
Metabolic Targets of Coenzyme Q10 in Mitochondria





Abstract
:1. Introduction
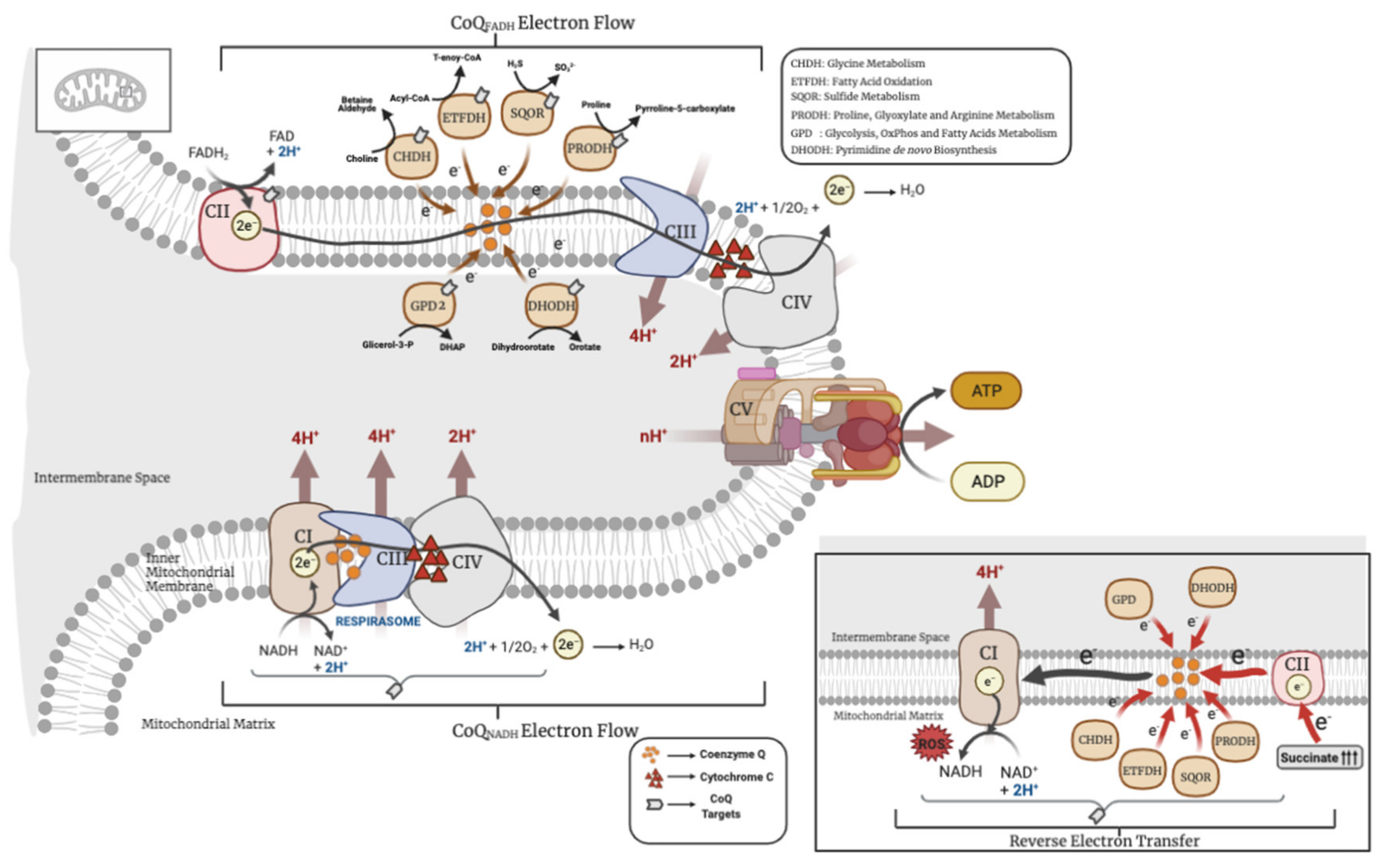
2. CoQ in the OxPhos System
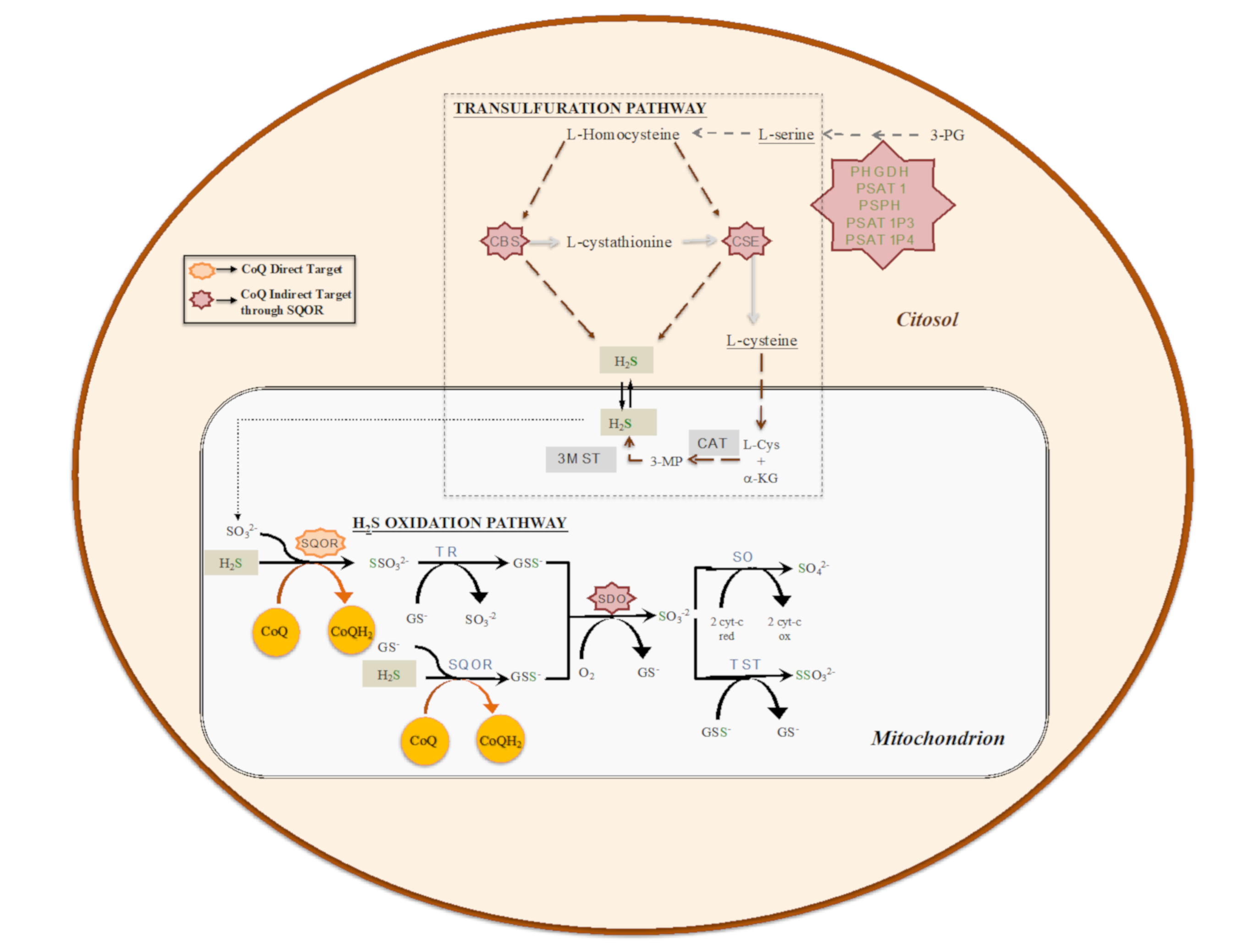
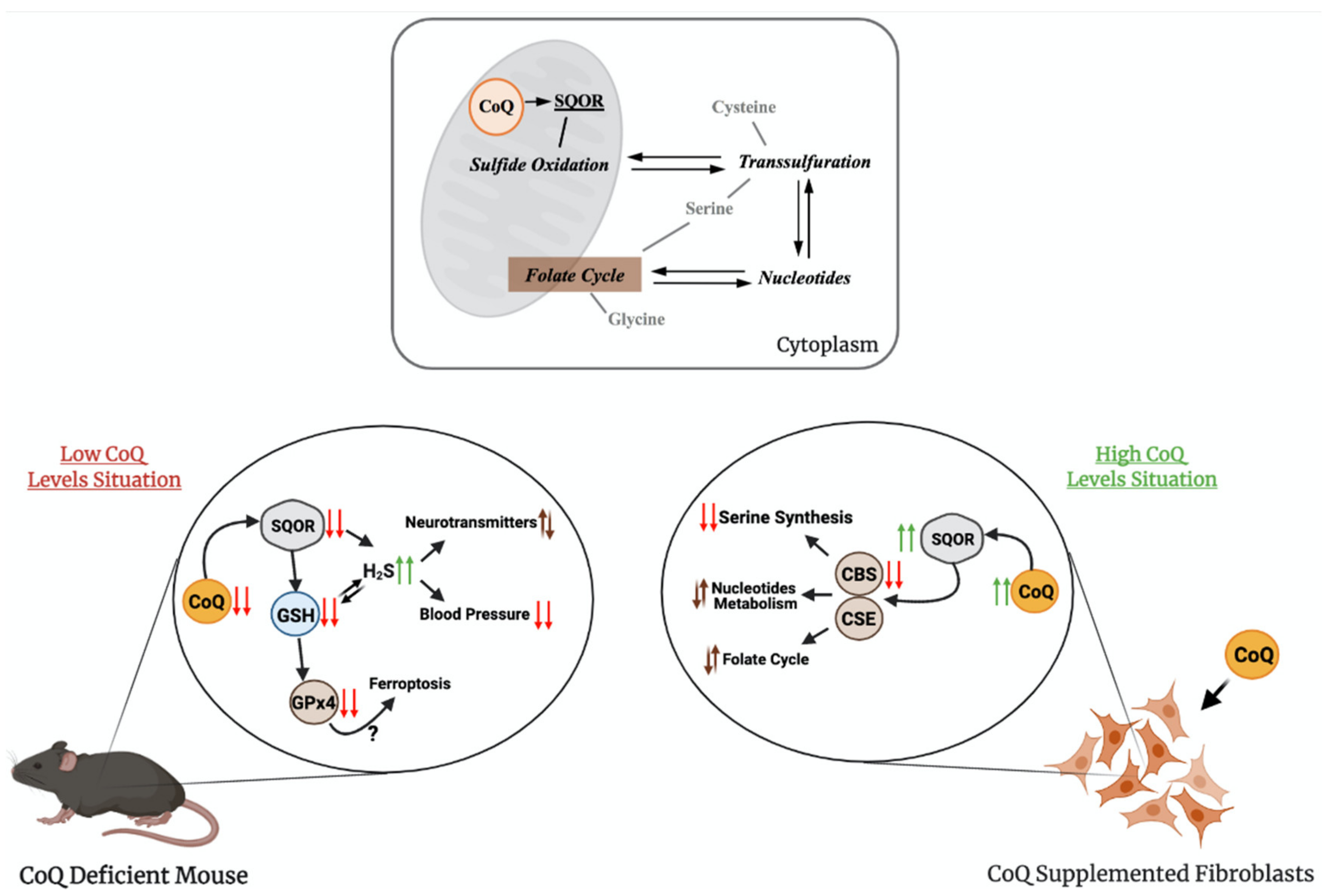
3. The Role of CoQ in the Regulation of Sulfide Metabolism and Others Linked Pathways
4. Other CoQ-Linked Reactions in Mitochondria
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of interest
References
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Garcia, J.A.; Guaras, A.; Acin-Perez, R.; Bullejos-Peregrin, J.; Lopez, A.; Escames, G.; Enriquez, J.A.; et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sanchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E4697–E4705. [Google Scholar] [CrossRef] [Green Version]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.P.; Medana, C.; Stainier, D.Y.; et al. Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 2015, 38, 145–156. [Google Scholar] [CrossRef]
- Kuhl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 2017, 6, e30952. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 2018, 7. [Google Scholar] [CrossRef]
- Awad, A.M.; Bradley, M.C.; Fernandez-Del-Rio, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hekimi, S. The Complexity of Making Ubiquinone. Trends Endocrinol. Metab. 2019, 30, 929–943. [Google Scholar] [CrossRef]
- Diaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; Gonzalez-Garcia, P.; Battino, M.; Lopez, L.C.; Varela-Lopez, A. The Paradox of Coenzyme Q10 in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Lopez, L.C.; Von-Moltke, J.; Naini, A.; Krishna, S.; Schuelke, M.; Salviati, L.; Navas, P.; DiMauro, S.; Hirano, M. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB J. 2008, 22, 1874–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Lopez, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010, 24, 3733–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Garone, C.; Emmanuele, V.; Tadesse, S.; Krishna, S.; Dorado, B.; Hirano, M. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 2013, 27, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Duberley, K.E.; Abramov, A.Y.; Chalasani, A.; Heales, S.J.; Rahman, S.; Hargreaves, I.P. Human neuronal coenzyme Q(10) deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2013, 36, 63–73. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef]
