
Reactive Oxygen Species-Induced TRPM2-Mediated Ca2+ Signalling in Endothelial Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
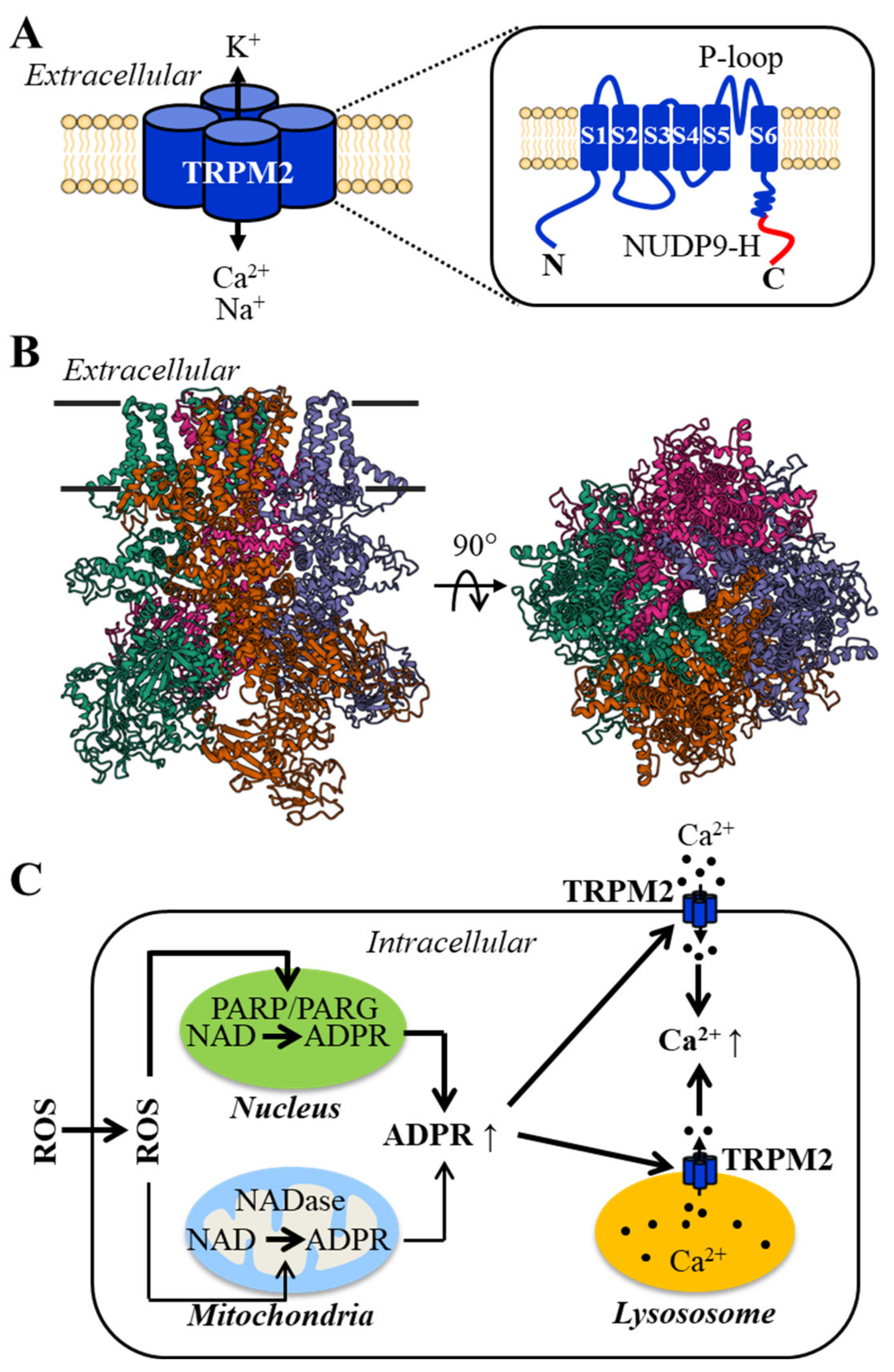
2. The Structural and Functional Properties of the TRPM2 Channel and Its Activation Mechanisms
3. TRPM2 Channel Expression in Endothelial Cells and Its Role in ROS-Induced Ca2+ Signaling
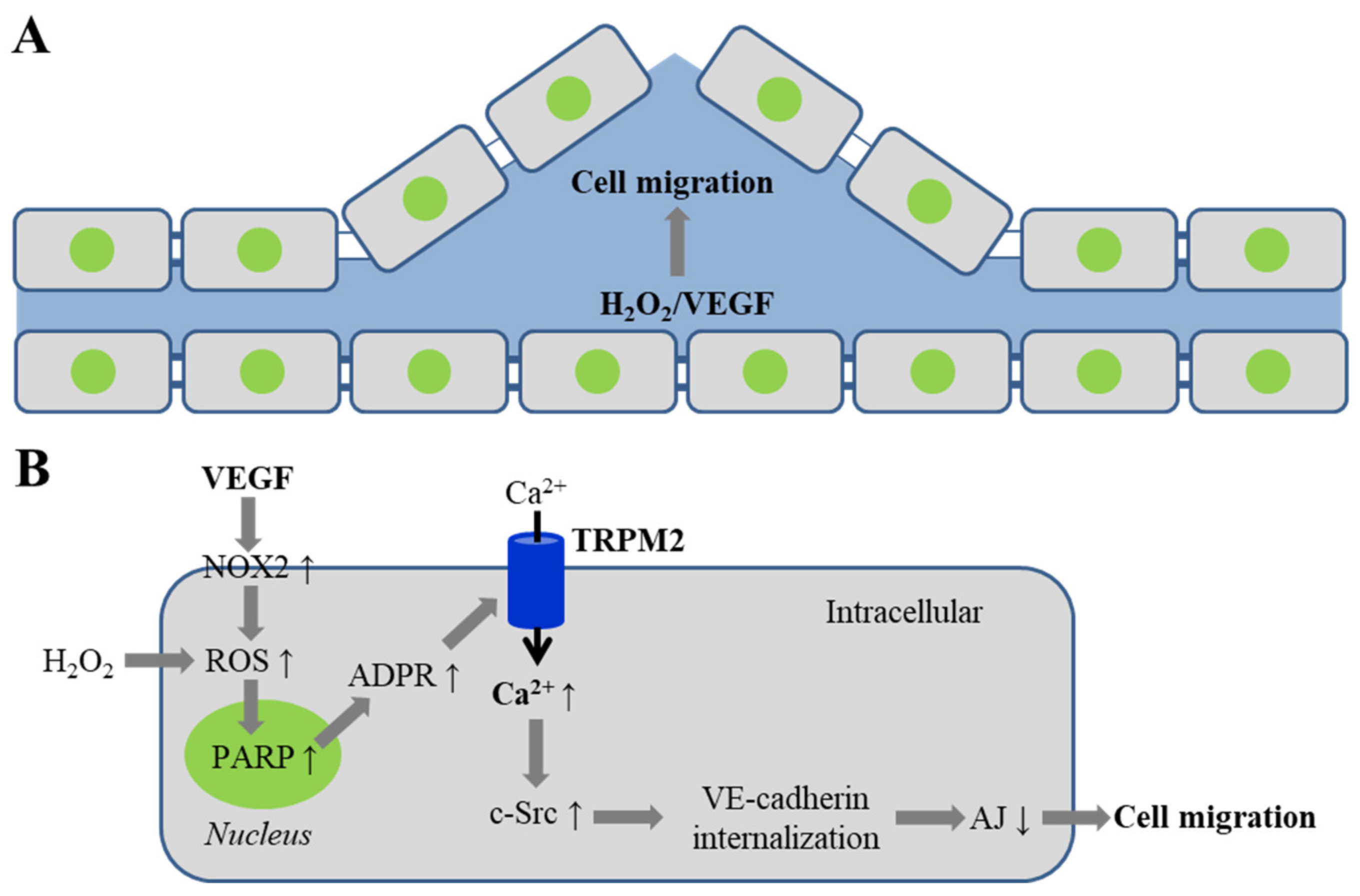
4. TRPM2 Channel in VEGF-Induced Angiogenesis and Post-Ischemic Neovascularization
5. TRPM2 Channel in Endothelial Dysfunction by Oxidative Stress-Inducing Pathological Factors
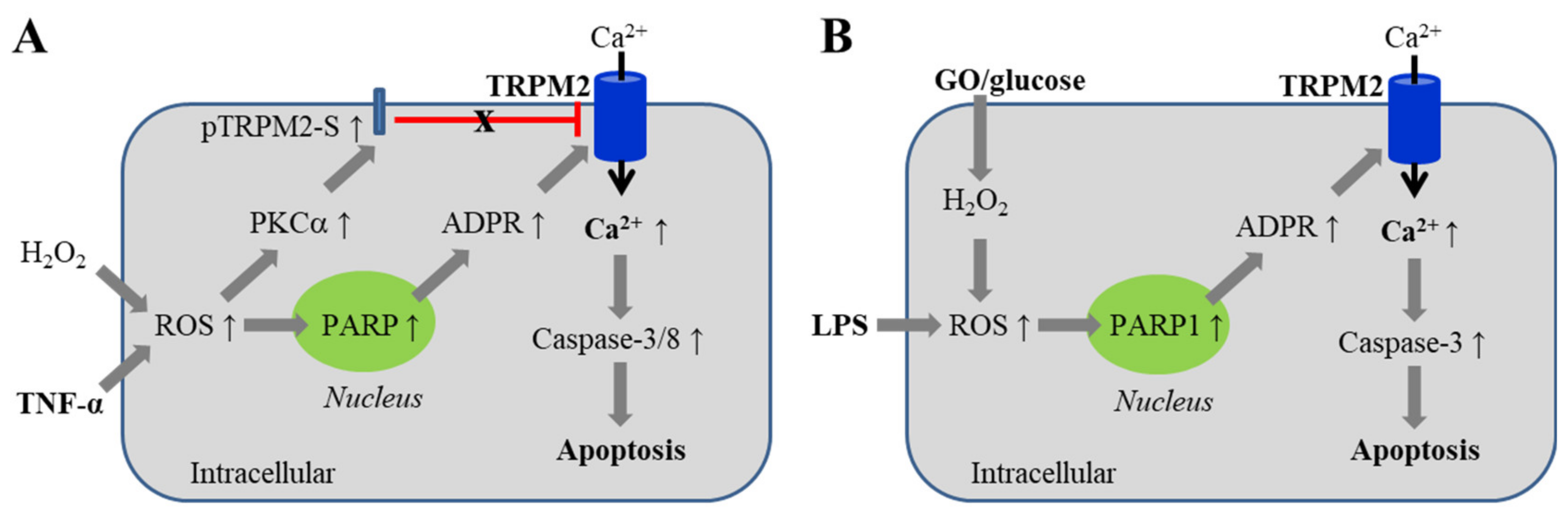
5.1. Endothelial Barrier Dysfunction via Inducing Cell Death
5.2. Endothelial Barrier Dysfunction via Disrupting Inter-Endothelial Junctions
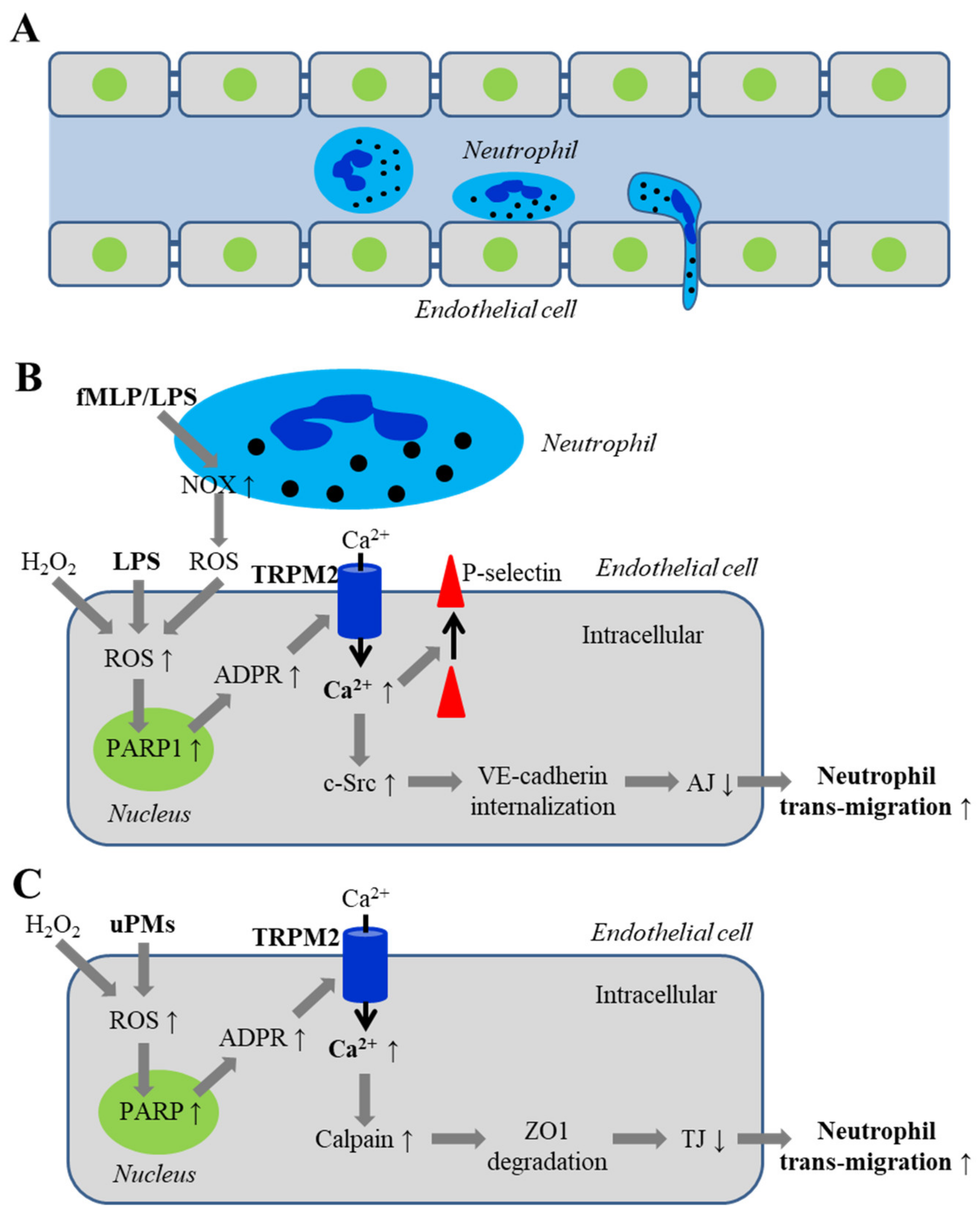
5.2.1. Disruption of Adherens Junctions by ROS and Oxidative Stress Associated with Infection
5.2.2. Disruption of Inter-Endothelial Tight Junctions Induced by Airborne Fine Particulate Matters
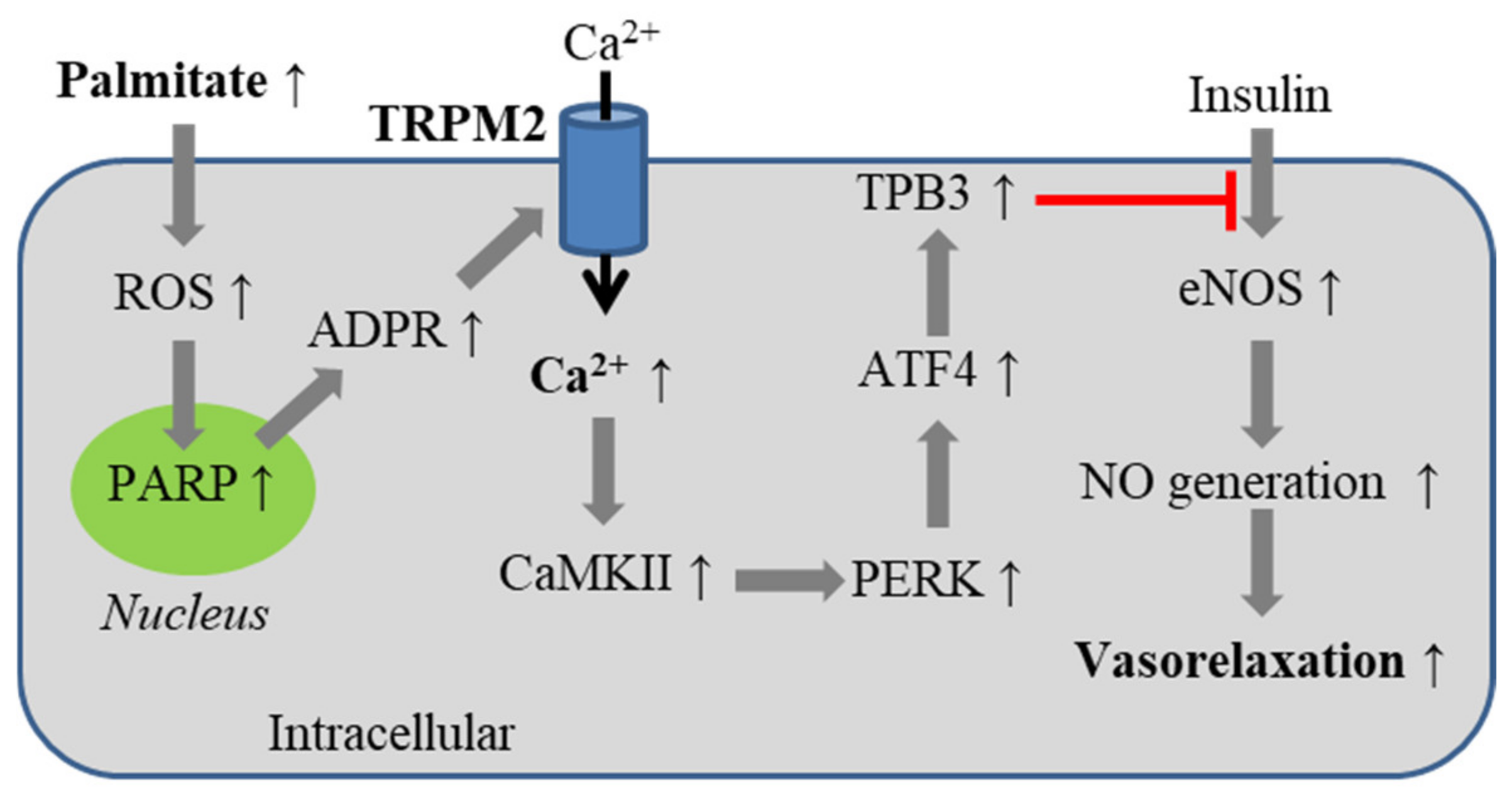
5.3. Obesity-Associated Endothelial Insulin Resistance
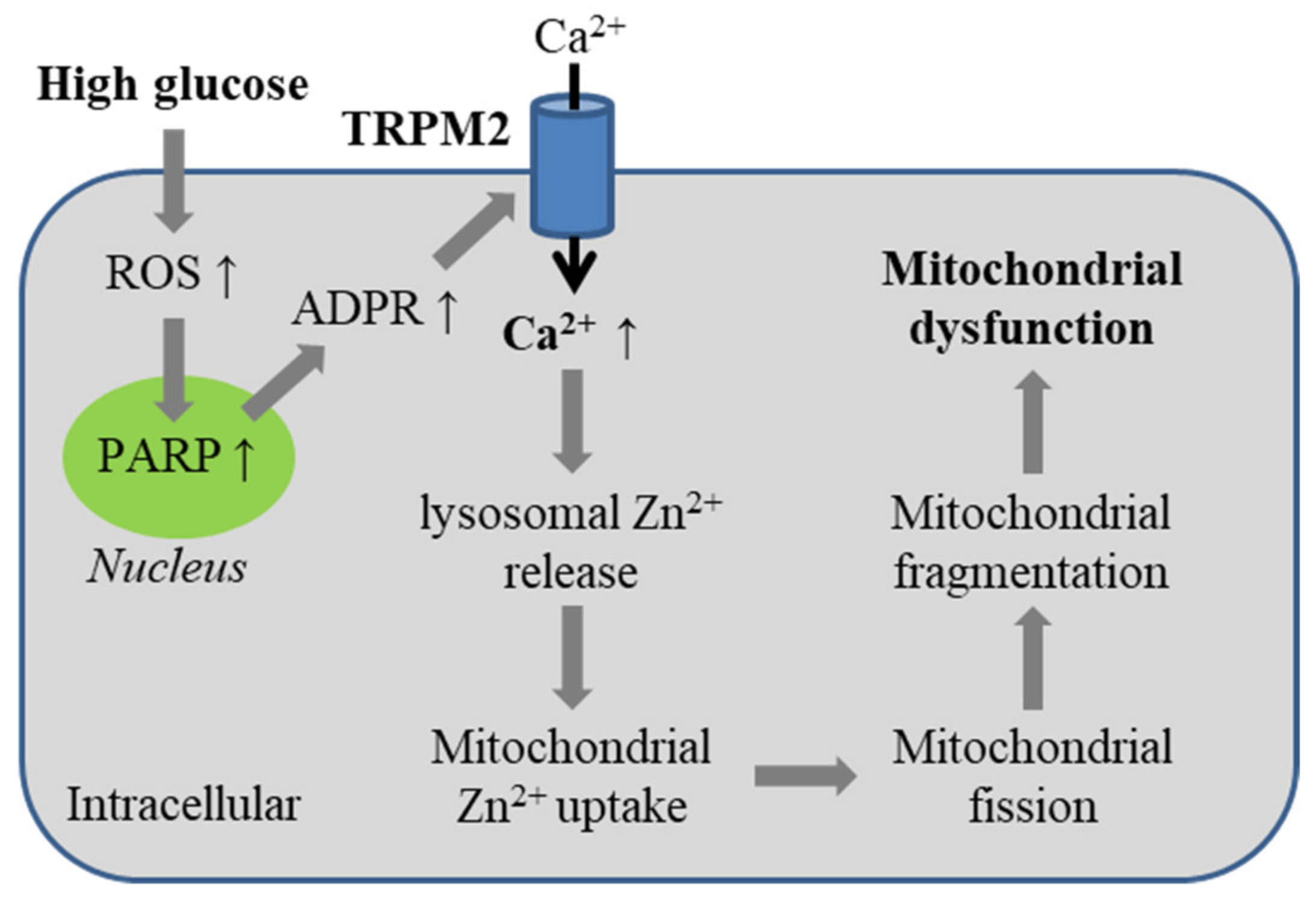
5.4. Diabetes-Related Oxidative Stress-Induced Alteration of Mitochondrial Dynamics
6. Conclusions, New Questions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nat. Cell Biol. 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Rafii, S.; Butler, J.M.; Ding, B.-S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Iv, C.D.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020, 37, 101696. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Cross-talk between NADPH oxidase and mitochondria: Role in ROS signaling and angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef]
- Deshpande, S.S.; Angkeow, P.; Huang, J.; Ozaki, M.; Irani, K. Rac1 inhibits TNF-α-induced endothelial cell apoptosis: Dual regulation by reactive oxygen species. FASEB J. 2000, 14, 1705–1714. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Talukder, M.A.H.; Gao, F. Oxidative stress and microvessel barrier dysfunction. Front. Physiol. 2020, 11, 472. [Google Scholar] [CrossRef]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Papaharalambus, C.A.; Griendling, K.K. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc. Med. 2007, 17, 48–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förstermann, U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pr. Neurol. 2008, 5, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kalogeris, T.; Korthuis, R.J. Reactive species-induced microvascular dysfunction in ischemia/reperfusion. Free. Radic. Biol. Med. 2019, 135, 182–197. [Google Scholar] [CrossRef]
