
Modulations of Cardiac Functions and Pathogenesis by Reactive Oxygen Species and Natural Antioxidants


Abstract
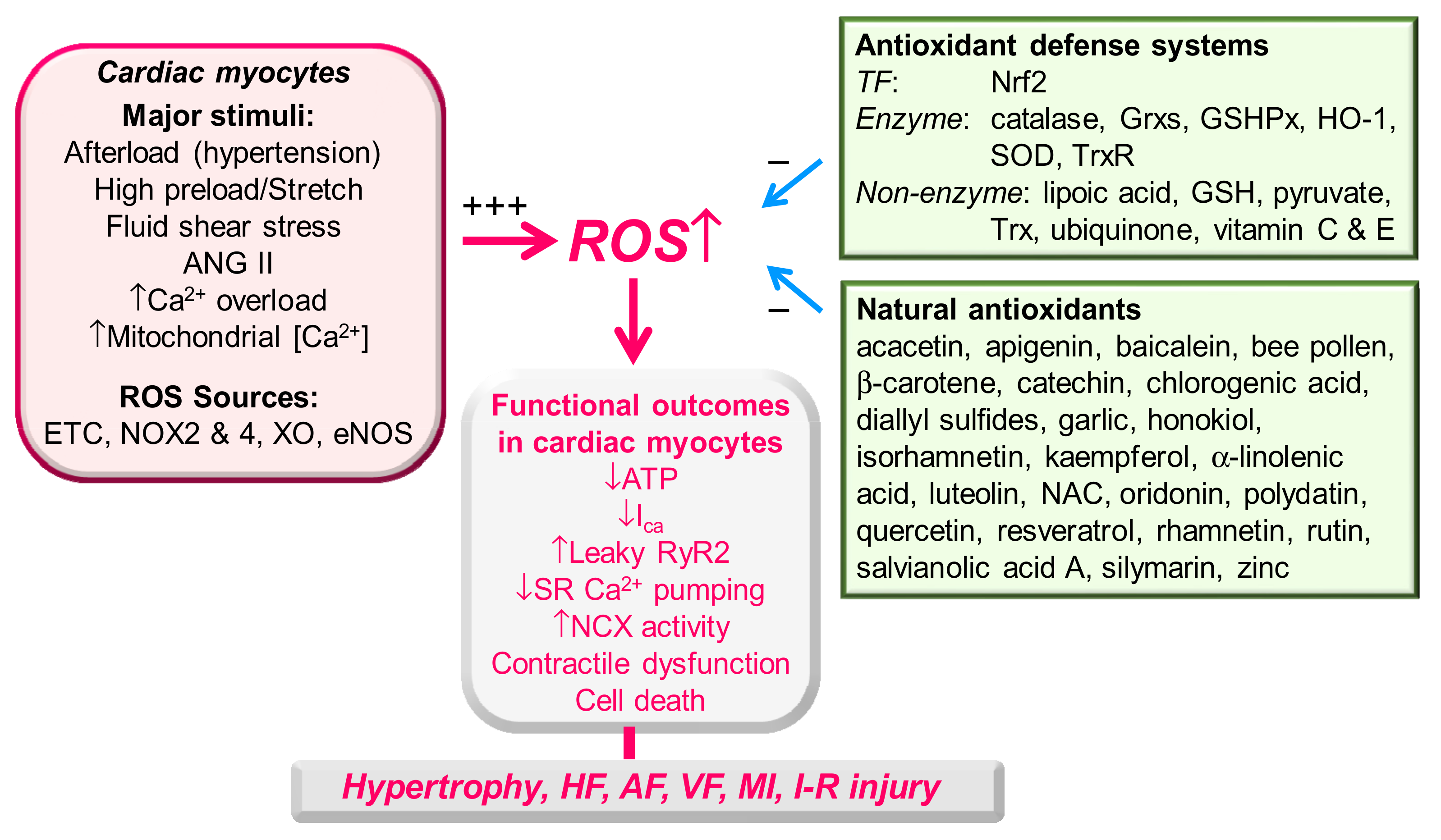
:1. Introduction
2. Oxidative Stress and Endogenous Antioxidants in Cardiac Muscle
2.1. Cardiac Oxidative Stress and Its Role in Ischemic Injury
2.2. Antioxidant Defense Systems
3. Regulation of Cardiac Ca2+ Signaling by Mitochondria and ROS in Health and Disease
3.1. Interaction between Cytosolic Ca2+ Signal and Mitochondria
3.2. Altered Ca2+-Signaling Proteins by ROS and Their Pathological Significance
4. Roles of ROS in Cardiac Mechanical Stress Response and Pathogenesis
5. Exogenous Natural Antioxidants to Protect Cardiac Muscle from Oxidative Stress
5.1. Flavonoids
5.2. Non-Flavonoids
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 3rd ed.; Clarendon Press: Oxford, UK, 1999. [Google Scholar]
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M.C.; Cross, C.E. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992, 119, 598–620. [Google Scholar]
- Sies, H. Oxidative stress: From basic research to clinical application. Am. J. Med. 1991, 91, 31–38. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dornll, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, G.; Liu, X.; Wang, W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J. Mol. Cell. Cardiol. 2014, 76, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: Influence of cardiac pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Cadenas, S.; Aragones, J.; Landazuri, M.O. Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc. Res. 2010, 88, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.C.; Wang, J.; Son, M.J.; Woo, S.H. Shear stress enhances Ca2+ sparks through Nox2-dependent mitochondrial reactive oxygen species generation in rat ventricular myocytes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1121–1131. [Google Scholar]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sanchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type a S-glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med. 2005, 38, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Wu, K.L.; Chang, A.Y.; Tai, M.H.; Chan, J.Y. Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension. Hypertension 2009, 53, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.X.; Yang, R.F.; Li, S.; Renshaw, A.O.; Li, Y.L.; Schultz, H.D.; Zimmerman, M.C. Mitochondria-produced superoxide mediates angiotensin II induced inhibition of neuronal potassium current. Am. J. Physiol. Cell Physiol. 2010, 298, C857–C865. [Google Scholar] [CrossRef] [Green Version]
- Flores-Arredondo, J.H.; García-Rivas, G.; Torre-Amione, G. Immune modulation in heart failure: Past challenges and future hopes. Curr. Heart Fail. Rep. 2011, 8, 28–37. [Google Scholar] [CrossRef]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, immunity, and hypertension. Hypertension 2011, 57, 132–140. [Google Scholar]
- Marvar, P.J.; Lob, H.; Vinh, A.; Zarreen, F.; Harrison, D.G. The central nervous system and inflammation in hypertension. Curr. Opin. Pharmacol. 2011, 11, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvar, P.J.; Thabet, S.R.; Guzik, T.J.; Lob, H.E.; McCann, L.A.; Weyand, C.; Gordon, F.J.; Harrison, D.G. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ. Res. 2010, 107, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Savoia, C.; Schiffrin, E.L. Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 2006, 15, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Tai, M.H.; Li, C.Y.; Chan, J.Y. Reduction in molecular synthesis or enzyme activity of superoxide dismutases and catalase contributes to oxidative stress and neurogenic hypertension in spontaneously hypertensive rats. Free Radic. Biol. Med. 2006, 40, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function—Role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Petroff, M.G.; Kim, S.H.; Pepe, S.; Dessy, C.; Marbán, E.; Balligand, J.L.; Sollott, S.J. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat. Cell Biol. 2001, 3, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Kinugawa, S.; Tsutsui, H.; Hayashidani, S.; Ide, T.; Suematsu, N.; Satoh, S.; Utsumi, H.; Takeshita, A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: Role of oxidative stress. Circ. Res. 2000, 87, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Garcia, J. Cardioprotection and Signaling Pathways; Springer: New York, NY, USA, 2011. [Google Scholar]
