
Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System

 , , ,
, , , 

Abstract
:1. Introduction
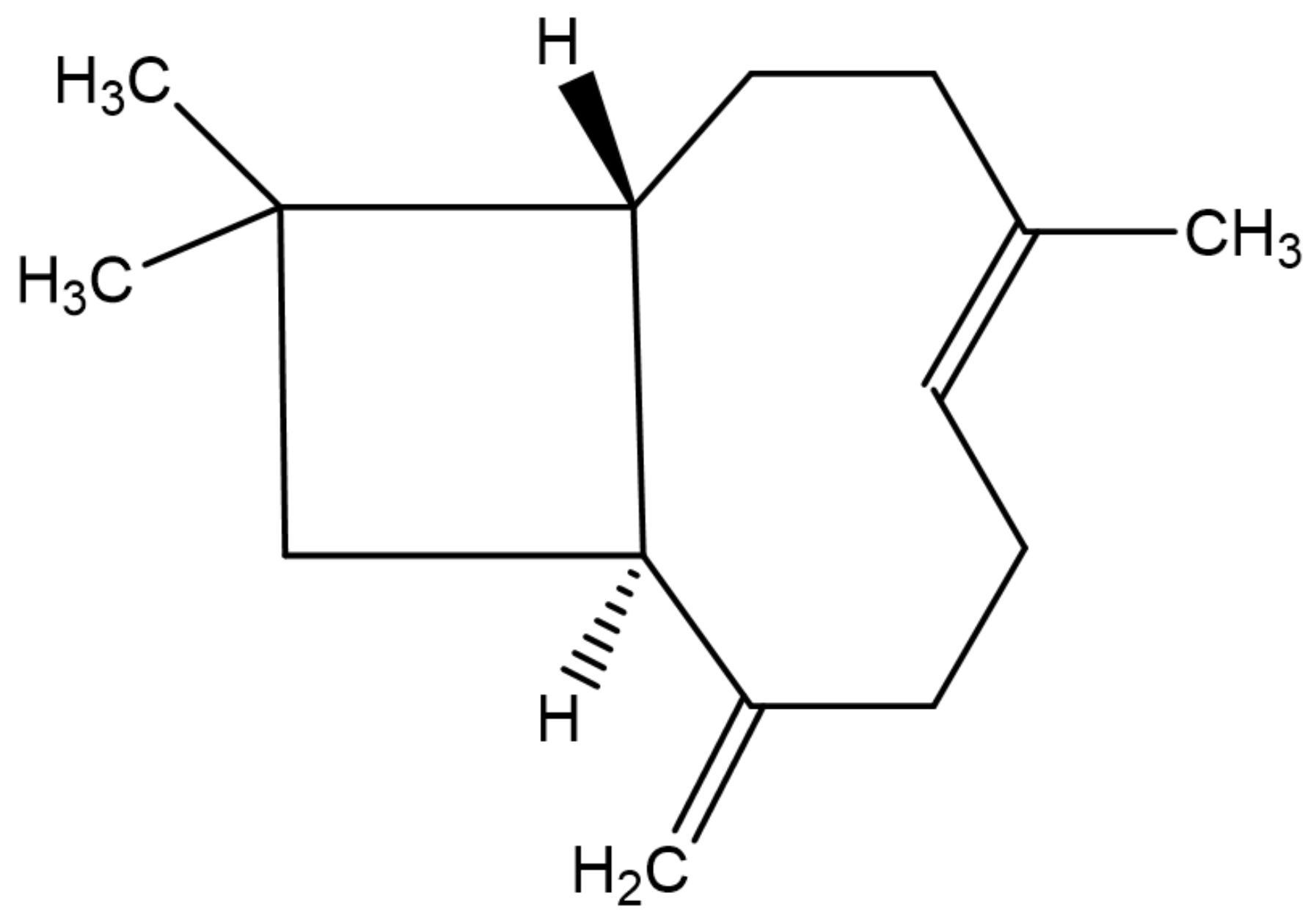
2. Chemistry and Vegetable Sources of β-Caryophyllene
3. Biological Activities of β-Caryophyllene
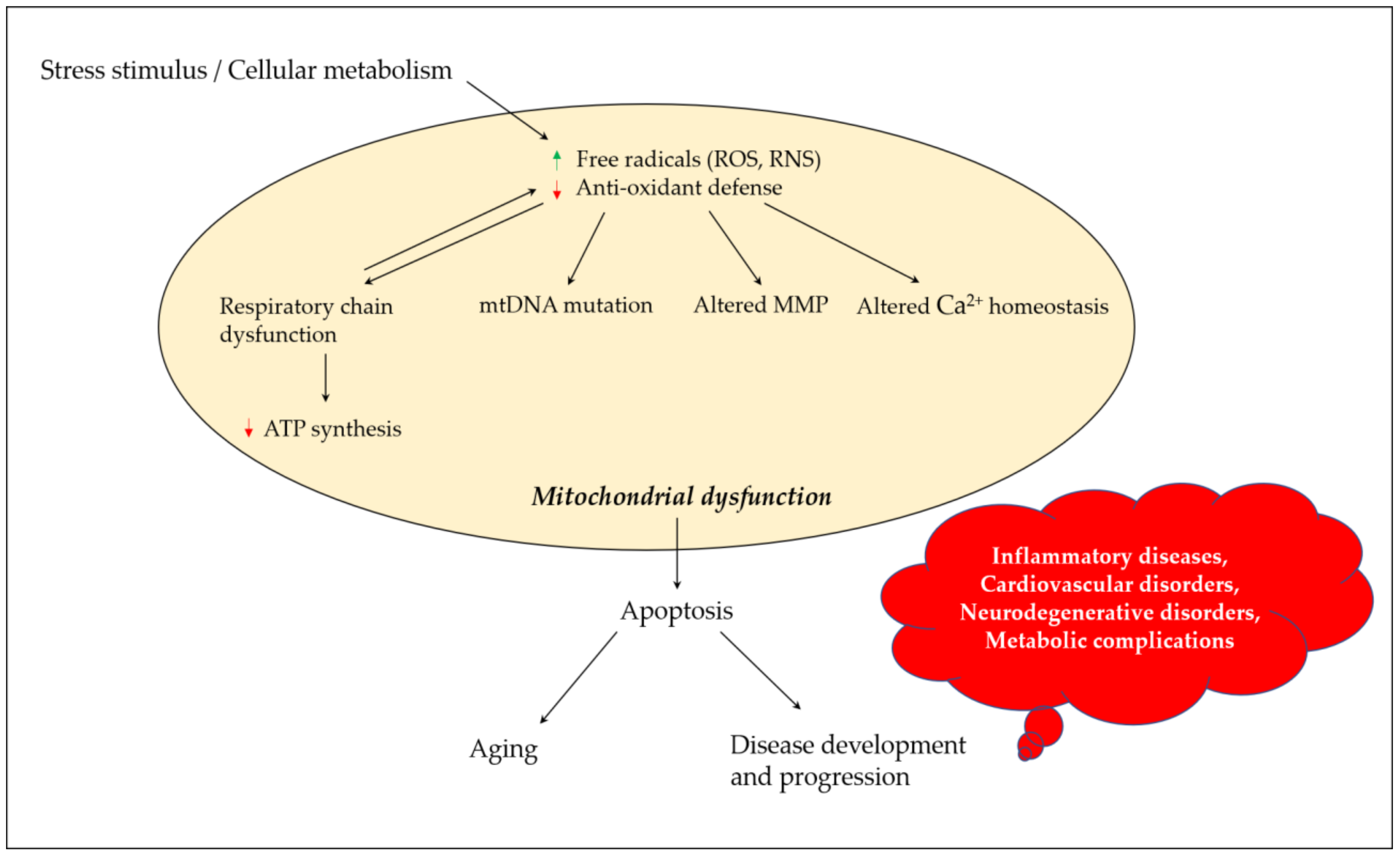
4. Mitochondrial Dysfunction and Neurodegeneration
4.1. Alzheimer’s Disease (AD)
4.2. Parkinson’s Disease (PD)
4.3. Multiple Sclerosis (MS)
4.4. Amyotrophic Lateral Sclerosis (ALS)
4.5. Huntington’s Disease (HD)
4.6. Other Neurological Disorders
5. Mitochondria-Based Therapies in Neurodegenerative Disorders
6. Neuroprotective Potential of β-Caryophyllene
7. β-Caryophyllene: Alteration of Oxidative Stress and Mitochondrial Dysfunction
8. Toxicological Aspects
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Facecchia, K.; Fochesato, L.A.; Ray, S.D.; Stohs, S.J.; Pandey, S. Oxidative Toxicity in Neurodegenerative Diseases: Role of Mitochondrial Dysfunction and Therapeutic Strategies. J. Toxicol. 2011, 2011, 683728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A. Mitochondrial Dysfunction & Neurological Disorders. Curr. Neuropharmacol. 2016, 14, 565. [Google Scholar] [PubMed] [Green Version]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS Generation by Mitochondria in Cells with Impaired Electron Transport Chain and Mitochondrial DNA Damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef]
- Chen, J.Q.; Yager, J.D.; Russo, J. Regulation of Mitochondrial Respiratory Chain Structure and Function by Estrogens/Estrogen Receptors and Potential Physiological/Pathophysiological Implications. Biochim. Biophys. Acta Mol. Cell Res. 2005, 1746, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezzi, D.; Zeviani, M. Assembly Factors of Human Mitochondrial Respiratory Chain Complexes: Physiology and Pathophysiology. Adv. Exp. Med. Biol. 2012, 748, 65–106. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A.; Wai, T. Mitochondrial DNA and the Mammalian Oocyte. Curr. Top. Dev. Biol. 2007, 77, 87–111. [Google Scholar] [CrossRef]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, Metabolic Disturbances, Oxidative Stress and the Kynurenine System, with Focus on Neurodegenerative Disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef]
- Porter, R.K.; Brand, M.D. Mitochondrial Proton Conductance and H+/O Ratio Are Independent of Electron Transport Rate in Isolated Hepatocytes. Biochem. J. 1995, 310, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, M.; Li, H.; Zhang, H.; Shi, Y.; Wei, F.; Liu, D.; Liu, K.; Chen, D. Accumulation of Nuclear and Mitochondrial DNA Damage in the Frontal Cortex Cells of Patients with HIV-Associated Neurocognitive Disorders. Brain Res. 2012, 1458, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.H.; Lu, C.Y.; Wei, C.Y.; Ma, Y.S.; Lee, H.C. Oxidative Stress in Human Aging and Mitochondrial Disease-Consequences of Defective Mitochondrial Respiration and Impaired Antioxidant Enzyme System. Chin. J. Physiol. 2001, 44, 1–11. [Google Scholar]
