharma, B.; Alhomida, A.S.; Siddiqi, N.J. Synergistic Effect of Quercetin and α-Lipoic Acid on Aluminium Chloride Induced Neurotoxicity in Rats. J. Toxicol. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, J.; Kabin, E.; Järving, I.; Bragina, O.; Tõugu, V.; Plitz, T.; Palumaa, P. Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. α-Lipoic acid as a potential anti-copper agent. Sci. Rep. 2018, 8, 1463. [Google Scholar] [CrossRef]
- Khalaf, A.A.; Zaki, A.R.; Galal, M.K.; Ogaly, H.A.; Ibrahim, M.A.; Hassan, A. The potential protective effect of α-lipoic acid against nanocopper particle–induced hepatotoxicity in male rats. Hum. Exp. Toxicol. 2016, 36, 881–891. [Google Scholar] [CrossRef]
- Bjørklund, G.; Crisponi, G.; Nurchi, V.M.; Cappai, R.; Djordjevic, A.B.; Aaseth, J. A Review on Coordination Properties of Thiol-Containing Chelating Agents Towards Mercury, Cadmium, and Lead. Molecules 2019, 24, 3247. [Google Scholar] [CrossRef] [Green Version]
- Androne, L.; Gavan, N.A.; Veresiu, I.A.; Orasan, R. In vivo effect of lipoic acid on lipid peroxidation in patients with diabetic neuropathy. Vivo 2000, 14, 327–330. [Google Scholar]
- Freitas, R. The evaluation of effects of lipoic acid on the lipid peroxidation, nitrite formation and antioxidant enzymes in the hippocampus of rats after pilocarpine-induced seizures. Neurosci. Lett. 2009, 455, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Haugaard, N.; Levin, R.M.; Surname, F. Regulation of the activity of choline acetyl transferase by lipoic acid. Mol. Cell. Biochem. 2000, 213, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Bussiere, J.R.; Hammond, R.S.; Montine, T.J.; Henson, E.; Jones, R.E.; Stackman, R.W. Chronic dietary α-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol. Aging 2007, 28, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—A 48 months follow-up analysis. J. Neural. Transm. Suppl. 2007, 72, 189–193. [Google Scholar]
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; de Bartolo, M.; Mauro, G.; Bosco, D. The Effect of Lipoic Acid Therapy on Cognitive Functioning in Patients with Alzheimer’s Disease. J. Neurodegener. Dis. 2013, 2013, 454253. [Google Scholar] [CrossRef]
- Rosini, M.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L.; Hrelia, P.; Minarini, A.; Tarozzi, A.; Melchiorre, C. Rational Approach to Discover Multipotent Anti-Alzheimer Drugs. J. Med. Chem. 2005, 48, 360–363. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Bartolini, M.; Tarozzi, A.; Matera, R.; Milelli, A.; Hrelia, P.; Andrisano, V.; Bolognesi, M.L.; Melchiorre, C. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 5435–5442. [Google Scholar] [CrossRef] [PubMed]
- Koufaki, M.; Kiziridi, C.; Nikoloudaki, F.; Alexis, M.N. Design and synthesis of 1,2-dithiolane derivatives and evaluation of their neuroprotective activity. Bioorg. Med. Chem. Lett. 2007, 17, 4223–4227. [Google Scholar] [CrossRef]
- Estrada, M.; Pérez, C.; Soriano, E.; Laurini, E.; Romano, M.; Pricl, S.; Morales-García, A.J.; Pérez-Castillo, A.; Rodríguez-Franco, M.I. New neurogenic lipoic-based hybrids as innovative Alzheimer’s drugs with sigma-1 agonism and beta-secretase inhibition. Future Med. Chem. 2016, 8, 1191–1207. [Google Scholar] [CrossRef]