- Krijnen, P.A.; Meischl, C.; Hack, C.E.; Meijer, C.J.; Visser, C.A.; Roos, D.; Niessen, H.W. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol. 2003, 56, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Hindi, S.M.; Shin, J.; Gallot, Y.S.; Straughn, A.R.; Simionescu-Bankston, A.; Hindi, L.; Xiong, G.; Friedland, R.P.; Kumar, A. MyD88 promotes myoblast fusion in a cell-autonomous manner. Nat. Commun. 2017, 8, 1624. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Garg, I.; Ashraf, M.Z. TLR signalling and association of TLR polymorphism with cardiovascular diseases. Vasc. Pharmacol. 2016, 87, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]
- Mallet, R.T.; Olivencia-Yurvati, A.H.; Bünger, R. Pyruvate enhancement of cardiac performance: Cellular mechanisms and clinical application. Exp. Biol. Med. 2018, 243, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Epstein, C.J. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar]
- Ardanaz, N.; Yang, X.P.; Cifuentes, M.E.; Haurani, M.J.; Jackson, K.W.; Liao, T.D.; Pagano, P.J. Lack of glutathione peroxidase 1 accelerates cardiac-specific hypertrophy and dysfunction in angiotensin II hypertension. Hypertension 2010, 55, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Chua, C.C.; Gao, J.; Chua, K.W.; Ho, Y.S.; Hamdy, R.C.; Chua, B.H. Prevention of ischemia/reperfusion-induced cardiac apoptosis and injury by melatonin is independent of glutathione peroxdiase 1. J. Pineal Res. 2009, 46, 235–241. [Google Scholar]
- Hu, C.; Zhang, H.; Qiao, Z.; Wang, Y.; Zhang, P.; Yang, D. Loss of thioredoxin 2 alters mitochondrial respiratory function and induces cardiomyocyte hypertrophy. Exp. Cell Res. 2018, 372, 61–72. [Google Scholar]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar]
- Giordano, F.J. J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant properties dedicated to nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Le, C.T.; Hollaar, L.; van der Valk, E.J.M.; van der Laarse, A. Buthionine sulfoximine reduces the protective capacity of myocytes to withstand peroxide-derived free radical attack. J. Mol. Cell. Cardiol. 1993, 25, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Chernov, N.N.; Saprin, A.N. Involvement of thio-, peroxi-, and glutaredoxins in cellular redoxdependent processes. Biochemistry 2008, 73, 1493–1510. [Google Scholar]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins–molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 as the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Elbirt, K.; Bonkovsky, H. Heme oxygenase: Recent advances in understanding its regulation and role. Proc. Assoc. Am. Physicians 1999, 111, 438–447. [Google Scholar] [CrossRef]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Mackern-Oberti, J.; Obreque, J.; Méndez, G.; Lianos, C.; Kalergis, A. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 182, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Montellano, P.R. The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 2000, 4, 221–227. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic reticulum anchored heme-oxygenase-1 faces the cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, N.G.; Tsenovoy, P.L.; McClung, J.; Drummond, G.S. Heme oxygenase: A target gene for anti-diabetic and obesity. Curr. Pharm. Des. 2008, 14, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Stocker, R.; Glazer, A.N.; Ames, B.N. Antioxidant activity of albumin-bound bilirubin. Proc. Natl. Acad. Sci. USA 1987, 84, 5918–5922. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Stocker, R. Free and albumin-bound bilirubin are efficient co-antioxidants for alphatocopherol, inhibiting plasma and low density lipoprotein lipid peroxidation. J. Biol. Chem. 1994, 269, 16712–16719. [Google Scholar] [CrossRef]
