
Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury

 ,
,  ,
,  , ,
, ,  and
and
Abstract
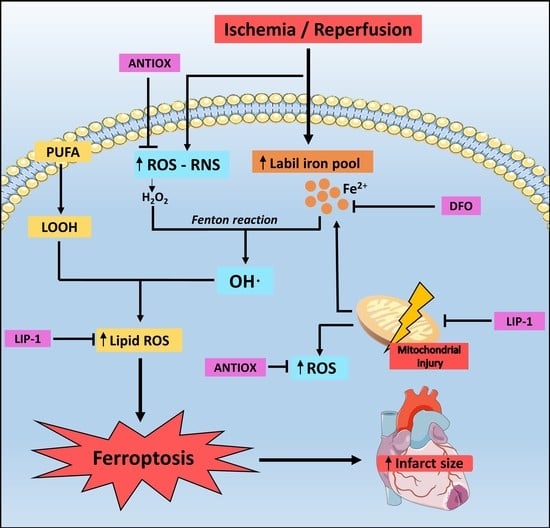
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Oxidative Stress
2.1. Production of Reactive Oxygen Species and Reactive Nitrogen Species
2.2. Antioxidant Systems
- -
- Scavenging of ROS or their precursors.
- -
- Inhibiting ROS production
- -
- Attenuating the catalysis of ROS generation via chelating metal ions
- -
- Enhancing endogenous antioxidant generation
- -
- Repairing the oxidative damage inflicted on the macromolecules
- -
- Reducing apoptotic cell death by up-regulating the anti-death gene Bcl-2.
3. Pathophysiology of Myocardial I/R Injury
3.1. Ischemia
3.2. Reperfusion
4. Iron Homeostasis
4.1. Intracellular Iron Regulation
4.2. Pathways of Iron Increase
4.2.1. Myocardial Hemorrhage
4.2.2. Degradation of Ferritin
4.2.3. Release from Enzymatic Iron-Sulfur Cluster
4.2.4. Polyol Pathway
4.2.5. Ferritin Heavy Chain
5. Ferroptosis
5.1. Class 1 Ferroptosis Inducers
5.2. Class 2 Ferroptosis Inducers
5.3. Lysosome and Ferroptosis
5.4. Ferroptosis and Relevance of the Cell Membrane
5.5. Autophagy-Induced Ferroptosis
5.6. Ferroptosis and Necroinflammation
5.7. Ferroptosis and Mitochondria in Cardiomyocytes
5.8. Cell Death Propagation and Ferroptosis
6. Does Ferroptosis Occur in the Ischemic Phase or Reperfusion Phase?
7. Therapies for Myocardial Reperfusion Injury
7.1. Vitamin E
7.2. Ascorbic Acid
7.3. n-3 Polyunsaturated Fatty Acid
7.4. Deferoxamine
7.5. N-Acetylcysteine
7.6. Nuclear Factor Erythroid 2-Related Factor 2
7.7. Mechanistic Target of Rapamycin
7.8. Heme Oxygenase-1
8. Ferroptosis-Based New Strategy to Reduce Infarct Size
8.1. Liproxstatin-1
8.1.1. Greater Effect of Lip-1 Compared to Others Drugs
8.1.2. Lipid Peroxide Radical Scavenger
8.1.3. Effect on Mitochondria
8.1.4. Anti Ferroptotic System Modulation and Production of Antioxidant Enzymes
8.1.5. Lip-1 Attenuates Acute Remote Organ Injury after I/R
8.1.6. Use in Humans and Concentrations
8.2. Other Ferroptosis Inhibitors
8.2.1. Baicalein
8.2.2. Mitochondrial-Targeted XJB-5-131
8.3. Considerations about Inhibiting Ferroptosis
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 14 May 2020).
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 15 December 2020).