- Navas, P.; Villalba, J.M.; Lenaz, G. Coenzyme Q-dependent functions of plasma membrane in the aging process. Age 2005, 27, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Barros, M.H.; Johnson, A.; Gin, P.; Marbois, B.N.; Clarke, C.F.; Tzagoloff, A. The Saccharomyces cerevisiae COQ10 gene encodes a START domain protein required for function of coenzyme Q in respiration. J. Biol. Chem. 2005, 280, 42627–42635. [Google Scholar] [CrossRef] [Green Version]
- Kemmerer, Z.A.; Robinson, K.P.; Schmitz, J.M.; Paulson, B.R.; Jochem, A.; Hutchins, P.D.; Coon, J.J.; Pagliarini, D.J. UbiB proteins regulate cellular CoQ distribution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Alcazar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Schagger, H. Blue-native gels to isolate protein complexes from mitochondria. Methods Cell Biol. 2001, 65, 231–244. [Google Scholar]
- Sousa, P.M.; Silva, S.T.; Hood, B.L.; Charro, N.; Carita, J.N.; Vaz, F.; Penque, D.; Conrads, T.P.; Melo, A.M. Supramolecular organizations in the aerobic respiratory chain of Escherichia coli. Biochimie 2011, 93, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Genova, M.L. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: Random collisions vs. solid state electron channeling. Am. J. Physiol. Cell Physiol. 2007, 292, C1221–C1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Guaras, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro-Lopez, M.; Pujol, C.; Martinez-Carrascoso, I.; Nunez, E.; Garcia-Marques, F.; Rodriguez-Hernandez, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szibor, M.; Gainutdinov, T.; Fernandez-Vizarra, E.; Dufour, E.; Gizatullina, Z.; Debska-Vielhaber, G.; Heidler, J.; Wittig, I.; Viscomi, C.; Gellerich, F.; et al. Bioenergetic consequences from xenotopic expression of a tunicate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148137. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.A.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e413. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Milliken, A.S.; Kulkarni, C.A.; Brookes, P.S. Acid enhancement of ROS generation by complex-I reverse electron transport is balanced by acid inhibition of complex-II: Relevance for tissue reperfusion injury. Redox Biol. 2020, 37, 101733. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Martin, J.M.; Salviati, L.; Trevisson, E.; Montini, G.; DiMauro, S.; Quinzii, C.; Hirano, M.; Rodriguez-Hernandez, A.; Cordero, M.D.; Sanchez-Alcazar, J.A.; et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007, 16, 1091–1097. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; Dimauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; de Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef] [Green Version]
- Levavasseur, F.; Miyadera, H.; Sirois, J.; Tremblay, M.L.; Kita, K.; Shoubridge, E.; Hekimi, S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J. Biol. Chem. 2001, 276, 46160–46164. [Google Scholar] [CrossRef] [Green Version]
- Nakai, D.; Yuasa, S.; Takahashi, M.; Shimizu, T.; Asaumi, S.; Isono, K.; Takao, T.; Suzuki, Y.; Kuroyanagi, H.; Hirokawa, K.; et al. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem. Biophys. Res. Commun. 2001, 289, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, P.; Grunler, J.; Mattsson, J.; Sindelar, P.J.; Nordlund, P.; Berthold, D.A. A new member of the family of di-iron carboxylate proteins. Coq7 (clk-1), a membrane-bound hydroxylase involved in ubiquinone biosynthesis. J. Biol. Chem. 2001, 276, 33297–33300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barriocanal-Casado, E.; Cueto-Urena, C.; Benabdellah, K.; Gutierrez-Guerrero, A.; Cobo, M.; Hidalgo-Gutierrez, A.; Rodriguez-Sevilla, J.J.; Martin, F.; Lopez, L.C. Gene Therapy Corrects Mitochondrial Dysfunction in Hematopoietic Progenitor Cells and Fibroblasts from Coq9R239X Mice. PLoS ONE 2016, 11, e0158344. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Vasta, V.; Hahn, S.; Gangoiti, J.A.; Opheim, E.; Sedensky, M.M.; Morgan, P.G. The role of DMQ(9) in the long-lived mutant clk-1. Mech. Ageing Dev. 2011, 132, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Hidalgo-Gutierrez, A.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef]
- Lopez, L.C.; Quinzii, C.M.; Area, E.; Naini, A.; Rahman, S.; Schuelke, M.; Salviati, L.; DiMauro, S.; Hirano, M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: Time- and compound-dependent effects. PLoS ONE 2010, 5, e11897. [Google Scholar] [CrossRef] [Green Version]