- Averilla, J.N.; Oh, J.; Kim, J.S. Carbon Monoxide Partially Mediates Protective Effect of Resveratrol Against UVB-Induced Oxidative Stress in Human Keratinocytes. Antioxidants 2019, 8, 432. [Google Scholar] [CrossRef] [Green Version]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef]
- Beuckelmann, D.J.; Wier, W.G. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J. Physiol. 1988, 405, 233–255. [Google Scholar] [CrossRef] [Green Version]
- Nabauer, M.; Callewaert, G.; Cleemann, L.; Morad, M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science 1989, 244, 800–803. [Google Scholar] [CrossRef]
- Niggli, E.; Lederer, W.J. Voltage-independent calcium release in heart muscle. Science 1990, 250, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Wier, W.G.; Egan, T.M.; Lopez-Lopez, J.R.; Balke, C.W. Local control of excitation-contraction coupling in rat heart cells. J. Physiol. 1994, 474, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannell, M.B.; Cheng, H.; Lederer, W.J. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys. J. 1994, 67, 1942–1956. [Google Scholar] [CrossRef] [Green Version]
- Shacklock, P.S.; Wier, W.G.; Balke, C.W. Local Ca2+ transients (Ca2+ sparks) originate at transverse tubules in rat heart cells. J. Physiol. 1995, 487, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Parker, I.; Zang, W.J.; Wier, W.G. Ca2+ sparks involving multiple Ca2+ release sites along Z-lines in rat heart cells. J. Physiol. 1996, 497, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negretti, N.; O’Neill, S.C.; Eisner, D.A. The relative contributions of different intracellular and sarcolemmal systems to relaxation in rat ventricular myocytes. Cardiovasc. Res. 1993, 27, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Bassani, J.W.; Bassani, R.A.; Bers, D.M. Relaxation in rabbit and rat cardiac cells: Species-dependent differences in cellular mechanisms. J. Physiol. 1994, 476, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Sommer, J.R.; Johnson, E.A. The ultra structure of cardiac muscle. In Handbook of Physiology, Section 2: The Cardiovascular System; Berne, R.M., Ed.; American Physiological Society: Washington, DC, USA, 1979; Volume 1, pp. 113–186. [Google Scholar]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- Traaseth, N.; Elfering, S.; Solien, J.; Haynes, V.; Giulivi, C. Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle. Biochim. Biophys. Acta 2004, 1658, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, E.; Maack, C. Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac exciatation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Mitochondrial Ca2+ and the heart. Cell Calcium 2008, 44, 77–91. [Google Scholar] [CrossRef]
- Ramesh, V.; Sharma, V.K.; Sheu, S.S.; Franzini-Armstrong, C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann. N. Y. Acad. Sci. 1998, 853, 341–344. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C. ER-mitochondria communication. How privileged? Physiology 2007, 22, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, J.J.; Jafri, M.S.; Winslow, R.L. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys. J. 1999, 77, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Michailova, A.; McCulloch, A. Model study of ATP and ADP buffering, transport of Ca2+ and Mg2+, and regulation of ion pumps in ventricular myocyte. Biophys. J. 2001, 81, 614–629. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Thomas, A.P.; Hajnóczky, G. Ca2+ marks: Miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 2380–2385. [Google Scholar] [CrossRef] [Green Version]
- Subedi, K.P.; Kim, J.C.; Kang, M.; Son, M.J.; Kim, Y.S.; Woo, S.H. Voltage-dependent anion channel 2 modulates resting Ca2+ sparks, but not action potential-induced Ca2+ signaling in cardiac myocytes. Cell Calcium 2011, 49, 136–143. [Google Scholar] [CrossRef]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuti, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyman, L.; Williams, G.S.B.; Khananshvili, D.; Sekler, I.; Lederer, W.J. NCXL: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013, 59, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Corral-Debrinski, M.; Shoffner, J.M.; Lott, M.T.; Wallace, D.C. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat. Res. 1992, 275, 169–180. [Google Scholar] [CrossRef]