- Ruiz-Meana, M.; García-Dorado, D. Pathophysiology of Ischemia-Reperfusion Injury: New Therapeutic Options for Acute Myocardial Infarction. Rev. Española Cardiol. Engl. Ed. 2009, 62, 199–209. [Google Scholar] [CrossRef]
- White, H.D.; Chew, D.P. Acute myocardial infarction. Lancet 2008, 372, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Dorado, D.; Rodríguez-Sinovas, A.; Ruiz-Meana, M.; Inserte, J. Protection Against Myocardial Ischemia-reperfusion Injury in Clinical Practice. Rev. Española Cardiol. Engl. Ed. 2014, 67, 394–404. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Juránek, I.; Bezek, Š. Controversy of free radical hypothesis: Reactive oxygen species—Cause or consequence of tissue injury. Gen. Physiol. Biophys. 2005, 24, 263–278. [Google Scholar]
- Rodrigo, R.; González, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 2011, 34, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Guichard, C.; Charles, R. Clinical pharmacology and therapeutic use of antioxidant vitamins. Fundam. Clin. Pharmacol. 2007, 21, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: Reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Parra, P.; Rodrigo, R. Novel Antioxidant Therapy against Myocardial Ischemia–Reperfusion Injury during Percutaneous Coronary Angioplasty. In Free Radicals and Diseases; Ahmad, R., Ed.; InTech: London, UK, 2016. [Google Scholar]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Ravingerová, T.; Kindernay, L.; Barteková, M.; Ferko, M.; Adameová, A.; Zohdi, V.; Bernátová, I.; Ferenczyová, K.; Lazou, A. The molecular mechanisms of iron metabolism and its role in cardiac dysfunction and cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889. [Google Scholar] [CrossRef]
- Rodrigo, R.; Prieto, J.C.; Castillo, R. Cardioprotection against ischaemia/reperfusion by vitamins C and E plus n-3 fatty acids: Molecular mechanisms and potential clinic applications. Clin. Sci. 2013, 124, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Libuy, M.; Feliú, F.; Hasson, D. Molecular basis of cardioprotective effect of antioxidant vitamins in myocardial infarction. BioMed Res. Int. 2013, 2013, 437613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparetto, C.; Malinverno, A.; Culacciati, D.; Gritti, D.; Prosperini, P.G.; Specchia, G.; Ricevuti, G. Antioxidant vitamins reduce oxidative stress and ventricular remodeling in patients with acute myocardial infarction. Int. J. Immunopathol. Pharmacol. 2005, 18, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Venardos, K.; Kaye, D. Myocardial Ischemia-Reperfusion Injury, Antioxidant Enzyme Systems, and Selenium: A Review. Curr. Med. Chem. 2007, 14, 1539–1549. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, Z.; Yao, J.; Zhao, G.; Fa, X.; Niu, J. Participation of protein kinase C in the activation of Nrf2 signaling by ischemic preconditioning in the isolated rabbit heart. Mol. Cell. Biochem. 2013, 372, 169–179. [Google Scholar] [CrossRef]
- Zhu, H.; Jia, Z.; Misra, B.R.; Zhang, L.; Cao, Z.; Yamamoto, M.; Trush, M.A.; Misra, H.P.; Li, Y. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc. Toxicol. 2008, 8, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar] [CrossRef]
- Neely, J.R.; Grotyohann, L.W. Role of glycolytic products in damage to ischemic myocardium. Dissociation of adenosine triphosphate levels and recovery of function of reperfused ischemic hearts. Circ. Res. 1984, 55, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, G.; Weisfeldt, M.L.; Jacobus, W.E.; Flaherty, J.T.; Hopkins, J. Evidence for a reversible oxygen radical-mediated component of reperfusion injury: Reduction by recombinant human superoxide dismutase administered at the time of reflow EXPERIMENTAL and clinical studies have indicated From the Department of Medicine, Div. Lab. Investig. Myocard. Reperfus. Circ. 1987, 75, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Avkiran, M.; Marber, M.S. Na+/H+ exchange inhibitors for cardioprotective therapy: Progress, problems and prospects. J. Am. Coll. Cardiol. 2002, 39, 747–753. [Google Scholar] [CrossRef]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic reticulum: The dynamic calcium governor of muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Joh, T. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. J. Clin. Biochem. Nutr. 2007, 40, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabo, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [CrossRef]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef] [Green Version]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D.Y. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 2008, 294, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Braunersreuther, V.; Montecucco, F.; Ashri, M.; Pelli, G.; Galan, K.; Frias, M.; Burger, F.; Quinderé, A.L.G.; Montessuit, C.; Krause, K.H.; et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Duilio, C.; Ambrosio, G.; Kuppusamy, P.; Dipaula, A.; Becker, L.C.; Zweier, J.L. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am. J. Physiol. Hear. Circ. Physiol. 2001, 280, 2649–2657. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Vivar, J.; Martasek, P.; Whitsett, J.; Joseph, J.; Kalyanaraman, B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: An EPR spin trapping study. Biochem. J. 2002, 362, 733–739. [Google Scholar] [CrossRef]
- Eddy, L.J.; Stewart, J.R.; Jones, H.P.; Engerson, T.D.; McCord, J.M.; Downey, J.M. Free radical-producing enzyme, xanthine oxidase, is undetectable in human hearts. Am. J. Physiol. Hear. Circ. Physiol. 1987, 253, 709–711. [Google Scholar] [CrossRef]
- Della Corte, E.; Gozzetti, G.; Novello, F.; Stirpe, F. Properties of the xanthine oxidase from human liver. BBA Enzymol. 1969, 191, 164–166. [Google Scholar] [CrossRef]
- Kim, J.S.; Jin, Y.; Lemasters, J.J. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Hear. Circ. Physiol. 2006, 290, 2024–2034. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.J.; Halestrapt, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 2003, 35, 339–341. [Google Scholar] [CrossRef] [Green Version]
- Kitakaze, M.; Takashima, S.; Funaya, H.; Minamino, T.; Node, K.; Shinozaki, Y.; Mori, H.; Hori, M. Temporary acidosis during reperfusion limits myocardial infarct size in dogs. Am. J. Physiol. Hear. Circ. Physiol. 1997, 272. [Google Scholar] [CrossRef]
- Javadov, S.; Purdham, D.M.; Zeidan, A.; Karmazyn, M. NHE-1 inhibition improves cardiac mitochondrial function through regulation of mitochondrial biogenesis during postinfarction remodeling. Am. J. Physiol. Circ. Physiol. 2006, 291, H1722–H1730. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Molkentin, J.D. Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. J. Mol. Cell. Cardiol. 2009, 46, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S. A Novel Conceptual Model for the Dual Role of FOF1-ATP Synthase in Cell Life and Cell Death. Biomol. Concepts 2020, 11, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Paterek, A.; Mackiewicz, U.; Mączewski, M. Iron and the heart: A paradigm shift from systemic to cardiomyocyte abnormalities. J. Cell. Physiol. 2019, 234, 21613–21629. [Google Scholar] [CrossRef]
- Gordan, R.; Wongjaikam, S.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.C.; Xie, L.H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: An update. Heart Fail. Rev. 2018, 23, 801–816. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [Green Version]
- Ghafourian, K.; Shapiro, J.S.; Goodman, L.; Ardehali, H. Iron and Heart Failure: Diagnosis, Therapies, and Future Directions. JACC Basic Transl. Sci. 2020, 5, 300–313. [Google Scholar] [CrossRef]
- Kumfu, S.; Chattipakorn, S.; Chinda, K.; Fucharoen, S.; Chattipakorn, N. T-type calcium channel blockade improves survival and cardiovascular function in thalassemic mice. Eur. J. Haematol. 2012, 88, 535–548. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Trivieri, M.G.; Khaper, N.; Liu, P.P.; Backx, P.H. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J. Mol. Med. 2006, 84, 349–364. [Google Scholar] [CrossRef]
- Carrick, D.; Haig, C.; Ahmed, N.; McEntegart, M.; Petrie, M.C.; Eteiba, H.; Hood, S.; Watkins, S.; Lindsay, M.M.; Davie, A.; et al. Myocardial hemorrhage after acute reperfused ST-segment-elevation myocardial infarction: Relation to microvascular obstruction and prognostic significance. Circ. Cardiovasc. Imaging 2016, 9, e004148. [Google Scholar] [CrossRef] [Green Version]