- Zhou, H.; Toan, S. Pathological roles of mitochondrial oxidative stress and mitochondrial dynamics in cardiac microvascular Ischemia/Reperfusion Injury. Biomolecules 2020, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Touyz, R.M. Redox signaling in hypertension. Cardiovasc. Res. 2006, 71, 247–258. [Google Scholar] [CrossRef]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Hear. Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef]
- Praticò, D. Antioxidants and endothelium protection. Atherosclerosis 2005, 181, 215–224. [Google Scholar] [CrossRef]
- Farías, J.G.; Molina, V.M.; Carrasco, R.A.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant therapeutic strategies for cardiovascular conditions associated with oxidative stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef]
- Sorriento, D.; De Luca, N.; Trimarco, B.; Iaccarino, G. The antioxidant therapy: New insights in the treatment of hypertension. Front. Physiol. 2018, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C. The Neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, K.; Wang, G.; Park, L. Endothelial dysfunction and amyloid-beta-induced neurovascular alterations. Cell. Mol. Neurobiol. 2016, 36, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic brain injury and Alzheimer’s Disease: The cerebrovascular link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Solis, E.; Hascup, K.N.; Hascup, E.R. Alzheimer’s Disease: The link between amyloid-beta and neurovascular dysfunction. J. Alzheimer’s Dis. 2020, 76, 1179–1198. [Google Scholar] [CrossRef] [PubMed]
- Underwood, E. The polluted brain. Science 2017, 355, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Kelly, F.J.; Fussell, J.C. Role of oxidative stress in cardiovascular disease outcomes following exposure to ambient air pollution. Free. Radic. Biol. Med. 2017, 110, 345–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangwar, R.S.; Bevan, G.H.; Palanivel, R.; Das, L.; Rajagopalan, S. Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol. 2020, 34, 101545. [Google Scholar] [CrossRef]
- Wang, L.; Wei, L.Y.; Ding, R.; Feng, Y.; Li, D.; Li, C.; Malko, P.; Mortadza, S.A.S.; Wu, W.; Yin, Y.; et al. Predisposition to Alzheimer’s and age-related brain pathologies by PM2.5 exposure: Perspective on the roles of oxidative stress and TRPM2 channel. Front. Physiol. 2020, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B. Ca2+ microdomains near plasma membrane Ca2+ channels: Impact on cell function. J. Physiol. 