- Tu, Y.-L.; Chen, Q.-H.; Wang, S.-N.; Uri, A.; Yang, X.-H.; Chu, J.-Q.; Chen, J.-K.; Luo, B.-L.; Chen, X.-H.; Wen, S.-J.; et al. Discovery of lipoic acid-4-phenyl-1H-pyrazole hybrids as novel bifunctional ROCK inhibitors with antioxidant activity. RSC Adv. 2016, 6, 58516–58520. [Google Scholar] [CrossRef]
- Jones, M.; Wang, J.; Harmon, S.; Kling, B.; Heilmann, J.; Gilmer, J.F. Novel Selective Butyrylcholinesterase Inhibitors Incorporating Antioxidant Functionalities as Potential Bimodal Therapeutics for Alzheimer’s Disease. Molecules 2016, 21, 440. [Google Scholar] [CrossRef] [Green Version]
- Jalili-Baleh, L.; Forootanfar, H.; Küçükkılınç, T.T.; Nadri, H.; Abdolahi, Z.; Ameri, A.; Jafari, M.; Ayazgok, B.; Baeeri, M.; Foroumadi, A.; et al. Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer’s disease based on coumarin and lipoic acid scaffolds. Eur. J. Med. Chem. 2018, 152, 600–614. [Google Scholar] [CrossRef]
- Pagoni, A.; Marinelli, L.; di Stefano, A.; Ciulla, M.; Turkez, H.; Mardinoglu, A.; Vassiliou, S.; Cacciatore, I. Novel anti-Alzheimer phenol-lipoyl hybrids: Synthesis, physico-chemical characterization, and biological evaluation. Eur. J. Med. Chem. 2020, 186, 111880. [Google Scholar] [CrossRef] [PubMed]
- Michalska, P.; Tenti, G.; Satriani, M.; Cores, A.; Ramos, M.T.; García, A.G.; Menéndez, J.C.; León, R. Aza-CGP37157-lipoic hybrids designed as novel Nrf2-inducers and antioxidants exert neuroprotection against oxidative stress and show neuroinflammation inhibitory properties. Drug Dev. Res. 2019, 81, 283–294. [Google Scholar] [CrossRef]
- Pachón-Angona, I.; Martin, H.; Chhor, S.; Oset-Gasque, M.J.; Refouvelet, B.; Marco-Contelles, J.; Ismaili, L. Synthesis of new ferulic/lipoic/comenic acid-melatonin hybrids as antioxidants and Nrf2 activators via Ugi reaction. Futur. Med. Chem. 2019, 11, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, L.; Liao, R.; Li, Q.; Pi, R.; Yang, X. N2L, a novel lipoic acid-niacin dimer protects HT22 cells against beta-amyloid peptide-induced damage through attenuating apoptosis. Metab. Brain Dis. 2019, 34, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Uppakara, K.; Jamornwan, S.; Duan, L.-X.; Yue, K.-R.; Sunrat, C.; Dent, E.W.; Wan, S.-B.; Saengsawang, W. Novel α-Lipoic Acid/3-n-Butylphthalide Conjugate Enhances Protective Effects against Oxidative Stress and 6-OHDA Induced Neuronal Damage. ACS Chem. Neurosci. 2020, 11, 1634–1642. [Google Scholar] [CrossRef]
- Syed, Y.Y. Correction to: Sodium Oligomannate: First Approval. Drugs 2020, 80, 445–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Zahrani, N.A.; El-Shishtawy, R.M.; Asiri, A.M. Recent developments of gallic acid derivatives and their hybrids in medicinal chemistry: A review. Eur. J. Med. Chem. 2020, 204, 112609. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Role of ROS | Ref. | Pathological Role of ROS | Ref. |
|---|---|---|---|
| Signalling between mitochondria and surrounding cells | [20] | mDNA damage, deletion, and mutation | [21] |
| Regulation of cellular proliferation, differentiation and apoptosis | [18,19] | Mitochondrial membrane permeability alteration and mitochondrial failure | [22,25,27,31] |
| Induction of MAPKs activation in cardiovascular system. | [20] | Lipid peroxidation | [27] |