- Hollensworth, S.B.; Shen, C.C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; Ledoux, S.P. Glial Cell Type-Specific Responses to Menadione-Induced Oxidative Stress. Free Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- van Houten, B.; Woshner, V.; Santos, J.H. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Voets, A.M.; Huigsloot, M.; Lindsey, P.J.; Leenders, A.M.; Koopman, W.J.H.; Willems, P.H.G.M.; Rodenburg, R.J.; Smeitink, J.A.M.; Smeets, H.J.M. Transcriptional Changes in OXPHOS Complex I Deficiency Are Related to Anti-Oxidant Pathways and Could Explain the Disturbed Calcium Homeostasis. Biochim. Biophys. Acta Mol. Basis. Dis. 2012, 1822, 1161–1168. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.D.R.; Castro, M.D.R.; Suarez, E.; Kraiselburd, E.; Isidro, A.; Paz, J.; Ferder, L.; Ayala-Torres, S. Aging Increases Mitochondrial DNA Damage and Oxidative Stress in Liver of Rhesus Monkeys. Exp. Gerontol. 2012, 47, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Alexeyev, M.F. Is There More to Aging than Mitochondrial DNA and Reactive Oxygen Species? FEBS J. 2009, 276, 5768–5787. [Google Scholar] [CrossRef] [Green Version]
- Andreazza, A.C.; Shoo, L.; Wang, J.F.; Trevor Young, L. Mitochondrial Complex I Activity and Oxidative Damage to Mitochondrial Proteins in the Prefrontal Cortex of Patients with Bipolar Disorder. Arch. Gen. Psychiatry 2010, 67, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Stewart, V.C.; Heales, S.J.R. Nitric Oxide-Induced Mitochondrial Dysfunction: Implications for Neurodegeneration. Free Radic. Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Mattson, M.R. Calcium and Neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef]
- Douarre, C.; Sourbier, C.; Dalla Rosa, I.; Brata Das, B.; Redon, C.E.; Zhang, H.; Neckers, L.; Pommier, Y. Mitochondrial Topoisomerase I Is Critical for Mitochondrial Integrity and Cellular Energy Metabolism. PLoS ONE 2012, 7, e41094. [Google Scholar] [CrossRef] [Green Version]
- Joshi, G.; Sultana, R.; Perluigi, M.; Butterfield, D.A. In Vivo Protection of Synaptosomes from Oxidative Stress Mediated by Fe2+/H2O2 or 2,2 Azobis-(2-Amidinopropane) Dihydrochloride by the Glutathione Mimetic Tricyclodecan-9-Yl Xanthogenate. Free Radic. Biol. Med. 2005, 38, 1023–1031. [Google Scholar] [CrossRef]
- Ross, W.N. Understanding Calcium Waves and Sparks in Central Neurons. Nat. Rev. Neurosci. 2012, 13, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Fidyt, K.; Fiedorowicz, A.; Strządała, L.; Szumny, A. β-Caryophyllene and β-Caryophyllene Oxide—Natural Compounds of Anticancer and Analgesic Properties. Cancer Med. 2016, 5, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Sarpietro, M.G.; di Sotto, A.; Accolla, M.L.; Castelli, F. Interaction of β-Caryophyllene and β-Caryophyllene Oxide with Phospholipid Bilayers: Differential Scanning Calorimetry Study. Thermochim. Acta 2015, 600, 28–34. [Google Scholar] [CrossRef]
- EssOilDB. Available online: http://www.nipgr.ac.in/Essoildb/ (accessed on 7 February 2021).