2008, 586, 3043–3054. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Di, A.; Mehta, D.; Malik, A.B. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium 2016, 60, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Bertero, E.; Maack, C. Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Feno, S.; Butera, G.; Reane, D.V.; Rizzuto, R.; Raffaello, A. Crosstalk between calcium and ROS in pathophysiological conditions. Oxidative Med. Cell. Longev. 2019, 2019, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, T.M.; Murphy, E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Jiang, L.-H.; Yang, W.; Zou, J.; Beech, D.J. TRPM2 channel properties, functions and therapeutic potentials. Expert Opin. Ther. Targets 2010, 14, 973–988. [Google Scholar] [CrossRef]
- Sumoza-Toledo, A.; Penner, R. TRPM2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.; Li, Y.; Perraud, A.-L. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res. 2013, 55, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Ru, X.; Yao, X. TRPM2: A multifunctional ion channel for oxidative stress sensing. Acta Physiol. Sin. 2014, 66, 7–15. [Google Scholar]
- Uchida, K.; Tominaga, M. The role of TRPM2 in pancreatic beta-cells and the development of diabetes. Cell Calcium 2014, 56, 332–339. [Google Scholar] [CrossRef]
- Mortadza, S.A.S.; Wang, L.; Li, D.; Jiang, L.-H. TRPM2 channel-mediated ROS-sensitive Ca2+ signaling mechanisms in immune cells. Front. Immunol. 2015, 6, 407. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.A.; Cheung, J.Y. TRPM2 protects against tissue damage following oxidative stress and ischaemia-reperfusion. J. Physiol. 2016, 594, 4181–4191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Gao, Y.; Bao, X.; Li, F.; Yao, W.; Feng, Z.; Yin, Y. TRPM2: A potential drug target to retard oxidative stress. Front. Biosci. 2017, 22, 1427–1438. [Google Scholar]
- Belrose, J.C.; Jackson, M.F. TRPM2: A candidate therapeutic target for treating neurological diseases. Acta Pharmacol. Sin. 2018, 39, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Turlova, E.; Feng, Z.-P.; Sun, H.-S. The role of TRPM2 channels in neurons, glial cells and the blood-brain barrier in cerebral ischemia and hypoxia. Acta Pharmacol. Sin. 2018, 39, 713–721. [Google Scholar] [CrossRef]
- Jiang, L.-H.; Li, X.; Mortadza, S.A.S.; Lovatt, M.; Yang, W. The TRPM2 channel nexus from oxidative damage to Alzheimer’s pathologies: An emerging novel intervention target for age-related dementia. Ageing Res. Rev. 2018, 47, 67–79. [Google Scholar] [CrossRef]