- Bulluck, H.; Rosmini, S.; Abdel-Gadir, A.; White, S.K.; Bhuva, A.N.; Treibel, T.A.; Fontana, M.; Ramlall, M.; Hamarneh, A.; Sirker, A.; et al. Residual Myocardial Iron Following Intramyocardial Hemorrhage during the Convalescent Phase of Reperfused ST-Segment-Elevation Myocardial Infarction and Adverse Left Ventricular Remodeling. Circ. Cardiovasc. Imaging 2016, 9, e004940. [Google Scholar] [CrossRef] [Green Version]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [Green Version]
- Berenshtein, E.; Vaisman, B.; Goldberg-Langerman, C.; Kitrossky, N.; Konijn, A.M.; Chevion, M. Roles of ferritin and iron in ischemic preconditioning of the heart. Mol. Cell. Biochem. 2002, 234–235, 283–292. [Google Scholar] [CrossRef]
- Chevion, M.; Jiang, Y.; Har-El, R.; Berenshtein, E.; Uretzky, G.; Kitrossky, N. Copper and iron are mobilized following myocardial ischemia: Possible predictive criteria for tissue injury. Proc. Natl. Acad. Sci. USA 1993, 90, 1102–1106. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.H.; Lightfoot, F.G.; Weglicki, W.B. Cardiac tissue iron: Effects on post-ischemic function and free radical production, and its possible role during preconditioning. Cell. Mol. Biol. 2000, 46, 1313–1327. [Google Scholar] [PubMed]
- Vernis, L.; El Banna, N.; Baïlle, D.; Hatem, E.; Heneman, A.; Huang, M.E. Fe-S Clusters Emerging as Targets of Therapeutic Drugs. Oxid. Med. Cell. Longev. 2017, 2017, 3647657. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Chung, S. Genetic Analysis of Aldose Reductase in Diabetic Complications. Curr. Med. Chem. 2005, 10, 1375–1387. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Bakr, S.; Ellery, C.A.; Oates, P.J.; Ramasamy, R. Sorbitol dehydrogenase: A novel target for adjunctive protection of ischemic myocardium. FASEB J. 2003, 17, 2331–2333. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wu, S.; Wong, T.M.; Chung, S.K.; Chung, S.S.M. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic. Biol. Med. 2008, 45, 602–610. [Google Scholar] [CrossRef]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef]
- Gale, C.P.; Metcalfe, E.; West, R.M.; Das, R.; Kilcullen, N.; Morrell, C.; Crook, R.; Batin, P.D.; Hall, A.S.; Barth, J.H. An Assessment of the concentration-related prognostic value of cardiac troponin i following acute coronary syndrome. Am. J. Cardiol. 2011, 108, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5896. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Pedro, J.; Angeli, F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; et al. Oxidized Arachidonic/Adrenic Phosphatidylethanolamines Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St. Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [Green Version]
- Sparvero, L.J.; Tian, H.; Amoscato, A.A.; Sun, W.-Y.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Kapralov, O.; Javadov, S.; He, R.-R.; Watkins, S.C.; et al. Direct Mapping of Phospholipid Ferroptotic Death Signals in Cells and Tissues by Gas Cluster Ion Beam Secondary Ion Mass Spec-trometry (GCIB-SIMS). Angew. Chemie Int. 2021. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Baba, Y.; Higa, J.K.; Shimada, B.K.; Horiuchi, K.M.; Suhara, T.; Kobayashi, M.; Woo, J.D.; Aoyagi, H.; Marh, K.S.; Kitaoka, H.; et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Hear. Circ. Physiol. 2018, 314, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Suhara, T.; Baba, Y.; Kawasaki, N.K.; Higa, J.K.; Matsui, T. Pathological Roles of Iron in Cardiovascular Disease. Curr. Drug Targets 2018, 19, 1068–1076. [Google Scholar] [CrossRef]
- Alu, A.; Han, X.; Ma, X.; Wu, M.; Wei, Y.; Wei, X. The role of lysosome in regulated necrosis. Acta Pharm. Sin. B 2020, 10, 1880–1903. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Stamenkovic, A.; O’Hara, K.A.; Nelson, D.C.; Maddaford, T.G.; Edel, A.L.; Maddaford, G.; Dibrov, E.; Aghanoori, M.; Kirshenbaum, L.A.; Fernyhough, P.; et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia/reperfusion injury. Am. J. Physiol. Circ. Physiol. 2021. [Google Scholar] [CrossRef]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. 2012, 17, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-Y.; Xiao, Z.-Z.; Ling, X.; Xu, R.-N.; Zhu, P.; Zheng, S.-Y. ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol. Med. 2021, 27, 1–14. [Google Scholar] [CrossRef]