- Moo, C.L.; Yang, S.K.; Osman, M.A.; Yuswan, M.H.; Loh, J.Y.; Lim, W.M.; Lim, S.H.E.; Lai, K.S. Antibacterial Activity and Mode of Action of β-Caryophyllene on Bacillus Cereus. Pol. J. Microbiol. 2020, 69, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, H.J.; Yang, J.Y.; Lee, M.H.; Kim, H.W.; Kwon, H.J.; Park, M.; Kim, S.K.; Park, S.Y.; Kim, S.H.; Kim, J.B. Inhibitory Effects of β-Caryophyllene on Helicobacter Pylori Infection in Vitro and in Vivo. Int. J. Mol. Sci. 2020, 21, 1008. [Google Scholar] [CrossRef] [Green Version]
- Ames-Sibin, A.P.; Barizão, C.L.; Castro-Ghizoni, C.V.; Silva, F.M.S.; Sá-Nakanishi, A.B.; Bracht, L.; Bersani-Amado, C.A.; Marçal-Natali, M.R.; Bracht, A.; Comar, J.F. β-Caryophyllene, the Major Constituent of Copaiba Oil, Reduces Systemic Inflammation and Oxidative Stress in Arthritic Rats. J. Cell. Biochem. 2018, 119, 10262–10277. [Google Scholar] [CrossRef] [PubMed]
- Machado, K.C.; Islam, M.T.; Ali, E.S.; Rouf, R.; Uddin, S.J.; Dev, S.; Shilpi, J.A.; Shill, M.C.; Reza, H.M.; Das, A.K.; et al. A Systematic Review on the Neuroprotective Perspectives of Beta-Caryophyllene. Phytother. Res. 2018, 32, 2376–2388. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. Cannabinoid Receptors: Where They Are and What They Do. J. Neuroendocrinol. 2008, 20, 10–14. [Google Scholar] [CrossRef]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.Z.; Xie, X.Q.; Altmann, K.H.; Karsak, M.; Zimmer, A. Beta-Caryophyllene Is a Dietary Cannabinoid. Proc. Natl. Acad. Sci. USA 2008, 105, 9099–9104. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, M.J.; Zimmer, E.R.; Shin, M.; Kang, M.S.; Fonov, V.S.; Mathieu, A.; Aliaga, A.; Kostikov, A.; do Carmo, S.; Dea, D.; et al. Multimodal Imaging in Rat Model Recapitulates Alzheimer’s Disease Biomarkers Abnormalities. J. Neurosci. 2017, 37, 12263–12271. [Google Scholar] [CrossRef] [Green Version]
- Kommaddi, R.P.; Das, D.; Karunakaran, S.; Nanguneri, S.; Bapat, D.; Ray, A.; Shaw, E.; Bennett, D.A.; Nair, D.; Ravindranath, V. Aβ Mediates F-Actin Disassembly in Dendritic Spines Leading to Cognitive Deficits in Alzheimer’s Disease. J. Neurosci. 2018, 38, 1085–1099. [Google Scholar] [CrossRef]
- Wallace, R.A.; Dalton, A.J. What Can We Learn from Study of Alzheimer’s Disease in Patients with Down Syndrome for Early-Onset Alzheimer’s Disease in the General Population? Alzheimer’s Res. Ther. 2011, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2020, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Xu, H.; Yu, H.; Zhang, N.; Ye, Y.; Estevez, A.G.; Deng, H.; Gibson, G.E. Inactivation and Reactivation of the Mitochondrial α-Ketoglutarate Dehydrogenase Complex. J. Biol. Chem. 2011, 286, 17640–17648. [Google Scholar] [CrossRef] [Green Version]
- Dumont, M.; Ho, D.J.; Calingasan, N.Y.; Xu, H.; Gibson, G.; Beal, M.F. Mitochondrial Dihydrolipoyl Succinyltransferase Deficiency Accelerates Amyloid Pathology and Memory Deficit in a Transgenic Mouse Model of Amyloid Deposition. Free Radic. Biol. Med. 2009, 7, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s Brains Harbor Somatic MtDNA Control-Region Mutations That Suppress Mitochondrial Transcription and Replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H. Mitochondrial Oxidative Damage in Aging and Alzheimer’s Disease: Implications for Mitochondrially Targeted Antioxidant Therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G.; Kayed, R. Common Structure and Toxic Function of Amyloid Oligomers Implies a Common Mechanism of Pathogenesis. Neurology 2006, 66, S74–S78. [Google Scholar] [CrossRef]
- Deshpande, A.; Mina, E.; Glabe, C.; Busciglio, J. Different Conformations of Amyloid β Induce Neurotoxicity by Distinct Mechanisms in Human Cortical Neurons. J. Neurosci. 2006, 26, 6011–6018. [Google Scholar] [CrossRef] [Green Version]
- Hansson, C.A.; Frykman, S.; Farmery, M.R.; Tjernberg, L.O.; Nilsberth, C.; Pursglove, S.E.; Ito, A.; Winblad, B.; Cowburn, R.F.; Thyberg, J.; et al. Nicastrin, Presenilin, APH-1, and PEN-2 Form Active Gamma-Secretase Complexes in Mitochondria. J. Biol. Chem. 2004, 279, 51654–51660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarasija, S.; Norman, K.R. Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis Elegans. Antioxidants 2018, 7, 111. [Google Scholar] [CrossRef] [Green Version]
- Herreman, A.; Serneels, L.; Annaert, W.; Collen, D.; Schoonjans, L.; de Strooper, B. Total Inactivation of γ–Secretase Activity in Presenilin-Deficient Embryonic Stem Cells. Nat. Cell Biol. 2000, 2, 461–462. [Google Scholar] [CrossRef]
- Li, Y.-M.; Xu, M.; Lai, M.-T.; Huang, Q.; Castro, J.L.; DiMuzio-Mower, J.; Harrison, T.; Lellis, C.; Nadin, A.; Neduvelil, J.G.; et al. Photoactivated γ-Secretase Inhibitors Directed to the Active Site Covalently Label Presenilin 1. Nature 2000, 405, 689–694. [Google Scholar] [CrossRef]
- Esler, W.P.; Kimberly, W.T.; Ostaszewski, B.L.; Diehl, T.S.; Moore, C.L.; Tsai, J.-Y.; Rahmati, T.; Xia, W.; Selkoe, D.J.; Wolfe, M.S. Transition-State Analogue Inhibitors of γ-Secretase Bind Directly to Presenilin-1. Nat. Cell Biol. 2000, 2, 428–434. [Google Scholar] [CrossRef]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. γ-Secretase Is a Membrane Protein Complex Comprised of Presenilin, Nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [Green Version]
- Ullah, H.; Khan, H. Anti-Parkinson Potential of Silymarin: Mechanistic Insight and Therapeutic Standing. Front. Pharmacol. 2018, 9, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, R.S.; Lewitt, P.A.; Ebert, M.H.; Pakkenberg, H.; Kopin, I.J. The Clinical Syndrome of Striatal Dopamine Deficiency: Parkinsonism Induced by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP). N. Engl. J. Med. 1985, 312, 1418–1421. [Google Scholar] [CrossRef]