- Sita, G.; Hrelia, P.; Graziosi, A.; Ravegnini, G.; Morroni, F. TRPM2 in the brain: Role in health and disease. Cells 2018, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Cho, P.S.; Yang, Y.D.; Hwang, S.W. Nociceptive roles of TRPM2 ion channel in pathologic pain. Mol. Neurobiol. 2018, 55, 6589–6600. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Mortadza, S.A.S.; McWilliam, J.; Jiang, L.-H. TRPM2 channel in microglia as a new player in neuroinflammation associated with a spectrum of central nervous system pathologies. Front. Pharmacol. 2019, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.; Mankoo, H.; Wei, L.; An, X.; Li, C.; Li, D.; Jiang, L. TRPM2 channel: A novel target for alleviating ischaemia-reperfusion, chronic cerebral hypo-perfusion and neonatal hypoxic-ischaemic brain damage. J. Cell. Mol. Med. 2020, 24, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Malko, P.; Jiang, L.-H. TRPM2 channel-mediated cell death: An important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions. Redox Biol. 2020, 37, 101755. [Google Scholar] [CrossRef]
- Nagamine, K.; Kudoh, J.; Minoshima, S.; Kawasaki, K.; Asakawa, S.; Ito, F.; Shimizu, N. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 1998, 54, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Harteneck, C.P.T.; Schultz, G. From worm to man: Three subfamilies of TRP channels. Trends Neurosci. 2000, 23, 159–166. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nat. Cell Biol. 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.-L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nat. Cell Biol. 2001, 411, 595–599. [Google Scholar] [CrossRef]
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ influx system mediated by LTRPC. Science 2001, 293, 1327–1330. [Google Scholar] [CrossRef]
- Du, J.; Xie, J.; Yue, L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. USA 2009, 106, 7239–7244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Roth, B.; Lü, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife 2019, 8, 50175. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, eaav4809. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Wang, L.; Fu, T.; Wu, H.; Hao, W. Mechanism of TRPM 2 channel gating revealed by cryo-EM. FEBS J. 2019, 286, 3333–3339. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85, 102111. [Google Scholar] [CrossRef]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef]