- Saiki, R.; Lunceford, A.L.; Shi, Y.; Marbois, B.; King, R.; Pachuski, J.; Kawamukai, M.; Gasser, D.L.; Clarke, C.F. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Renal Physiol. 2008, 295, F1535–F1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuna-Castroviejo, D.; Lopez, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta 2014, 1842, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Gutierrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Diaz-Casado, M.E.; Sanchez-Maldonado, L.; Romero, M.; Sayed, R.K.; Prehn, C.; Escames, G.; Duarte, J.; et al. beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Luna-Sanchez, M.; Diaz-Casado, E.; Barca, E.; Tejada, M.A.; Montilla-Garcia, A.; Cobos, E.J.; Escames, G.; Acuna-Castroviejo, D.; Quinzii, C.M.; Lopez, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef]
- Di Meo, I.; Lamperti, C.; Tiranti, V. Mitochondrial diseases caused by toxic compound accumulation: From etiopathology to therapeutic approaches. EMBO Mol. Med. 2015, 7, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Banerjee, R. Sulfur as a signaling nutrient through hydrogen sulfide. Annu. Rev. Nutr. 2014, 34, 171–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modis, K.; Coletta, C.; Erdelyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [Green Version]
- Kabil, H.; Kabil, O.; Banerjee, R.; Harshman, L.G.; Pletcher, S.D. Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc. Natl. Acad. Sci. USA 2011, 108, 16831–16836. [Google Scholar] [CrossRef] [Green Version]
- Hine, C.; Mitchell, J.R. Calorie restriction and methionine restriction in control of endogenous hydrogen sulfide production by the transsulfuration pathway. Exp. Gerontol. 2015, 68, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.J.; Duan, C. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014, 6, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Trevino-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Garcia, P.; Hidalgo-Gutierrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sanchez-Hernandez, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.W.; Elias, A.F.; Kuster, A.; Laugwitz, L.; Larson, A.A.; Landry, A.P.; Ellwood-Digel, L.; Mirsky, D.M.; Dimmock, D.; Haven, J.; et al. Pathogenic variants in SQOR encoding sulfide:quinone oxidoreductase are a potentially treatable cause of Leigh disease. J. Inherit. Metab. Dis. 2020, 43, 1024–1036. [Google Scholar] [CrossRef] [Green Version]
- Mottawea, W.; Chiang, C.K.; Muhlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef]