| Influence on pro-survival transcription factors (i.g. Nrf2 and NF-κB). | [21] | ETC enzymes malfunctions | [29] |
| Adaption and regulation of hypoxia | [20] | Promotion of inflammation | [16] |
| Regulation of immune functions | [19,20] | Metallostasis and metal accumulation | [40,42] |
| Induction of autophagy | [20] | Proteostasis and misfolded proteins clearance impairment | [51,53] |
| Entry | Scaffolds Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 1 | 1-aminoindan beared with polyamine scaffold |  | Neuroprotection against NMDA toxicity and ischemia damages No neurotoxicity | [85] |
| 2 | 1,4-benzoquinone and polyamine structure of caproctamine |  | ↓Aβ aggregation ↓tau phosphorylation ↑antioxidant activity ↓AChE ↓BACE-1 | [87] |
| 3 | Ferulic acid-memoquin hybrids |  | ↓AChE ↓BuChE ↓self-induced Aβ1-42 aggregation no cytotoxicity in SH-SY5Y cells good BBB predicted permeability | [88] |
| 4 | Genistein with polyamines |  | ↓AChE ↓BuChE Fe3+/Cu2+/Zn2+ chelation no HepG-2 cell cytotoxicity | [89] |
| 5 | 3,5-dibenzylidenepiperidin-4- one functionalized with spermine |  | ↓Aβ42 aggregation no antioxidant properties in T67 cells neuroprotection and no cytoxicity in vitro | [90] |
| 6 | Dicaffeoylsper-midine cyclized derivatives |  | Antioxidant activity ↑memory and learning in fruit flies model | [92] |
| Entry | Scaffolds Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 7 | Tacrine linked with ferulic acid |  | ↓ Aβ-aggregation ↓ ROS production ↓ AChE ↑ cognitive functions ↑ SOD/ChAT | [110] |
| 8 | Tacrine and functionalized ferulic acid |  | ↓ Aβ-self aggregation ↓ AChE ↓ BuChE ↑ memory no hepatotoxicity | [111] |
| 9 | Ferulic and caffeic merged with serotonin |  | ↑ antioxidant activity ↓ BACE-1 | [112] |
| 10 | Aromatic amides and esters of caffeic acid |  | ↓MAO-A/MAO-B ↑ antioxidant activity | [113] |
| 11 | Hydroxycinnamic acids and NBP (donepezil) |  | ↓MAO-A/MAO-B ↓AChE ↓BuChE ↑ antioxidant activity | [114] |
| 12 | Hydroxycinnamic scaffolds and DBMA (AP2238) |  | ↓MAO-A/MAO-B ↓AChE ↓BuChE ↑ antioxidant activity | [114] |
| 13 | Caffeic acid with hydrophobic moieties |  | ↓Aβ1-40 self-aggregation ↑ antioxidant activity neuroprotection in SH-SY5Y cells | [115] |
| 14 | Rivastigmine with GA |  | ↑ antioxidant activity Cu2+ chelating properties ↓ChEs ↓Aβ self-aggregation neuroprotective effects in vitro no cytotoxicity | [116] |
| 15 | Caffeic acid and diallyl sulfide |  | ↓Aβ42 self-aggregation ↑cytoprotection against H2O2-induced damages ↓p53 alteration induced by Aβ | [117] |
| 16 | Ferulic core merged with 1,2,3,4-tetrahydroisoquinoline and (benzyl(ethyl)amino)butoxy scaffold |  | ↑antioxidant activity ↓AChE ↓BuChE ↓ MAO-A/MAO-B ↓ Aβ self-aggregation ↑ self-induced Aβ1-42 fibrils disaggregation ↑neuroprotective effect in SH-5YSY cells ↑autophagy in U87 cells ↑motility in Zebrafish model ↓ Aβ1-40-induced vascular injury in Zebrafish model ↑In vivo cognitive functions | [118] |
| 17 | Ferulic acid merged with 1,3,4-oxadiazole scaffold |  | ↓ChEs ↓BACE-1 ↓ Aβ self-aggregation ↓ Aβ AChE-induced aggregation neuroprotective effects in vitro ↑In vivo cognitive functions | [119] |