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct gating of the TRPM2 channel by cADPR via specific interactions with the ADPR binding pocket. Cell Rep. 2019, 27, 3684–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegert, R.; Riekehr, W.M.; Guse, A.H. Does cyclic ADP-Ribose (cADPR) activate the non-selective cation channel TRPM2? Front. Immunol. 2020, 11, 2018. [Google Scholar] [CrossRef] [PubMed]
- Fliegert, R.; Gasser, A.; Guse, A. Regulation of calcium signalling by adenine-based second messengers. Biochem. Soc. Trans. 2007, 35, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Wehage, E.; Eisfeld, J.; Heiner, I.; Jüngling, E.; Zitt, C.; Lückhoff, A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. J. Biol. Chem. 2002, 277, 23150–23156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chu, X.; Tong, Q.; Cheung, J.Y.; Conrad, K.; Masker, K.; Miller, B.A. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J. Biol. Chem. 2003, 278, 16222–16229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonfria, E.; Marshall, I.C.B.; Benham, C.D.; Boyfield, I.; Brown, J.D.; Hill, K.; Hughes, J.P.; Skaper, S.D.; McNulty, S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol. 2004, 143, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Marshall, I.C.B.; Boyfield, I.; Skaper, S.D.; Hughes, J.P.; Owen, D.E.; Zhang, W.; Miller, B.A.; Benham, C.D.; McNulty, S.E. Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 2005, 95, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Buelow, B.; Song, Y.; Scharenberg, A.M. The poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 2008, 283, 24571–24583. [Google Scholar] [CrossRef] [Green Version]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 channel in mediating H2O2-Induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Kashio, M.; Sokabe, T.; Shintaku, K.; Uematsu, T.; Fukuta, N.; Kobayashi, N.; Mori, Y.; Tominaga, M. Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. USA 2012, 109, 6745–6750. [Google Scholar] [CrossRef] [Green Version]
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-beta-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.-L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [Green Version]
- Kozai, D.; Ogawa, N.; Mori, Y. Redox regulation of transient receptor potential channels. Antioxid. Redox Signal. 2014, 21, 971–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, N.; Kurokawa, T.; Mori, Y. Sensing of redox status by TRP channels. Cell Calcium 2016, 60, 115–122. [Google Scholar] [CrossRef]