- Phillips, C.M.; Zatarain, J.R.; Nicholls, M.E.; Porter, C.; Widen, S.G.; Thanki, K.; Johnson, P.; Jawad, M.U.; Moyer, M.P.; Randall, J.W.; et al. Upregulation of Cystathionine-beta-Synthase in Colonic Epithelia Reprograms Metabolism and Promotes Carcinogenesis. Cancer Res. 2017, 77, 5741–5754. [Google Scholar] [CrossRef] [Green Version]
- Nikkanen, J.; Forsstrom, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamaki, J.; Roivainen, A.; Marjamaki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Forsstrom, S.; Jackson, C.B.; Carroll, C.J.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.M.; Khan, N.A.; et al. Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 2019, 30, 1040–1054.e1047. [Google Scholar] [CrossRef]
- Krug, A.K.; Gutbier, S.; Zhao, L.; Poltl, D.; Kullmann, C.; Ivanova, V.; Forster, S.; Jagtap, S.; Meiser, J.; Leparc, G.; et al. Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP+. Cell Death Dis. 2014, 5, e1222. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkila, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjosund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.R.; Ong, S.E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016, 5. [Google Scholar] [CrossRef]
- Ziosi, M.; Di Meo, I.; Kleiner, G.; Gao, X.H.; Barca, E.; Sanchez-Quintero, M.J.; Tadesse, S.; Jiang, H.; Qiao, C.; Rodenburg, R.J.; et al. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med. 2017, 9, 96–111. [Google Scholar] [CrossRef]
- Luna-Sanchez, M.; Hidalgo-Gutierrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, A.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2017, 9, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, F.L. Metabolic supplementation with orotic acid and magnesium orotate. Cardiovasc. Drugs Ther. 1998, 12 (Suppl. s2), 147–152. [Google Scholar] [CrossRef]
- Montero, R.; Sanchez-Alcazar, J.A.; Briones, P.; Navarro-Sastre, A.; Gallardo, E.; Bornstein, B.; Herrero-Martin, D.; Rivera, H.; Martin, M.A.; Marti, R.; et al. Coenzyme Q10 deficiency associated with a mitochondrial DNA depletion syndrome: A case report. Clin. Biochem. 2009, 42, 742–745. [Google Scholar] [CrossRef]