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. BioMed Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef] [Green Version]
- Hannenhalli, S.; Kaestner, K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, M.; Land, W.G.; Tonnus, W.; Hugo, C.P.; Linkermann, A. Origin and consequences of necroinflammation. Physiol. Rev. 2018, 98, 727–780. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Ichim, G.; Green, D.R. Die another way-non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [Green Version]
- Proneth, B.; Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019, 26, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Martín-Fernández, B.; Gredilla, R. Mitochondria and oxidative stress in heart aging. Age 2016, 38, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol. Res. 2021, 166, 105466. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Škárka, L.; Ošťádal, B. Mitochondrial membrane potential in cardiac myocytes. Physiol. Res. 2002, 51, 425–434. [Google Scholar]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Van Remmen, H.; Frohlich, V.; Lechleiter, J.; Richardson, A.; Ran, Q. Gpx4 protects mitochondrial ATP generation against oxidative damage. Biochem. Biophys. Res. Commun. 2007, 356, 893–898. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [Green Version]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020, 22, 1042–1048. [Google Scholar] [CrossRef]
- Riegman, M.; Bradbury, M.S.; Overholtzer, M. Population Dynamics in Cell Death: Mechanisms of Propagation. Trends Cancer 2019, 5, 558–568. [Google Scholar] [CrossRef]
- Tang, L.J.; Luo, X.J.; Tu, H.; Chen, H.; Xiong, X.M.; Li, N.S.; Peng, J. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn. Schmiedebergs. Arch. Pharmacol. 2021, 394, 401–410. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef]
- Chang, H.; Wu, R.; Shang, M.; Sato, T.; Chen, C.; Shapiro, J.S.; Liu, T.; Thakur, A.; Sawicki, K.T.; Prasad, S.V.; et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 2016, 8, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Jian, Y.; Lesnefsky, E.J. Incremental iron overload during reperfusion progressively augments oxidative injury. Chin. Med. J. Engl. 1996, 109, 450–458. [Google Scholar]
- Heger, M.; Reiniers, M.J.; Van Golen, R.F. Mitochondrial metabolomics unravel the primordial trigger of ischemia/reperfusion injury. Gastroenterology 2015, 148, 1071–1073. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.; Evelson, P.; Gelpi, R.J. Protecting the heart from ischemia/reperfusion injury: An update on remote ischemic preconditioning and postconditioning. Curr. Opin. Cardiol. 2017, 32, 784–790. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Autoxidation of Biological Molecules. 1. The Antioxidant Activity of Vitamin E and Related Chain-Breaking Phenolic Antioxidants in Vitro. J. Am. Chem. Soc. 1981, 103, 6472–6477. [Google Scholar] [CrossRef]
- Huang, J.; Weinstein, S.J.; Yu, K.; Männistö, S.; Albanes, D. Relationship between serum alpha-tocopherol and overall and cause-specific mortality a 30-year prospective cohort analysis. Circ. Res. 2019, 125, 29–40. [Google Scholar] [CrossRef]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. α-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef]
- Saleh, N.K.; Saleh, H.A. Protective effects of vitamin E against myocardial ischemia/reperfusion injury in rats. Saudi Med. J. 2010, 31, 142–147. [Google Scholar]
- Tripathi, Y.; Hegde, B.M. Effect of α-tocopherol pretreatment on infarct size following of 90 minutes ischemia and 4 hours of reperfusion in dogs. Indian J. Physiol. Pharmacol. 1997, 41, 241–247. [Google Scholar] [PubMed]