- Yamamoto, S.; Shimizu, S. Significance of TRP channels in oxidative stress. Eur. J. Pharmacol. 2016, 793, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial transient receptor potential channels and vascular remodeling: Extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2020, 10, 1618. [Google Scholar] [CrossRef] [Green Version]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci. Signal. 2009, 2, ra23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef] [Green Version]
- Manna, P.T.; Munsey, T.S.; Abuarab, N.; Li, F.; Asipu, A.; Howell, G.; Sedo, A.; Yang, W.; Naylor, J.; Beech, D.J.; et al. TRPM2-mediated intracellular Zn2+ release triggers pancreatic beta-cell death. Biochem. J. 2015, 466, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.H.; McNaughton, P.A. The TRPM2 ion channel is required for sensitivity to warmth. Nature 2016, 536, 460–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Wang, H.; Kamm, G.B.; Pohle, J.; Reis, F.D.C.; Heppenstall, P.; Wende, H.; Siemens, J. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science 2016, 353, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil activation of endothelial cell-expressed TRPM2 mediates transendothelial neutrophil migration and vascular injury. Circ. Res. 2017, 121, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.-Z.; Mao, H.-J.; Jiang, L.-H. Conserved cysteine residues in the pore region are obligatory for human TRPM2 channel function. Am. J. Physiol. Physiol. 2006, 291, C1022–C1028. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-Z.; Zhong, W.; Watson, N.M.; Dickerson, E.; Wake, J.D.; Lindow, S.W.; Newton, C.J.; Atkin, S. Fluvastatin reduces oxidative damage in human vascular endothelial cells by upregulating Bcl-J. Thromb. Haemost. 2008, 6, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Abuarab, N.; Munsey, T.S.; Jiang, L.-H.; Lin-Hua, J.; Sivaprasadarao, A. High glucose–induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Liu, Y.-L.; Ye, F.; Xie, J.-W.; Zeng, J.-W.; Qin, L.; Xue, J.; Wang, Y.-T.; Guo, K.-M.; Ma, M.-M.; et al. Free fatty acid-induced H2O2 activates TRPM2 to aggravate endothelial insulin resistance via Ca2+-dependent PERK/ATF4/TRB3 cascade in obese mice. Free. Radic. Biol. Med. 2019, 143, 288–299. [Google Scholar] [CrossRef]
- Wang, T.; Wang, L.; Moreno-Vinasco, L.; Lang, G.D.; Siegler, J.H.; Mathew, B.; Usatyuk, P.V.; Samet, J.M.; Geyh, A.S.; Breysse, P.N.; et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 2012, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Yau, H.Y.; Wong, W.Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H2O2-induced cell apoptosis in endothelial cells. PLoS ONE 2012, 7, e43186. [Google Scholar] [CrossRef]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.; Carmeliet, P. Blood-vessel formation: Bridges that guide and unite. Nature 2010, 465, 697–699. [Google Scholar] [CrossRef]