- Santorelli, F.M.; Mak, S.C.; El-Schahawi, M.; Casali, C.; Shanske, S.; Baram, T.Z.; Madrid, R.E.; DiMauro, S. Maternally inherited cardiomyopathy and hearing loss associated with a novel mutation in the mitochondrial tRNA(Lys) gene (G8363A). Am. J. Hum. Genet. 1996, 58, 933–939. [Google Scholar]
- Arbustini, E.; Diegoli, M.; Fasani, R.; Grasso, M.; Morbini, P.; Banchieri, N.; Bellini, O.; Bello, B.D.; Pilotto, A.; Magrini, G.; et al. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am. J. Pathol. 1998, 153, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Grad, L.I.; Sayles, L.C.; Lemire, B.D. Introduction of an additional pathway for lactate oxidation in the treatment of lactic acidosis and mitochondrial dysfunction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2005, 102, 18367–18372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, S. The failing heart: An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Tandler, B.; Ye, J.; Slabe, T.J.; Turkaly, J.; Hoppel, C.L. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am. J. Physiol. 1997, 273, H1544–H1554. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Di Lisa, F.; Carpi, A.; Giorgio, V.; Bernardi, P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Mikami, A.; Imoto, K.; Tanabe, T.; Niidome, T.; Mori, Y.; Takeshima, H.; Narumiya, S.; Numa, S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature 1989, 340, 230–233. [Google Scholar] [CrossRef]
- Fearon, I.M.; Palmer, A.C.; Balmforth, A.J.; Ball, S.G.; Varadi, G.; Peers, C. Modulation of recombinant human cardiac L-type Ca2+ channel α1C subunits by redox agents and hypoxia. J. Physiol. 1999, 514, 629–637. [Google Scholar] [CrossRef]
- Hu, H.; Chiamvimonvat, N.; Yamagishi, T.; Marban, E. Direct inhibition of expressed cardiac L-type Ca2+ channels by S-nitrosothiol nitric oxide donors. Circ. Res. 1997, 81, 742–752. [Google Scholar] [PubMed]
- Campbell, D.L.; Stamler, J.S.; Strauss, H.C. Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J. Gen. Physiol. 1996, 108, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Hove-Madsen, L.; Llach, A.; Bayes-Genís, A.; Roura, S.; Font, E.R.; Arís, A.; Cinca, J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 2004, 110, 1358–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, T.H.; Maier, L.S.; Sossalla, S. The ryanodine receptor leak: How a tattered receptor plunges the failing heart into crisis. Heart Fail. Rev. 2013, 18, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Eu, J.P.; Meissner, G.; Stamler, J.S. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 1998, 279, 234–237. [Google Scholar] [CrossRef]
- Pessah, I.N.; Kim, K.H.; Feng, W. Redox sensing properties of the ryanodine receptor complex. Front. Biosci. 2002, 7, a72–a79. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, C.; Donoso, P.; Carrasco, M.A. The ryanodine receptors Ca2+ release channels: Cellular redox sensors? IUBMB Life 2005, 57, 315–322. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, J.; Wei, C.; Li, K.; Xie, W.; Wang, Y.; Cheng, H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 2008, 77, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Trimm, J.L.; Salama, G.; Abramson, J.J. Sulfhydryl oxidation induces rapid calcium release from sarcoplasmic reticulum vesicles. J. Biol. Chem. 1986, 261, 16092–16098. [Google Scholar] [CrossRef]
- Boraso, A.; Williams, A.J. Modification of the gating of the cardiac sarcoplasmic reticulum Ca2+-release channel by H2O2 and dithiothreitol. Am. J. Physiol. 1994, 267, H1010–H1016. [Google Scholar]
- Marengo, J.J.; Hidalgo, C.; Bull, R. Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys. J. 1998, 74, 1263–1277. [Google Scholar] [CrossRef] [Green Version]