- Hu, X.X.; Fu, L.; Li, Y.; Lin, Z.B.; Liu, X.; Wang, J.F.; Chen, Y.X.; Wang, Z.P.; Zhang, X.; Ou, Z.J.; et al. The cardioprotective effect of Vitamin E (alpha-tocopherol) is strongly related to age and gender in mice. PLoS ONE 2015, 10, e0137405. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The pharmacokinetics of vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.L.; Paris, H.L.; Beals, J.W.; Binns, S.E.; Giordano, G.R.; Scalzo, R.L.; Schweder, M.M.; Blair, E.; Bell, C. Liposomal-encapsulated Ascorbic Acid: Influence on Vitamin C Bioavailability and Capacity to Protect against Ischemia–Reperfusion Injury. Nutr. Metab. Insights 2016, 9, NMI.S39764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Li, H.; Liang, L.; Jin, T.; Zhang, G.; Bradley, J.L.; Peberdy, M.A.; Ornato, J.P.; Wijesinghe, D.S.; Tang, W. Effects of ω-3 PUFA and ascorbic acid combination on post-resuscitation myocardial function. Biomed. Pharmacother. 2021, 133. [Google Scholar] [CrossRef] [PubMed]
- Siscovick, D.S.; Barringer, T.A.; Fretts, A.M.; Wu, J.H.Y.; Lichtenstein, A.H.; Costello, R.B.; Kris-Etherton, P.M.; Jacobson, T.A.; Engler, M.B.; Alger, H.M.; et al. Omega-3 Polyunsaturated Fatty Acid (Fish Oil) Supplementation and the Prevention of Clinical Cardiovascular Disease: A Science Advisory from the American Heart Association. Circulation 2017, 135, 867–884. [Google Scholar] [CrossRef]
- Farías, J.G.; Carrasco-Pozo, C.; Carrasco Loza, R.; Sepúlveda, N.; Álvarez, P.; Quezada, M.; Quiñones, J.; Molina, V.; Castillo, R.L. Polyunsaturated fatty acid induces cardioprotection against ischemia-reperfusion through the inhibition of NF-kappaB and induction of Nrf2. Exp. Biol. Med. 2017, 242, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.W.; Tong, J.; Yan, Y.S.; Chen, Q.Q.; Zhao, X.P. ω-3 polyunsaturated fatty acid postconditioning protects the isolated perfused rat heart from ischemia-reperfusion injury. CardioRenal Med. 2018, 8, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, J.; Anton, A.W. Deferoxamine. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557654/ (accessed on 18 August 2020).
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Kusakari, Y.; Xiao, C.-Y.; Inouye, B.T.; Takahashi, M.; Scherrer-Crosbie, M.; Rosenzweig, A.; Hara, K.; Matsui, T. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Hear. Circ. Physiol. 2012, 303, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, T.; Higa, J.K.; Aoyagi, H.; Yorichika, N.; Shimada, B.K.; Matsui, T. Cardiac mTOR rescues the detrimental effects of diet-induced obesity in the heart after ischemia-reperfusion. Am. J. Physiol. Hear. Circ. Physiol. 2015, 308, H1530–H1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayeva, M.; Khechaduri, A.; Puig, S.; Chang, H.-C.; Patial, S.; Blackshear, P.J.; Ardehali, H. mTOR Regulates Cellular Iron Homeostasis through Tristetraprolin. Cell Metab. 2012, 16, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Hamid, T.; Keith, R.J.; Zhou, G.; Partridge, C.R.; Xiang, X.; Kingery, J.R.; Lewis, R.K.; Li, Q.; Rokosh, D.G.; et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 2010, 121, 1912–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satyanarayanan, S.K.; Shih, Y.H.; Chien, Y.C.; Huang, S.Y.; Gałecki, P.; Kasper, S.; Chang, J.P.C.; Su, K.P. Anti-Oxidative Effects of Melatonin Receptor Agonist and Omega-3 Polyunsaturated Fatty Acids in Neuronal SH-SY5Y Cells: Deciphering Synergic Effects on Anti-Depressant Mechanisms. Mol. Neurobiol. 2018, 55, 7271–7284. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, R.C.; Binu, P.; Arathi, P.; Nair, R.H. L-ascorbic acid and α-tocopherol attenuate arsenic trioxide-induced toxicity in H9c2 cardiomyocytes by the activation of Nrf2 and Bcl2 transcription factors. Toxicol. Mech. Methods 2018, 28, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Margison, K.; Pratt, D.A. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem. Biol. 2017, 12, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.Y.; Pang, Y.L.; Li, W.X.; Zhao, C.X.; Zhang, Y.; Wang, X.; Ning, G.Z.; Kong, X.H.; Liu, C.; Yao, X.; et al. Liproxstatin-1 is an effective inhibitor of oligodendrocyte ferroptosis induced by inhibition of glutathione peroxidase 4. Neural Regen. Res. 2021, 16, 561–566. [Google Scholar] [CrossRef]
- Park, E.J.; Park, Y.J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol. Lett. 2019, 303, 55–66. [Google Scholar] [CrossRef]