- Udan, R.S.; Culver, J.C.; Dickinson, M.E. Understanding vascular development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Dardik, A. A Murine model of hind limb ischemia to study angiogenesis and arteriogenesis. Methods Mol. Biol. 2018, 1717, 135–143. [Google Scholar] [CrossRef]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tojo, T.; Ushio-Fukai, M.; Yamaoka-Tojo, M.; Ikeda, S.; Patrushev, N.; Alexander, R.W. Role of gp91phox(Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation 2005, 111, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Urao, N.; Inomata, H.; Razvi, M.; Kim, H.W.; Wary, K.; McKinney, R.; Fukai, T.; Ushio-Fukai, M. Role of Nox2-based NADPH oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. Circ. Res. 2008, 103, 212–220. [Google Scholar] [CrossRef]
- Bentley, K.; Franco, C.A.; Philippides, A.; Blanco, R.; Dierkes, M.; Gebala, V.; Stanchi, F.; Jones, M.K.; Aspalter, I.M.; Cagna, G.; et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat. Cell Biol. 2014, 16, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Burridge, K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J. Biol. Chem. 2009, 284, 25602–25611. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.-P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel role of reactive oxygen species–activated trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arter. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; O’Neill, L.A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 2016, 46, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Hecquet, C.M.; Zhang, M.; Mittal, M.; Vogel, S.M.; Di, A.; Gao, X.; Bonini, M.G.; Malik, A.B. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ. Res. 2014, 114, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chen, R.; Sera, F.; Vicedo-Cabrera, A.M.; Guo, Y.; Tong, S.; Coelho, M.S.Z.S.; Saldiva, P.H.N.; Lavigne, E.; Matus, P.; et al. Ambient particulate air pollution and daily mortality in 652 cities. N. Engl. J. Med. 2019, 381, 705–715. [Google Scholar] [CrossRef]
- Heusinkveld, H.J.; Wahle, T.; Campbell, A.; Westerink, R.H.; Tran, L.; Johnston, H.; Stone, V.; Cassee, F.R.; Schins, R.P. Neurodegenerative and neurological disorders by small inhaled particles. Neurotoxicology 2016, 56, 94–106. [Google Scholar] [CrossRef]