- Gen, W.; Tani, M.; Takeshita, J.; Ebihara, Y.; Tamaki, K. Mechanisms of Ca2+ overload induced by extracellular H2O2 in quiescent isolated rat cardiomyocytes. Basic Res. Cardiol. 2001, 96, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Isaeva, E.V.; Shkryl, V.M.; Shirokova, N. Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J. Physiol. 2005, 565, 855–872. [Google Scholar] [CrossRef]
- Goldhaber, J.I.; Liu, E. Excitation-contraction coupling in single guinea-pig ventricular myocytes exposed to hydrogen peroxide. J. Physiol. 1994, 477, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Blatter, L.A. Inositol-1,4,5-trisphosphate-dependent Ca2+ signalling in cat atrial excitation-contraction coupling and arrhythmias. J. Physiol. 2004, 555, 607–615. [Google Scholar] [CrossRef]
- Mackenzie, L.; Bootman, M.D.; Laine, M.; Berridge, M.J.; Thuring, J.; Holmes, A.; Li, W.; Lipp, P. The role of inositol 1,4,5-trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J. Physiol. 2002, 541, 395–409. [Google Scholar] [CrossRef]
- Kim, J.C.; Woo, S.H. Shear stress induces a longitudinal Ca2+ wave via autocrine activation of P2Y1 purinergic signalling in rat atrial myocytes. J. Physiol. 2015, 593, 5091–5109. [Google Scholar] [CrossRef] [Green Version]
- Renard-Rooney, D.C.; Joseph, S.K.; Seitz, M.B.; Thomas, A.P. Effect of oxidized glutathione and temperature on inositol 1,4,5-trisphosphate binding in permeabilized hepatocytes. Biochem. J. 1995, 310, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplin, A.I.; Snyder, S.H.; Linden, D.J. Reduced nicotinamide adenine dinucleotide-selective stimulation of inositol 1,4,5-trisphosphate receptors mediates hypoxic mobilization of calcium. J. Neurosci. 1996, 16, 2002–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.J. Sulfhydryl group modification of sarcoplasmic reticulum membranes. Biochemistry 1976, 15, 4492–4496. [Google Scholar] [CrossRef]
- Morris, T.E.; Sulakhe, P.V. Sarcoplasmic reticulum Ca2+-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic. Biol. Med. 1997, 22, 37–47. [Google Scholar] [CrossRef]
- Xu, K.Y.; Zweier, J.L.; Becker, L.C. Hydroxyl radical inhibits sarcoplasmic reticulum Ca2+-ATPase function by direct attack on the ATP binding site. Circ. Res. 1997, 80, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, D.A.; Ottolia, M.; Lu, L.; Lu, Y.; Philipson, K.D. A new topological model of the cardiac sarcolemmal Na+-Ca2+ exchanger. J. Biol. Chem. 1999, 274, 910–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, J.P.; Bailey, C.A.; Hale, C.C. Redox modification of sodium-calcium exchange activity in cardiac sarcolemmal vesicles. J. Biol. Chem. 1986, 261, 4948–4955. [Google Scholar] [CrossRef]
- Sedova, M.; Dedkova, E.N.; Blatter, L.A. Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am. J. Physiol. Cell Physiol. 2006, 291, C840–C850. [Google Scholar] [CrossRef]
- Bell, C.J.; Bright, N.A.; Rutter, G.A.; Griffiths, E.J. ATP regulation in adult rat cardiomyocytes: Time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J. Biol. Chem. 2006, 281, 28058–28067. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.A.; Garcia, M.C.; Sharma, V.K.; Young, K.C.; Matlib, M.A.; Sheu, S.S. Mitochondria regulate inactivation of L-type Ca2+ channels in rat heart. J. Physiol. 2001, 536, 387–396. [Google Scholar] [CrossRef]
- Zima, A.V.; Pabbidi, M.R.; Lipsius, S.L.; Blatter, L.A. Effects of mitochondrial uncoupling on Ca2+ signaling during excitation-contraction coupling in atrial myocytes. Am. J. Physiol Heart Circ. Physiol. 2013, 304, H983–H993. [Google Scholar] [CrossRef] [Green Version]
- Lakatta, E.G. Cardiovascular regulatory mechanisms in advanced age. Physiol. Rev. 1993, 73, 413–467. [Google Scholar] [CrossRef]
- Nazir, S.A.; Lab, M.J. Mechanoelectric feedback and atrial arrhythmias. Cardiovasc. Res. 1996, 32, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Sato, R.; Koumi, S. Characterization of the stretch-activated chloride channel in isolated human atrial myocytes. J. Membr. Biol. 1998, 163, 67–76. [Google Scholar] [CrossRef]