- William Langston, J.; Forno, L.S.; Rebert, C.S.; Irwin, I. Selective Nigral Toxicity after Systemic Administration of 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyrine (MPTP) in the Squirrel Monkey. Brain Res. 1984, 292, 390–394. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial Approaches for Neuroprotection. Ann. N. Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondria Take Center Stage in Aging and Neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial Dysfunction in Parkinson’s Disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Horowitz, M.P.; Greenamyre, J.T. Gene-Environment Interactions in Parkinson’s Disease: The Importance of Animal Modeling. Clin. Pharmacol. Ther. 2010, 88, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Kathrin Lutz, A.; Exner, N.; Fett, M.E.; Schleke, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of Parkin or PINK1 Function Increases Drp1-Dependent Mitochondrial Fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin-Induced Mitophagy in the Pathogenesis of Parkinson Disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef] [Green Version]
- Varin, M.; Bentea, E.; Michotte, Y.; Sarre, S. Oxidative Stress in Genetic Mouse Models of Parkinsons Disease. Oxid. Med. Cell. Longev. 2012, 2012, 624925. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, G.; Xu, Z. Mitochondrial Dysfunction and Its Role in Motor Neuron Degeneration in ALS. Mitochondrion 2005, 5, 77–87. [Google Scholar] [CrossRef]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What Can We Learn about Parkinson’s Disease Pathobiology? Parkinson’s Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and P97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase P97 Is Essential for Outer Mitochondrial Membrane Protein Turnover. Mol. Biol. Cell 2011, 22, 291–300. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice Lacking Alpha-Synuclein Are Resistant to Mitochondrial Toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple Sclerosis—A Review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, H.; Tovchiga, O.; Daglia, M.; Khan, H. Modulating Gut Microbiota: An Emerging Approach in the Prevention and Treatment of Multiple Sclerosis. Curr. Neuropharmacol. 2021, 19. (epub ahead of print). [Google Scholar] [CrossRef]
- Ullah, H.; Khan, H. Epigenetic drug development for autoimmune and inflammatory diseases. In Histone Modifications in Therapy; Castelo-Branco, P., Jeronimo, C., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 395–413. [Google Scholar]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Neurodegenerative Diseases and Therapeutic Strategies Using Iron Chelators. J. Trace Elem. Med. Biol. 2015, 31, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Rottlaender, A.; Kuerten, S. Stepchild or Prodigy? Neuroprotection in Multiple Sclerosis (MS) Research. Int. J. Mol. Sci. 2007, 16, 14850–14865. [Google Scholar] [CrossRef] [Green Version]
- Verber, N.; Shaw, P.J. Biomarkers in Amyotrophic Lateral Sclerosis: A Review of New Developments. Curr. Opin. Neurol. 2020, 33, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Baltazar, M.T.; Dinis-Oliveira, R.J.; de Lourdes Bastos, M.; Tsatsakis, A.M.; Duarte, J.A.; Carvalho, F. Pesticides Exposure as Etiological Factors of Parkinson’s Disease and Other Neurodegenerative Diseases-A Mechanistic Approach. Toxicol. Lett. 2014, 230, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.V.; Halliday, G. Missing Pieces in the Parkinson’s Disease Puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, V.S.; Fenech, M. Mutations That Affect Mitochondrial Functions and Their Association with Neurodegenerative Diseases. Mutat. Res. Rev. Mutat. Res. 2014, 759, 1–13. [Google Scholar] [CrossRef]
- Sasaki, S.; Warita, H.; Murakami, T.; Shibata, N.; Komori, T.; Abe, K.; Kobayashi, M.; Iwata, M. Ultrastructural Study of Aggregates in the Spinal Cord of Transgenic Mice with a G93A Mutant SOD1 Gene. Acta Neuropathol. 2005, 109, 247–255. [Google Scholar] [CrossRef]
- Israelson, A.; Arbel, N.; da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.A.; Rademakers, R.; Neumann, M. TDP-43 and FUS in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R.A. A New Subtype of Frontotemporal Lobar Degeneration with FUS Pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; de Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2012, 2, 352–363. [Google Scholar]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L.; et al. TDP-43 Induces Mitochondrial Damage and Activates the Mitochondrial Unfolded Protein Response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanidis, S.; Kattan, F.-G.; Doxakis, E. Unraveling the Pathways to Neuronal Homeostasis and Disease: Mechanistic Insights into the Role of RNA-Binding Proteins and Associated Factors. Int. J. Mol. Sci. 2018, 19, 2280. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.-J.; Wang, D.; Wei, X.; Xia, X.-G. FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. PLoS Genet. 2011, 7, e1002011. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with Hsp60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [Green Version]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial Abnormalities and Disruption of the Neuromuscular Junction Precede the Clinical Phenotype and Motor Neuron Loss in HFUSWT Transgenic Mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial Oxidative Stress Causes Mitochondrial Fragmentation via Differential Modulation of Mitochondrial Fission-Fusion Proteins. FEBS J. 2011, 278, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Ullah, H.; Tundis, R.; Belwal, T.; Devkota, H.P.; Daglia, M.; Cetin, Z.; Saygili, E.I.; Campos, M.d.G.; Capanoglu, E.; et al. Dietary Flavonoids in the Management of Huntington’s Disease: Mechanism and Clinical Perspective. eFood 2020, 1, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Carmichael, J. Huntington’s Disease: Molecular Basis of Neurodegeneration. Expert Rev. Mol. Med. 2003, 5, 1–21. [Google Scholar] [CrossRef]