- Maher, B.A.; Ahmed, I.A.M.; Karloukovski, V.; MacLaren, D.A.; Foulds, P.G.; Allsop, D.; Mann, D.M.A.; Torres-Jardón, R.; Calderon-Garciduenas, L. Magnetite pollution nanoparticles in the human brain. Proc. Natl. Acad. Sci. USA 2016, 113, 10797–10801. [Google Scholar] [CrossRef] [Green Version]
- Bencsik, A.; Lestaevel, P.; Canu, I.G. Nano- and neurotoxicology: An emerging discipline. Prog. Neurobiol. 2018, 160, 45–63. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, J.; Dong, S.; Cai, X.; Simaiti, A.; Yang, X.; Zhu, X.; Luo, J.; Jiang, L.-H.; Du, B.; et al. Silica nanoparticles induce lung inflammation in mice via ROS/PARP/TRPM2 signaling-mediated lysosome impairment and autophagy dysfunction. Part. Fibre Toxicol. 2020, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.; Michel, L.Y.M.; Balligand, J.-L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, T.A.; Lundberg, J.O.; Weitzberg, E.; Carlström, M. Modulation of mitochondria and NADPH oxidase function by the nitrate-nitrite-NO pathway in metabolic disease with focus on type 2 diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165811. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial dynamics in diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nat. Cell Biol. 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jiang, L.H. Multiple molecular mechanisms form a positive feedback loop driving amyloid beta42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis. 2018, 9, 195. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jiang, L.-H. A critical role of the transient receptor potential melastatin 2 channel in a positive feedback mechanism for reactive oxygen species-induced delayed cell death. J. Cell. Physiol. 2019, 234, 3647–3660. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, R.; Yin, Y.-L.; Jiang, L.-H. Reactive Oxygen Species-Induced TRPM2-Mediated Ca2+ Signalling in Endothelial Cells. Antioxidants 2021, 10, 718. https://doi.org/10.3390/antiox10050718
Ding R, Yin Y-L, Jiang L-H. Reactive Oxygen Species-Induced TRPM2-Mediated Ca2+ Signalling in Endothelial Cells. Antioxidants. 2021; 10(5):718. https://doi.org/10.3390/antiox10050718
Chicago/Turabian StyleDing, Ran, Ya-Ling Yin, and Lin-Hua Jiang. 2021. "Reactive Oxygen Species-Induced TRPM2-Mediated Ca2+ Signalling in Endothelial Cells" Antioxidants 10, no. 5: 718. https://doi.org/10.3390/antiox10050718
APA StyleDing, R., Yin, Y.-L., & Jiang, L.-H. (2021). Reactive Oxygen Species-Induced TRPM2-Mediated Ca2+ Signalling in Endothelial Cells. Antioxidants, 10(5), 718. https://doi.org/10.3390/antiox10050718