- Tavi, P.; Han, C.; Weckström, M. Mechanisms of stretch-induced changes in [Ca2+]i in rat atrial myocytes. Circ. Res. 1998, 83, 1165–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamkin, A.; Kiseleva, I.; Wagner, K.D.; Bohm, J.; Theres, H.; Günther, J.; Scholz, H. Characterization of stretch-activated ion currents in isolated atrial myocytes from human hearts. Pflügers Archiv. 2003, 446, 339–346. [Google Scholar] [CrossRef]
- Allen, D.G.; Nichols, C.G.; Smith, G.L. The effects of changes in muscle length during diastole on the calcium transient in ferret ventricular muscle. J. Physiol. 1988, 406, 359–370. [Google Scholar] [CrossRef]
- Le Guennec, J.Y.; White, E.; Gannier, F.; Argibay, J.A.; Garnier, D. Stretch-induced increase of resting intracellular calcium concentration in single guinea-pig ventricular myocytes. Exp. Physiol. 1991, 76, 975–978. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.G.; Kurihara, S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J. Physiol. 1982, 327, 79–94. [Google Scholar] [CrossRef]
- Alvarez, B.V.; Pérez, N.G.; Ennis, I.L.; Camilión de Hurtado, M.C.; Cingolani, H.E. Mechanisms underlying the increase in force and Ca2+ transient that follow stretch of cardiac muscle: A possible explanation of the Anrep effect. Circ. Res. 1999, 85, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, H.E.; Alvarez, B.V.; Ennis, I.L.; de Hurtado, M.C.C. Stretch-induced alkalinization of feline papillary muscle: An autocrine-paracrine system. Circ. Res. 1998, 83, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Clerk, A.; Sugden, P.H. Activation of protein kinase cascades in the heart by hypertrophic G protein-coupled receptor agonists. Am. J. Cardiol. 1999, 83, 64H–69H. [Google Scholar] [CrossRef]
- Saward, L.; Zahradka, P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ. Res. 1997, 81, 249–257. [Google Scholar] [CrossRef]
- Nattel, S. New ideas about atrial fibrillation 50 years on. Nature 2002, 415, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maytin, M.; Siwik, D.A.; Ito, M.; Xiao, L.; Sawyer, D.B.; Liao, R.; Colucci, W.S. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox. Circulation 2004, 109, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Brewer, A.C.; Schröder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, M.J.C.; Price, K.R. Analytical problems in the study of flavonoid compounds in onions. Food Chem. 1996, 57, 113–117. [Google Scholar] [CrossRef]
- Rocha-Guzmán, N.E.; Herzog, A.; González-Laredo, R.F.; Ibarra-Pérez, F.J.; Zambrano-Galván, G.; Gallegos-Infante, J.A. Antioxidant and antimutagenic activity of phenolic compounds in three different colour groups of common bean cultivars. Food Chem. 2007, 103, 521–527. [Google Scholar] [CrossRef]
- Hanasaki, Y.; Ogawa, S.; Fnkui, S. The correlation between active oxygens scavenging and antioxidative effects of flavonoids. Free Radic. Biol. 1994, 16, 845–850. [Google Scholar] [CrossRef]
- Afanas’ev, I.B.; Dorozhko, A.I.; Brodskii, A.V.; Kostyuk, V.A.; Potapovitch, A.I. Chelating and free radical scavenging mechanisms of inhibitory action of rutin and quercetin in lipid peroxidation. Biochem. Pharmacol. 1989, 38, 1763–1769. [Google Scholar] [CrossRef]
- Wictome, M.; Michelangeli, F.; Lee, A.G.; East, J.M. The inhibitors thapsigargin and 2,5-di(tert-butyl)-1,4-benzohydroquinone favour the E2 form of the Ca2+, Mg2+-ATPase. FEBS Lett. 1992, 304, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Dyer, J.L.; Khan, S.Z.; Bilmen, J.G.; Hawtin, S.R.; Wheatley, M.; Javed, M.U.; Michelangeli, F. Curcumin: A new cell-permeant inhibitor of the inositol 1,4,5-trisphosphate receptor. Cell Calcium 2002, 31, 45–52. [Google Scholar]
- Shoshan, V.; MacLennan, D.H. Quercetin interaction with the (Ca2+ + Mg2+)-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 1981, 256, 887–892. [Google Scholar] [CrossRef]
- Ogunbayo, O.A.; Harris, R.M.; Waring, R.H.; Kirk, C.J.; Michelangeli, F. Inhibition of the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase by flavonoids: Aquantitative structure-activity relationship study. IUBMB Life 2008, 60, 853–858. [Google Scholar] [CrossRef]