- Montero, R.; Grazina, M.; Lopez-Gallardo, E.; Montoya, J.; Briones, P.; Navarro-Sastre, A.; Land, J.M.; Hargreaves, I.P.; Artuch, R.; Coenzyme, Q.D.S.G. Coenzyme Q10 deficiency in mitochondrial DNA depletion syndromes. Mitochondrion 2013, 13, 337–341. [Google Scholar] [CrossRef]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019, 29, 399–416.e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta 2010, 1797, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Palmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.C.; Ohkuma, A.; Hayashi, Y.K.; Lopez, L.C.; Hirano, M.; Nonaka, I.; Noguchi, S.; Chen, L.H.; Jong, Y.J.; Nishino, I. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul. Disord. 2009, 19, 212–216. [Google Scholar] [CrossRef]
- Summitt, C.B.; Johnson, L.C.; Jonsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015, 466, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Moxley, M.A.; Tanner, J.J.; Becker, D.F. Steady-state kinetic mechanism of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch. Biochem. Biophys. 2011, 516, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Rauchova, H.; Battino, M.; Fato, R.; Lenaz, G.; Drahota, Z. Coenzyme Q-pool function in glycerol-3-phosphate oxidation in hamster brown adipose tissue mitochondria. J. Bioenerg. Biomembr. 1992, 24, 235–241. [Google Scholar] [CrossRef]
- Barrett, M.C.; Dawson, A.P. The reaction of choline dehydrogenase with some electron acceptors. Biochem. J. 1975, 151, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Echtay, K.S.; Winkler, E.; Frischmuth, K.; Klingenberg, M. Uncoupling proteins 2 and 3 are highly active H+ transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc. Natl. Acad. Sci. USA 2001, 98, 1416–1421. [Google Scholar] [CrossRef] [Green Version]
- Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef]
- Jaburek, M.; Garlid, K.D. Reconstitution of recombinant uncoupling proteins: UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 2003, 278, 25825–25831. [Google Scholar] [CrossRef] [Green Version]
- Esteves, T.C.; Echtay, K.S.; Jonassen, T.; Clarke, C.F.; Brand, M.D. Ubiquinone is not required for proton conductance by uncoupling protein 1 in yeast mitochondria. Biochem. J. 2004, 379, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Sluse, F.E.; Jarmuszkiewicz, W.; Navet, R.; Douette, P.; Mathy, G.; Sluse-Goffart, C.M. Mitochondrial UCPs: New insights into regulation and impact. Biochim. Biophys. Acta 2006, 1757, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Miyoshi, H.; Leverve, X.; Bernard, P.; Fontaine, E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic. Res. 2002, 36, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E.; Bernardi, P. Progress on the mitochondrial permeability transition pore: Regulation by complex I and ubiquinone analogs. J. Bioenerg. Biomembr. 