- Siddiqui, A.; Rivera-Sánchez, S.; Castro, M.D.R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA Damage Is Associated with Reduced Mitochondrial Bioenergetics in Huntington’s Disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, H.V.S.; Chow, A.M.; Kerman, K. Recent Advances in Biosensors for Neurodegenerative Disease Detection. Trends Anal. Chem. 2016, 79, 363–370. [Google Scholar] [CrossRef]
- Trushina, E.; McMurray, C.T. Oxidative Stress and Mitochondrial Dysfunction in Neurodegenerative Diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.; Beal, M.F. Potential for Creatine and Other Therapies Targeting Cellular Energy Dysfunction in Neurological Disorders. Ann. Neurol. 2001, 49, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Andres, R.H.; Ducray, A.D.; Schlattner, U.; Wallimann, T.; Widmer, H.R. Functions and Effects of Creatine in the Central Nervous System. Brain Res. Bull. 2008, 76, 329–343. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial Creatine Kinase in Human Health and Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.; Schulze, A. Health Implications of Creatine: Can Oral Creatine Supplementation Protect against Neurological and Atherosclerotic Disease? Neuroscience 2002, 112, 243–260. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; Beal, M.F. Creatine and Its Potential Therapeutic Value for Targeting Cellular Energy Impairment in Neurodegenerative Diseases. Neuromolecular Med. 2008, 10, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Kato, N.; Kato, T. Effects of Creatine on Mental Fatigue and Cerebral Hemoglobin Oxygenation. Neurosci. Res. 2002, 42, 279–285. [Google Scholar] [CrossRef]
- Rae, C.; Digney, A.L.; McEwan, S.R.; Bates, T.C. Oral Creatine Monohydrate Supplementation Improves Brain Performance: A Double-Blind, Placebo-Controlled, Cross-over Trial. Proc. R. Soc. B Biol. Sci. 2003, 270, 2147–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksenov, M.; Aksenova, M.; Butterfield, D.A.; Markesbery, W.R. Oxidative Modification of Creatine Kinase BB in Alzheimer’s Disease Brain. J. Neurochem. 2000, 74, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.B.; Sweeney, G. Creatine Supplementation Increases Glucose Oxidation and AMPK Phosphorylation and Reduces Lactate Production in L6 Rat Skeletal Muscle Cells. J. Physiol. 2004, 555, 409–421. [Google Scholar] [CrossRef]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP Kinase Is Required for Mitochondrial Biogenesis in Skeletal Muscle in Response to Chronic Energy Deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.; Koch, W.; Elstner, M.; Schombacher, Y.; Bender, J.; Moeschl, M.; Gekeler, F.; Müller-Myhsok, B.; Gasser, T.; Tatsch, K.; et al. Creatine Supplementation in Parkinson Disease: A Placebo-Controlled Randomized Pilot Trial. Neurology 2006, 67, 1262–1264. [Google Scholar] [CrossRef]
- Hass, C.J.; Collins, M.A.; Juncos, J.L. Resistance Training with Creatine Monohydrate Improves Upper-Body Strength in Patients with Parkinson Disease: A Randomized Trial. Neurorehabil. Neural Repair 2007, 21, 107–115. [Google Scholar] [CrossRef]
- Matthews, R.T.; Ferrante, R.J.; Klivenyi, P.; Yang, L.; Klein, A.M.; Mueller, G.; Kaddurah-Daouk, R.; Beal, M.F. Creatine and Cyclocreatine Attenuate MPTP Neurotoxicity. Exp. Neurol. 1999, 157, 142–149. [Google Scholar] [CrossRef]
- Ravina, B.; Kieburtz, K.; Tilley, B.; Shannon, K.; Tanner, C.; Frederick Wooten, G.; Racette, B.; Deppen, P.; Dewey, R.B.; Hayward, B.; et al. A Randomized, Double-Blind, Futility Clinical Trial of Creatine and Minocycline in Early Parkinson Disease. Neurology 2006, 66, 664–671. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Blamire, A.M.; Manners, D.N.; Rajagopalan, B.; Styles, P.; Schapira, A.H.V.; Warner, T.T. High-Dose Creatine Therapy for Huntington Disease: A 2-Year Clinical and MRS Study. Neurology 2005, 64, 1655–1656. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington Disease Is Safe, Tolerable, Bioavailable in Brain and Reduces Serum 8OH2′dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef]
- Klivenyi, P.; Ferrante, R.J.; Matthews, R.T.; Bogdanov, M.B.; Klein, A.M.; Andreassen, O.A.; Mueller, G.; Wermer, M.; Kaddurah-Daouk, R.; Beal, M.F. Neuroprotective Effects of Creatine in a Transgenic Animal Model of Amyotrophic Lateral Sclerosis. Nat. Med. 1999, 5, 347–350. [Google Scholar] [CrossRef]
- Drory, V.E.; Gross, D. No Effect of Creatine on Respiratory Distress in Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2002, 3, 43–46. [Google Scholar] [CrossRef]
- Groeneveld, G.J.; Veldink, J.H.; van der Tweel, I.; Kalmijn, S.; Beijer, C.; de Visser, M.; Wokke, J.H.J.; Franssen, H.; van den Berg, L.H. A Randomized Sequential Trial of Creatine in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2003, 53, 437–445. [Google Scholar] [CrossRef]
- Shefner, J.M.; Cudkowicz, M.E.; Schoenfeld, D.; Conrad, T.; Taft, J.; Chilton, M.; Urbinelli, L.; Qureshi, M.; Zhang, H.; Pestronk, A.; et al. A Clinical Trial of Creatine in ALS. Neurology 2004, 63, 1656–1661. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Hirano, M. Coenzyme Q and Mitochondrial Disease. Dev. Disabil. Res. Rev. 2010, 16, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P. Coenzyme Q10 as a Therapy for Mitochondrial Disease. Int. J. Biochem. Cell Biol. 2014, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondrial Dysfunction and Oxidative Damage in Alzheimer’s and Parkinson’s Diseases and Coenzyme Q 10 as a Potential Treatment. J. Bioenerg. Biomembr. 2004, 36, 381–386. [Google Scholar] [CrossRef]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme Q10 Prevents Apoptosis by Inhibiting Mitochondrial Depolarization Independently of Its Free Radical Scavenging Property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an Obligatory Cofactor for Uncoupling Protein Function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L.; Diano, S.; Leranth, C.; Garcia-Segura, L.M.; Cowley, M.A.; Shanabrough, M.; Elsworth, J.D.; Sotonyi, P.; Roth, R.H.; Dietrich, E.H.; et al. Coenzyme Q Induces Nigral Mitochondrial Uncoupling and Prevents Dopamine Cell Loss in a Primate Model of Parkinson’s Disease. Endocrinology 2003, 144, 2757–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsucci, D.; Mancuso, M.; Ienco, E.C.; LoGerfo, A.; Siciliano, G. Targeting Mitochondrial Dysfunction and Neurodegeneration by Means of Coenzyme Q10 and Its Analogues. Curr. Med. Chem. 2011, 18, 4053–4064. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A Review on Mitochondrial Restorative Mechanism of Antioxidants in Alzheimer’s Disease and Other Neurological Conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Kader, R.; Hauptmann, S.; Keil, U.; Scherping, I.; Leuner, K.; Eckert, A.; Müller, W.E. Stabilization of Mitochondrial Function by Ginkgo Biloba Extract (EGb 761). Pharmacol. Res. 2007, 56, 493–502. [Google Scholar] [CrossRef]