- Yoshino, K.; Hara, Y.; Sano, M.; Tomita, I. Antioxidative effects of black tea theaflavins and thearubigin on lipid peroxidation of rat liver homogenates induced by tert-butyl hydroperoxide. Biol. Pharm. Bull. 1994, 17, 146–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salah, N.; Miller, N.J.; Paganga, G.; Tijburg, L.; Bolwell, G.P.; Riceevans, C. Polyphenolic flavanols as scavengers of aqueous phase radicals and as chain-breaking antioxidants. Arch. Biochem. Biophys. 1995, 322, 339–346. [Google Scholar] [CrossRef]
- Stangl, V.; Dreger, H.; Stangl, K.; Lorenz, M.. Molecular targets of tea polyphenols in the cardiovascular system. Cardiovasc. Res. 2007, 73, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Mukhtar, H. Tea polyphenols for health promotion. Life Sci. 2007, 81, 519–533. [Google Scholar]
- Townsend, P.A.; Scarabelli, T.M.; Pasini, E.; Gitti, G.; Menegazzi, M.; Suzuki, H.; Stephanou, A. Epigallocatechin-3-gallate inhibits STAT-1 activation and protects cardiac myocytes from ischemia/reperfusion-induced apoptosis. FASEB J. 2004, 18, 1621–1623. [Google Scholar] [CrossRef]
- Sheng, R.; Gu, Z.L.; Xie, M.L.; Zhou, W.X.; Guo, C.Y. EGCG inhibits cardiomyocyte apoptosis in pressure overload-induced cardiac hypertrophy and protects cardiomyocytes from oxidative stress in rats. Acta Pharmacol. Sin. 2007, 28, 191–201. [Google Scholar] [CrossRef]
- Ha, T.; Hua, F.; Liu, X.; Ma, J.; McMullen, J.R.; Shioi, T. Lipopolysaccharide-induced myocardial protection against ischaemia/reperfusion injury is mediated through a PI3K/Akt-dependent mechanism. Cardiovasc. Res. 2008, 78, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Shang, P.; Li, D. Luteolin: A flavonoid that has multiple cardio-protective effects and its molecular mechanisms. Front. Pharmacol. 2017, 8, 692. [Google Scholar]
- Qi, L.; Pan, H.; Li, D.; Fang, F.; Chen, D.; Sun, H. Luteolin improves contractile function and attenuates apoptosis following ischemia–reperfusion in adult rat cardiomyocytes. Eur. J. Pharmacol. 2011, 668, 201–207. [Google Scholar]
- Jain, A.K.; Mehra, N.K.; Swarnakar, N.K. Role of Antioxidants for the Treatment of Cardiovascular Diseases: Challenges and Opportunities. Curr. Pharm. Des. 2015, 21, 4441–4455. [Google Scholar] [CrossRef]
- Carrizzo, A.; Izzo, C.; Forte, M.; Sommella, E.; Di Pietro, P.; Venturini, E.; Ciccarelli, M.; Galasso, G.; Rubattu, S.; Campiglia, P.; et al. A Novel Promising Frontier for Human Health: The Beneficial Effects of Nutraceuticals in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 8706. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Peng, X.; Luo, Y.; You, J.; Yin, D.; Xu, Q.; He, M. Quercetin protects cardiomyocytes against doxorubicin-induced toxicity by suppressing oxidative stress and improving mitochondrial function via 14-3-3γ. Toxicol. Mech. Methods 2019, 29, 344–354. [Google Scholar]
- Santos, M.S.; Oliveira, E.D.; Santos-Miranda, A.; Cruz, J.S.; Gondim, A.N.S.; Menezes-Filho, J.E.R.; Vasconcelos, C.M.L. Dissection of the effects of quercetin on mouse myocardium. Basic Clin. Pharmacol. Toxicol. 2017, 120, 550–559. [Google Scholar]
- Van den Hoek, T.L.; Becker, L.B.; Shao, Z.; Li, C.; Schumacker, P.T. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 1998, 273, 18092–18098. [Google Scholar] [CrossRef] [Green Version]
- Bel, A.; Ricci, M.; Piquet, J.; Bruneval, P.; Perier, M.-C.; Gagnieu, C.; Fabiani, J.-N.; Menasché, P. Prevention of postcardiopulmonary bypass pericardial adhesions by a new resorbable collagen membrane. Interact. Cardio Vasc. Thorac. Surg. 2012, 14, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Shaughnessy, K.S.; Boswall, I.A.; Scanlan, A.P.; Gottschall-Pass, K.T.; Sweeney, M.I. Diets containing blueberry extract lower blood pressure in spontaneously hypertensive stroke-prone rats. Nutr. Res. 2009, 29, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Cortes, S.F.; Valadares, Y.M.; de Oliveira, A.B.; Lemos, V.S.; Barbosa, M.P.T.; Braga, F.C. Mechanism of endothelium-dependent vasodilation induced by a proanthocyanidin-rich fraction from Ouratea semiserrata. Planta Med. 2002, 68, 412–415. [Google Scholar] [CrossRef]