| Entry | Scaffolds Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 18 | Urolithin scaffold with rivastigmine portion |  | ↓AChE ↓BuChE | [136] |
| 19 | Urolithin scaffold with donepezil-like moieties |  | ↓AChE ↓BuChE | [136] |
| 20 | Donepezil-like urolithin and tetrahydrourolithin derivatives |  | ↓AChE ↓BuChE ↓ AChE induced Aβ aggregation | [137] |
| 21 | Nitro- and bromo-derivatives of urolithins |  | CK2 inhibition Selectivity in other kinases panel | [140] |
| 22 | Tetrahydrourolithin scaffold linked with donepezil moiety |  | AChE/BuChE inhibition MAO-B inhibition BBB permeability no cytotoxicity in brain and liver cells | [141] |
| Entry | Scaffold Combination | Structure | Effects | Ref. |
|---|---|---|---|---|
| 23 | Lipoic acid and tacrine |  | ↑ROS protection ↓AChE ↓BuChE ↓ AChE-induced Aβ aggregation | [160] |
| 24 | Dopamine and LA linked by tetrazole ring |  | ↑antioxidant activity neuroprotection in vitro | [162] |
| 25 | LA-NBP and LA-DBMA conjugation |  | ↓AChE ↓BuChE ↓BACE-1 ↑antioxidant activity σ1R agonism good BBB permeability prediction neuroprotection in vitro | [163] |
| 26 | LA-4-Phenyl-1H-pyrazole derivatives |  | ROCK1/ROCK2 inhibition ↓ROS ↑GSH vasorelaxant activity | [164] |
| 27 | Lipoic isosorbide-2-benzylcarbamate |  | ↓ROS ↓BuChE ↓cytotoxicity in treated HT-22 cells | [165] |
| 28 | LA and coumarin scaffold linked bridged with triazole |  | ↓AChE ↓BuChE ↓ Aβ peptide aggregation ↓intracellular ROS neuroprotection against H2O2− or Aβ1-42-induced cytotoxicity in SH-SY5Y cell lines Selective Cu/Fe chelation | [166] |
| 29 | FA/CA-LA hybrids |  | ↓Aβ1-42-induced neurotoxicity in SH-SY5Y cells ↑protection in H2O2-insulted cells no cytotoxicity | [167] |
| 30 | Lipoic-functionalized benzodiazepine |  | ↑ROS scavenging ↑Nrf2-ARE pathway ↑HO-1/GCLc neuroprotection in in vitro model no cytotoxicity no hepatotoxicity | [168] |
| 31 | Lipoic-melatonin hybrids |  | ↑ROS scavenging ↑Nrf2-ARE pathway antioxidant activity and neuroprotection in vitro no cytotoxicity | [169] |
| 32 | LA-niacin hybrids |  | ↓Aβ1-42-induced cytotoxicity in HT22 cells ↓mitochondrial dysfunctions ↓intracellular ROS ↑SOD, CAT, GPx ↓apoptosis in Aβ1-42treated cells | [170] |
| 33 | LA-3-n-butylphthalide amide |  | ↓intracellular ROS ↑direct ROS-scavenger ↓ H2O2-induced cell death ↑GSH ↓ H2O2-induced damage in cortical neurons ↓6-OHDA-induced neuronal damage in SH-5YSY cells | [171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacci, A.; Runfola, M.; Sestito, S.; Rapposelli, S. Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases. Antioxidants 2021, 10, 367. https://doi.org/10.3390/antiox10030367
Bacci A, Runfola M, Sestito S, Rapposelli S. Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases. Antioxidants. 2021; 10(3):367. https://doi.org/10.3390/antiox10030367
Chicago/Turabian StyleBacci, Andrea, Massimiliano Runfola, Simona Sestito, and Simona Rapposelli. 2021. "Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases" Antioxidants 10, no. 3: 367. https://doi.org/10.3390/antiox10030367
APA StyleBacci, A., Runfola, M., Sestito, S., & Rapposelli, S. (2021). Beyond Antioxidant Effects: Nature-Based Templates Unveil New Strategies for Neurodegenerative Diseases. Antioxidants, 10(3), 367. https://doi.org/10.3390/antiox10030367