- Eckert, A.; Keil, U.; Scherping, I.; Hauptmann, S.; Müller, W.E. Stabilization of Mitochondrial Membrane Potential and Improvement of Neuronal Energy Metabolism by Ginkgo Biloba Extract EGb 761. Ann. N. Y. Acad. Sci. 2005, 1056, 474–485. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic Effects of Coenzyme Q10 and Remacemide in Transgenic Mouse Models of Huntington’s Disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Shaw, P.J.; de Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Liu, J. The Effects and Mechanisms of Mitochondrial Nutrient α-Lipoic Acid on Improving Age-Associated Mitochondrial and Cognitive Dysfunction: An Overview. Neurochem. Res. 2008, 33, 194–203. [Google Scholar] [CrossRef]
- Cole, G.M.; Ma, Q.L.; Frautschy, S.A. Omega-3 Fatty Acids and Dementia. Prostaglandins Leukot. Essent. Fatty Acids 2009, 81, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, G.P.; Chang, S.; Eckmann, J.; Copanaki, E.; Hagl, S.; Hener, U.; Müller, W.E.; Kögel, D. Liposome-Incorporated DHA Increases Neuronal Survival by Enhancing Non-Amyloidogenic APP Processing. Biochim. Biophys. Acta Biomembr. 2011, 1808, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Barceló-Coblijn, G.; Kitajka, K.; Puskás, L.G.; Hogyes, E.; Zvara, A.; Hackler, L.; Farkas, T. Gene Expression and Molecular Composition of Phospholipids in Rat Brain in Relation to Dietary N-6 to n-3 Fatty Acid Ratio. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2003, 1632, 72–79. [Google Scholar] [CrossRef]
- Stanley, W.C.; Khairallah, R.J.; Dabkowski, E.R. Update on Lipids and Mitochondrial Function: Impact of Dietary n-3 Polyunsaturated Fatty Acids. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Irrera, N.; D’ascola, A.; Pallio, G.; Bitto, A.; Mannino, F.; Arcoraci, V.; Rottura, M.; Ieni, A.; Minutoli, L.; Metro, D.; et al. β-Caryophyllene Inhibits Cell Proliferation through a Direct Modulation of CB2 Receptors in Glioblastoma Cells. Cancers 2020, 12, 1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.J.; Gao, M.; Gao, F.F.; Su, Q.X.; Wu, J. Brain Cannabinoid Receptor 2: Expression, Function and Modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Gao, M.; Liu, Q.R.; Bi, G.H.; Li, X.; Yang, H.J.; Gardner, E.L.; Wu, J.; Xi, Z.X. Cannabinoid CB2 Receptors Modulate Midbrain Dopamine Neuronal Activity and Dopamine-Related Behavior in Mice. Proc. Natl. Acad. Sci. USA 2014, 111, E5007–E5015. [Google Scholar] [CrossRef] [Green Version]
- Stempel, A.V.; Stumpf, A.; Zhang, H.Y.; Özdoğan, T.; Pannasch, U.; Theis, A.K.; Otte, D.M.; Wojtalla, A.; Rácz, I.; Ponomarenko, A.; et al. Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron 2016, 90, 795–809. [Google Scholar] [CrossRef] [Green Version]
- den Boon, F.S.; Chameau, P.; Schaafsma-Zhao, Q.; van Aken, W.; Bari, M.; Oddi, S.; Kruse, C.G.; Maccarrone, M.; Wadman, W.J.; Werkmana, T.R. Excitability of Prefrontal Cortical Pyramidal Neurons Is Modulated by Activation of Intracellular Type-2 Cannabinoid Receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 3534–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viveros-Paredes, J.M.; González-Castañeda, R.E.; Gertsch, J.; Chaparro-Huerta, V.; López-Roa, R.I.; Vázquez-Valls, E.; Beas-Zarate, C.; Camins-Espuny, A.; Flores-Soto, M.E. Neuroprotective Effects of β-Caryophyllene against Dopaminergic Neuron Injury in a Murine Model of Parkinson’s Disease Induced by MPTP. Pharmaceuticals 2017, 10, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, D.A.; El-Fayoumi, H.M.; Mahmoud, M.F. β-Caryophyllene Alleviates Diet-Induced Neurobehavioral Changes in Rats: The Role of CB2 and PPAR-γ Receptors. Biomed. Pharmacother. 2019, 110, 145–154. [Google Scholar] [CrossRef]
- Cheng, Y.; Dong, Z.; Liu, S. Β-caryophyllene Ameliorates the Alzheimer-like Phenotype in APP/PS1 Mice through CB2 Receptor Activation and the PPARγ Pathway. Pharmacology 2014, 94, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alberti, T.B.; Marcon, R.; Bicca, M.A.; Raposo, N.R.B.; Calixto, J.B.; Dutra, R.C. Essential Oil from Pterodon Emarginatus Seeds Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating Th1/Treg Cell Balance. J. Ethnopharmacol. 2014, 155, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Alberti, T.B.; Barbosa, W.L.R.; Vieira, J.L.F.; Raposo, N.R.B.; Dutra, R.C. (−)-β-Caryophyllene, a CB2 Receptor-Selective Phytocannabinoid, Suppresses Motor Paralysis and Neuroinflammation in a Murine Model of Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 691. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.A.G.; Martins, N.M.; Sisti, F.M.; Fernandes, L.S.; Ferreira, R.S.; de Freitas, O.; Santos, A.C. The Cannabinoid Beta-Caryophyllene (BCP) Induces Neuritogenesis in PC12 Cells by a Cannabinoid-Receptor-Independent Mechanism. Chem. Biol. Interact. 2017, 261, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lv, Y.; Tian, X.; Lou, J.; An, R.; Zhang, Q.; Li, M.; Xu, L.; Dong, Z. Neuroprotective Effect of β-Caryophyllene on Cerebral Ischemia-Reperfusion Injury via Regulation of Necroptotic Neuronal Death and Inflammation: In Vivo and In Vitro. Front. Neurosci. 2017, 11, 583. [Google Scholar] [CrossRef]