- Lin, D.; Cui, B.; Ren, J.; Ma, J. Regulation of VDAC1 contributes to the cardioprotective effects of penehyclidine hydrochloride during myocardial ischemia/reperfusion. Exp. Cell Res. 2018, 367, 257–263. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. In Apoptotic and Non-Apoptotic Cell Death; Nagata, S., Nakano, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; Volume 403, pp. 143–170. ISBN 0260-2938. [Google Scholar]
- Wu, M.; Xu, L.G.; Li, X.; Zhai, Z.; Shu, H.B. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 2002, 277, 25617–25623. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Moraes, L.B.; Murakami, A.H.F.; Fontes, B.; Poggetti, R.S.; Van Rooijen, N.; Younes, R.N.; Heimbecker, A.M.C.; Birolini, D. Gut ischemia/reperfusion induced acute lung injury is an alveolar macrophage dependent event. J. Trauma Inj. Infect. Crit. Care 2008, 64, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, J.D.; Kenyon, V.A.; Holman, T.R. Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorganic Med. Chem. 2006, 14, 4295–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Song, X.; Sun, X.; Huang, J.; Zhong, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 2016, 473, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, H.; Yang, Y.; Wang, R.; Wang, Y.; Wu, C.; Du, G. Baicalein administered in the subacute phase ameliorates ischemia-reperfusion-induced brain injury by reducing neuroinflammation and neuronal damage. Biomed. Pharmacother. 2019, 117, 109102. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.M.; Fidan, E.; Yang, Q.; Anthonymuthu, T.S.; New, L.A.; Meyer, E.A.; Wang, H.; Kochanek, P.M.; Dixon, C.E.; Kagan, V.E.; et al. Ferroptosis Contributes to Neuronal Death and Functional Outcome after Traumatic Brain Injury. Crit. Care Med. 2019, 47, 410–418. [Google Scholar] [CrossRef]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Sun, Q.Y.; Zhou, H.H.; Jin, W.L.; Mao, X.Y. Baicalein exerts neuroprotective effects in FeCl3-induced posttraumatic epileptic seizures via suppressing ferroptosis. Front. Pharmacol. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Xun, Z.; Rivera-Sánchez, S.; Ayala-Peña, S.; Lim, J.; Budworth, H.; Skoda, E.M.; Robbins, P.D.; Niedernhofer, L.J.; Wipf, P.; McMurray, C.T. Targeting of XJB-5-131 to Mitochondria Suppresses Oxidative DNA Damage and Motor Decline in a Mouse Model of Huntington’s Disease. Cell Rep. 2012, 2, 1137–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wipf, P.; Xiao, J.; Jiang, J.; Belikova, N.A.; Tyurin, V.A.; Fink, M.P.; Kagan, V.E. Mitochondrial targeting of selective electron scavengers: Synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J. Am. Chem. Soc. 2005, 127, 12460–12461. [Google Scholar] [CrossRef]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent. Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef]
- Escobales, N.; Nuñez, R.E.; Jang, S.; Parodi-Rullan, R.; Ayala-Peña, S.; Sacher, J.R.; Skoda, E.M.; Wipf, P.; Frontera, W.; Javadov, S. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell. Cardiol. 2014, 77, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart during Ischemia-Reperfusion. Antioxidants Redox Signal. 2017, 27, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Bebber, C.M.; Müller, F.; Clemente, L.P.; Weber, J.; von Karstedt, S. Ferroptosis in cancer cell biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lillo-Moya, J.; Rojas-Solé, C.; Muñoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants 2021, 10, 667. https://doi.org/10.3390/antiox10050667
Lillo-Moya J, Rojas-Solé C, Muñoz-Salamanca D, Panieri E, Saso L, Rodrigo R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants. 2021; 10(5):667. https://doi.org/10.3390/antiox10050667
Chicago/Turabian StyleLillo-Moya, José, Catalina Rojas-Solé, Diego Muñoz-Salamanca, Emiliano Panieri, Luciano Saso, and Ramón Rodrigo. 2021. "Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury" Antioxidants 10, no. 5: 667. https://doi.org/10.3390/antiox10050667
APA StyleLillo-Moya, J., Rojas-Solé, C., Muñoz-Salamanca, D., Panieri, E., Saso, L., & Rodrigo, R. (2021). Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants, 10(5), 667. https://doi.org/10.3390/antiox10050667