1999, 31, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E.; Ichas, F.; Bernardi, P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Biol. Chem. 1998, 273, 25734–25740. [Google Scholar] [CrossRef] [Green Version]
- Belliere, J.; Devun, F.; Cottet-Rousselle, C.; Batandier, C.; Leverve, X.; Fontaine, E. Prerequisites for ubiquinone analogs to prevent mitochondrial permeability transition-induced cell death. J. Bioenerg. Biomembr. 2012, 44, 207–212. [Google Scholar] [CrossRef]
- Devun, F.; Walter, L.; Belliere, J.; Cottet-Rousselle, C.; Leverve, X.; Fontaine, E. Ubiquinone analogs: A mitochondrial permeability transition pore-dependent pathway to selective cell death. PLoS ONE 2010, 5, e11792. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tissue | ||||
|---|---|---|---|---|
| Protein | Kidneys | Heart | Cerebellum | |
| SQOR | −1.2755 | −1.1292 | ||
| GPD2 | −0.0407 | 0.2659 | 0.0454 | |
| ETFDH | 0.1131 | −0.1072 | 0.0665 | |
| CHDH | 0.1572 | |||
| DHODH | 0.2237 | |||
| PRODH | 0.4076 | 0.1372 | 0.5226 | |
| PRODH2 | 0.6400 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hidalgo-Gutiérrez, A.; González-García, P.; Díaz-Casado, M.E.; Barriocanal-Casado, E.; López-Herrador, S.; Quinzii, C.M.; López, L.C. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants 2021, 10, 520. https://doi.org/10.3390/antiox10040520
Hidalgo-Gutiérrez A, González-García P, Díaz-Casado ME, Barriocanal-Casado E, López-Herrador S, Quinzii CM, López LC. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants. 2021; 10(4):520. https://doi.org/10.3390/antiox10040520
Chicago/Turabian StyleHidalgo-Gutiérrez, Agustín, Pilar González-García, María Elena Díaz-Casado, Eliana Barriocanal-Casado, Sergio López-Herrador, Catarina M. Quinzii, and Luis C. López. 2021. "Metabolic Targets of Coenzyme Q10 in Mitochondria" Antioxidants 10, no. 4: 520. https://doi.org/10.3390/antiox10040520
APA StyleHidalgo-Gutiérrez, A., González-García, P., Díaz-Casado, M. E., Barriocanal-Casado, E., López-Herrador, S., Quinzii, C. M., & López, L. C. (2021). Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants, 10(4), 520. https://doi.org/10.3390/antiox10040520