- Klauke, A.L.; Racz, I.; Pradier, B.; Markert, A.; Zimmer, A.M.; Gertsch, J.; Zimmer, A. The Cannabinoid CB2 Receptor-Selective Phytocannabinoid Beta-Caryophyllene Exerts Analgesic Effects in Mouse Models of Inflammatory and Neuropathic Pain. Eur. Neuropsychopharmacol. 2014, 24, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Fotio, Y.; el Alaoui, A.A.; Borruto, A.M.; Acciarini, S.; Giordano, A.; Ciccocioppo, R. Efficacy of a Combination of N-Palmitoylethanolamide, Beta-Caryophyllene, Carnosic Acid, and Myrrh Extract on Chronic Neuropathic Pain: A Preclinical Study. Front. Pharmacol. 2019, 10, 711. [Google Scholar] [CrossRef]
- Chávez-Hurtado, P.; González-Castañeda, R.E.; Beas-Zarate, C.; Flores-Soto, M.E.; Viveros-Paredes, J.M. β-Caryophyllene Reduces DNA Oxidation and the Overexpression of Glial Fibrillary Acidic Protein in the Prefrontal Cortex and Hippocampus of d-Galactose-Induced Aged BALB/c Mice. J. Med. Food 2020, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Ojha, S.; Javed, H.; Azimullah, S.; Haque, M.E. β-Caryophyllene, a Phytocannabinoid Attenuates Oxidative Stress, Neuroinflammation, Glial Activation, and Salvages Dopaminergic Neurons in a Rat Model of Parkinson Disease. Mol. Cell. Biochem. 2016, 418, 59–70. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [Green Version]
- Assis, L.C.; Straliotto, M.R.; Engel, D.; Hort, M.A.; Dutra, R.C.; de Bem, A.F. β-Caryophyllene Protects the C6 Glioma Cells against Glutamate-Induced Excitotoxicity through the Nrf2 Pathway. Neuroscience 2014, 279, 220–231. [Google Scholar] [CrossRef]
- Tian, X.; Peng, J.; Zhong, J.; Yang, M.; Pang, J.; Lou, J.; Li, M.; An, R.; Zhang, Q.; Xu, L.; et al. β-Caryophyllene Protects in Vitro Neurovascular Unit against Oxygen-Glucose Deprivation and Re-Oxygenation-Induced Injury. J. Neurochem. 2016, 39, 757–768. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, C.C.; de Oliveira, C.V.; Grigoletto, J.; Ribeiro, L.R.; Funck, V.R.; Grauncke, A.C.B.; de Souza, T.L.; Souto, N.S.; Furian, A.F.; Menezes, I.R.A.; et al. Anticonvulsant Activity of β-Caryophyllene against Pentylenetetrazol-Induced Seizures. Epilepsy Behav. 2016, 56, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, J.; Cao, G.; Li, R.; Liu, J.; Dong, Z.; Xu, L. β-Caryophyllene Attenuates Focal Cerebral Ischemia-Reperfusion Injury by Nrf2/HO-1 Pathway in Rats. Neurochem. Res. 2016, 41, 1291–1304. [Google Scholar] [CrossRef]
- Fontes, L.B.A.; Dias, D.; dos, S.; Aarestrup, B.J.V.; Aarestrup, F.M.; Filho, A.A.D.S.; Corrêa, J.O.d.A. β-Caryophyllene Ameliorates the Development of Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice. Biomed. Pharmacother. 2017, 91, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ma, W.; Du, J. β-Caryophyllene (BCP) Ameliorates MPP+ Induced Cytotoxicity. Biomed. Pharmacother. 2018, 103, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Postu, P.A.; Sadiki, F.Z.; el Idrissi, M.; Cioanca, O.; Trifan, A.; Hancianu, M.; Hritcu, L. Pinus Halepensis Essential Oil Attenuates the Toxic Alzheimer’s Amyloid Beta (1-42)-Induced Memory Impairment and Oxidative Stress in the Rat Hippocampus. Biomed. Pharmacother. 2019, 112, 108673. [Google Scholar] [CrossRef] [PubMed]
- Abuhamdah, S.; Abuhamdah, R.; Howes, M.-J.R.; Al-Olimat, S.; Ennaceur, A.; Chazot, P.L. Pharmacological and Neuroprotective Profile of an Essential Oil Derived from Leaves of Aloysia Citrodora Palau. J. Pharm. Pharmacol. 2015, 67, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.K.; Halder, S.; Khanna, N.; Tandon, O.P.; Sharma, K.K. The Effect of the Essential Oil of Eugenia Caryophyllata in Animal Models of Depression and Locomotor Activity. Nutr. Neurosci. 2013, 16, 233–238. [Google Scholar] [CrossRef]
- Kumar, A.; Aggrawal, A.; Pottabathini, R.; Singh, A. Possible Neuroprotective Mechanisms of Clove Oil against Icv-Colchicine Induced Cognitive Dysfunction. Pharmacol. Rep. 2016, 68, 764–772. [Google Scholar] [CrossRef]
- Beserra-Filho, J.I.A.; de Macêdo, A.M.; Leão, A.H.F.F.; Bispo, J.M.M.; Santos, J.R.; de Oliveira-Melo, A.J.; Menezes, P.D.P.; Duarte, M.C.; de Souza Araújo, A.A.; Silva, R.H.; et al. Eplingiella Fruticosa Leaf Essential Oil Complexed with β-Cyclodextrin Produces a Superior Neuroprotective and Behavioral Profile in a Mice Model of Parkinson’s Disease. Food Chem. Toxicol. 2019, 124, 17–29. [Google Scholar] [CrossRef]
- Garabadu, D.; Singh, D. Ocimum Basilicum Attenuates Ethidium Bromide-Induced Cognitive Deficits and Pre-Frontal Cortical Neuroinflammation, Astrogliosis and Mitochondrial Dysfunction in Rats. Metab. Brain Dis. 2020, 35, 483–495. [Google Scholar] [CrossRef]
- Sajjadi, S.E. Analysis of the Essential Oils of Two Cultivated Basil (Ocimum basilicum L.) from Iran. DARU J. Pharm. Sci. 2006, 14, 128–130. [Google Scholar]
- Leporini, M.; Bonesi, M.; Loizzo, M.R.; Passalacqua, N.G.; Tundis, R. The Essential Oil of Salvia Rosmarinus Spenn. From Italy as a Source of Health-Promoting Compounds: Chemical Profile and Antioxidant and Cholinesterase Inhibitory Activity. Plants 2020, 9, 798. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, D.; Levy, R.; Carroll, B. Toxicological Evaluation of β-Caryophyllene Oil: Subchronic Toxicity in Rats. Int. J. Toxicol. 2016, 35, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.L.d.S.; Machado, K.C.; da Silva, A.P.; dos, S.C.L.; Feitosa, C.M.; de Castro Almeida, F.R. Non-Clinical Toxicity of β-Caryophyllene, a Dietary Cannabinoid: Absence of Adverse Effects in Female Swiss Mice. Regul. Toxicol. Pharmacol. 2018, 92, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Bahi, A.; al Mansouri, S.; al Memari, E.; al Ameri, M.; Nurulain, S.M.; Ojha, S. β-Caryophyllene, a CB2 Receptor Agonist Produces Multiple Behavioral Changes Relevant to Anxiety and Depression in Mice. Physiol. Behav. 2014, 135, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Molina-Jasso, D.; Álvarez-González, I.; Madrigal-Bujaidar, E. Clastogenicity of Beta-Caryophyllene in Mouse. Biol. Pharm. Bull. 2009, 32, 520–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francomano, F.; Caruso, A.; Barbarossa, A.; Fazio, A.; la Torre, C.; Ceramella, J.; Mallamaci, R.; Saturnino, C.; Iacopetta, D.; Sinicropi, M.S. β-Caryophyllene: A Sesquiterpene with Countless Biological Properties. Appl. Sci. 2019, 9, 5420. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Myslivečková, Z.; Szotáková, B.; Špičáková, A.; Lněničková, K.; Ambrož, M.; Kubíček, V.; Krasulová, K.; Anzenbacher, P.; Skálová, L. The Inhibitory Effects of β-Caryophyllene, β-Caryophyllene Oxide and α-Humulene on the Activities of the Main Drug-Metabolizing Enzymes in Rat and Human Liver in Vitro. Chem. Biol. Interact. 2017, 278, 123–128. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Botanical Name | Family | Active Parts | Percentage 1 |
|---|---|---|---|
| Ocimum basilicum L. | Lamiaceae | Leaf | 0.3–3.1 |
| Cinnamomum species | Lauraceae | Leaf/bark a | 0.2–35.9 a |
| Piper nigrum L. | Piperaceae | Berries/Leaf/stem b | 3.3–46 b |
| Syzygium aromaticum (L.) Merr. and L.M. Perry | Myrtaceae | Floral bud | 3.2 |
| Cannabis sativa L. | Cannabaceae | Whole plant (fresh material) | 3–16.2 |
| Lavandula angustifolia Mill. | Lamiaceae/labiatae | Flower and stem | 1.08 |
| Lavandula angustifolia Mill. | Lamiaceae/labiatae | Whole plant | 0.3 |
| Origanum vulgare L. | Lamiaceae/labiatae | Leaf/Stem/Flower/Whole plantc | 0.4–24.5 c |
| Rosmarinus officinalis L. | Lamiaceae | Aerial parts | 0.5–13.6 |
| Study Model | Extract or Compound (Dose/Concentration) | Study Outcomes | References |
|---|---|---|---|
| BALB/c mice, with d-galactose induced aging | β-caryophyllene (10 mg/kg/day, p.o. for 4 weeks) | ↓ DNA oxidation and overexpression of glial fibrillary acidic proteins in the prefrontal cortex and hippocampus. | [151] |
| Rats, with PD | β-caryophyllene (50 mg/kg/day, i.p. for 4 weeks) | ↑ GSH, SOD and CAT. Inhibit lipid peroxidation. ↓ IL-1β, IL-6, and TNF-α levels. ↓ COX-2 and iNOS expression. ↓ glial activation and rescued dopaminergic neurons. | [152] |
| Rats with PD | β-caryophyllene (50 mg/kg/day, i.p.) for 4 weeks | ↓ pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) and inflammatory mediators (NF-κB, COX-2, and iNOS). ↑ glutathione, SOD and CAT. ↓ lipid peroxidation. | [153] |
| C6 glioma cell line | β-caryophyllene (0.5 and 1.0 μM) | ↑ cellular antioxidant responses via CB2 receptor dependent Nrf2 activation. ↓ ROS production. Restored MMP. | [154] |
| Neurovascular unit (BMECs, neurons and astrocytes) | β-caryophyllene (10 μmol/L) | ↓ BBB permeability, neuronal apoptosis, oxidative stress damage, inflammatory cytokines. ↓ metalloproteinase-9 expression/activity, Bax expression. ↑ expression of claudin-5, occludin, ZO-1, GAP-43, and Bcl-2. | [155] |
| Adult male Sprague–Dawley rats, with focal cerebral ischemia | β-caryophyllene (34, 102 and 306 mg/kg/day, p.o.). | ↑ Nrf2 and HO-1 expression. Restored SOD and CAT activity and expression. | [157] |
| C57BL/6 mice, with autoimmune encephalomyelitis | β-caryophyllene (25 and 50 mg/kg/day, p.o.). | ↓ H2O2, IFN-γ, TNF-α, IL-17 and NO. | [158] |
| Human neuroblastoma SH-SY5Y cells | β-caryophyllene (1 and 2.5 μM) | Restored reduction in MMP. ↑ intracellular GSH and GPx activity. ↓ Caspase-3 and Bax.Restored Bcl-2 expression Suppressed HO-1 activation and JNK phosphorylation. | [159] |
| Wistar rats-male with Aβ (1-42)-induced memory impairment | Pinus halepensis essential oil (1 and 3%). | ↓ hippocampal AChE activity. ↑ hippocampal antioxidant markers (SOD, CAT, GPx and GSH). ↓ malondialdehyde (MDA) levels. | [160] |
| CAD neuroblastoma cell lines | Aloysia citrodora Palau essential oil (0.01 and 0.001 mg/mL) | ↓ H2O2 (250 μM) and Aβ (10 μM) induced neurotoxicity. Fe2+ chelation in vitro. | [161] |
| Rats, with ICV colchicine induced memory impairment | Syzygium aromaticum (L.) Merr. and L.M. Perry (0.05 mL/kg and 0.1 mL/kg) | ↓ AChE activity, lipid peroxidation levels, and nitrite concentrations. Restored activities of GSH and mitochondrial respiratory enzyme complex (I–IV). | [163] |
| Male mice, with PD | Eplingiella fruticosa leaf essential oil. (5 mg/kg/day, p.o. for 40 days) | ↓ membrane lipid peroxide levels in the striatum. ↑ dopamine levels in the striatum and substantia nigra pars compacta. | [164] |
| Male adult Wistar albino rats, with induced MS like manifestations | Ocimum basilicum L. essential oil (100 and 200 μL/kg) | ↓ proinflammatory cytokines (TNF-α and IL-6) in prefrontal cortex. ↓ astrogliosis by increasing GFAP and Iba-1 levels in prefrontal cortex. ↑ mitochondrial function, integrity, respiratory control rate and ATP production. ↓ mitochondria-dependent apoptosis in prefrontal cortex of rats. | [165] |
| In vitro, antioxidant and AChE inhibition assays | Salvia rosmarinus Spenn. essential oil. | Strong antioxidant effects (DPPH, ABTS, FRAP and β-carotene bleaching tests). Significant AChE inhibition. | [167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, H.; Di Minno, A.; Santarcangelo, C.; Khan, H.; Daglia, M. Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System. Antioxidants 2021, 10, 546. https://doi.org/10.3390/antiox10040546
Ullah H, Di Minno A, Santarcangelo C, Khan H, Daglia M. Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System. Antioxidants. 2021; 10(4):546. https://doi.org/10.3390/antiox10040546
Chicago/Turabian StyleUllah, Hammad, Alessandro Di Minno, Cristina Santarcangelo, Haroon Khan, and Maria Daglia. 2021. "Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System" Antioxidants 10, no. 4: 546. https://doi.org/10.3390/antiox10040546
APA StyleUllah, H., Di Minno, A., Santarcangelo, C., Khan, H., & Daglia, M. (2021). Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System. Antioxidants, 10(4), 546. https://doi.org/10.3390/antiox10040546