- Andriambeloson, E.; Magnier, C.; Haan-Archipoff, G.; Lobstein, A.; Anton, R.; Beretz, A.; Stoclet, J.C.; Andriantsitohaina, R. Natural dietary polyphenolic compounds cause endothelium-dependent vasorelaxation in rat thoracic aorta. J. Nutr. 1998, 128, 2324–2333. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, D.; Zhang, Y.; Sun, R.; Xia, M. Anthocyanin increases adiponectin secretion and protects against diabetes-related endothelial dysfunction. Am. J. Physiol Endocrinol. Metab. 2014, 306, E975–E988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Vazquez, F.J.; Ibarra-Alvarado, C.; Rojas-Molina, A.; Rojas-Molina, J.I.; Yahia, E.M.; Rivera-Pastrana, D.M.; Rojas-Molina, A.; Zavala-Sánchez, M.A. Nutraceutical value of black cherry Prunus serotina Ehrh. fruits: Antioxidant and antihypertensive properties. Molecules 2013, 18, 14597–14612. [Google Scholar] [CrossRef]
- Gui-Rong, L.; Wang, H.B.; Qin, G.W.; Jin, M.W.; Tang, Q.; Sun, H.Y.; Du, X.L.; Deng, X.L.; Zhang, X.H.; Chen, J.B.; et al. Acacetin, a Natural Flavone, Selectively Inhibits Human Atrial Repolarization Potassium Currents and Prevents Atrial Fibrillation in Dogs. Circulation 2008, 117, 2449–2457. [Google Scholar]
- Yuan, Y.; Meng, L.; Zhou, Y.; Lu, N. Genetic polymorphism of angiotensin-converting enzyme and hypertrophic cardiomyopathy risk: A systematic review and meta-analysis. Medicine 2017, 96, e8639. [Google Scholar] [CrossRef]
- Kim, D.S.; Ha, K.C.; Kwon, D.Y.; Kim, M.S.; Kim, H.R.; Chae, S.W.; Chae, H.J. Kaempferol protects ischemia/reperfusion-induced cardiac damage through the regulation of endoplasmic reticulum stress. Immunopharmacol. Immunotoxicol. 2008, 30, 257–270. [Google Scholar]
- Kim, D.S.; Kwon, D.Y.; Kim, M.S.; Kim, H.K.; Lee, Y.C.; Park, S.J. The involvement of endoplasmic reticulum stress in flavonoid-induced protection on cardiac cell death caused by ischaemia/reperfusion. J. Pharm. Pharmacol. 2010, 62, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, C.; Zhang, L.; Lv, J.; Ni, H. Rutin alleviates hypoxia/reoxygenation-induced injury in myocardial cells by up-regulating SIRT1 expression. Chem. Biol. Interact. 2019, 297, 44–49. [Google Scholar] [CrossRef]
- Lin, Q.; Chen, X.Y.; Zhang, J.; Yuan, Y.L.; Zhao, W.; Wei, B. Upregulation of SIRT1 contributes to the cardioprotective effect of rutin against myocardial ischemia reperfusion injury in rats. J. Funct. Foods 2018, 46, 227–236. [Google Scholar] [CrossRef]
- Chu, J.X.; Li, G.M.; Gao, X.J.; Wang, J.X.; Han, S.Y. Buckwheat rutin inhibits Ang II-induced cardiomyocyte hypertrophy via blockade of CaN-dependent signal pathway. Iran. J. Pharm. Res. 2014, 13, 1347–1355. [Google Scholar]
- Gao, J.P.; Chen, C.X.; Gu, W.L. Effects of polydatin on attenuating ventricular remodeling in isoproterenol-induced mouse and pressure-overload rat models. Fitoterapia 2010, 81, 953–960. [Google Scholar] [CrossRef]
- Zhang, L.P.; Yang, C.Y.; Wang, Y.P. Protective effect of polydatin against ischemia/reperfusion injury in rat heart. Acta Physiol. Sin. 2008, 60, 161–168. [Google Scholar]
- Deng, J.; Liu, W.; Wang, Y.; Dong, M.; Zheng, M.; Liu, J. Polydatin modulates Ca2+ handling, excitation–contraction coupling and β-adrenergic signaling in rat ventricular myocytes. J. Mol. Cell. Cardiol. 2012, 53, 646–656. [Google Scholar] [CrossRef]
- Lee, K.P.; Kim, J.E.; Park, W.H. Cytoprotective effect of rhamnetin on miconazole-induced H9c2 cell damage. Nutr. Res. Pract. 2015, 9, 586–591. [Google Scholar]
- Park, E.S.; Kang, J.C.; Jang, Y.C.; Park, J.S.; Jang, S.Y.; Kim, D.E.; Shin, H.S. Cardioprotective effects of rhamnetin in H9c2 cardiomyoblast cells under H2O2-induced apoptosis. J. Ethnopharmacol. 2014, 153, 552–560. [Google Scholar]
- Zhou, Z.; Zhang, Y.; Lin, L.; Zhou, J. Apigenin suppresses the apoptosis of H9C2 rat cardiomyocytes subjected to myocardial ischemia-reperfusion injury via upregulation of the PI3K/Akt pathway. Mol. Med. Rep. 2018, 18, 1560–1570. [Google Scholar]
- Zhu, Z.Y.; Gao, T.; Huang, Y.; Xue, J.; Xie, M.L. Apigenin ameliorates hypertension-induced cardiac hypertrophy and down-regulates cardiac hypoxia inducible factor-lα in rats. Food Funct. 2016, 7, 1992–1998. [Google Scholar]
- Li-Weber, M. New therapeutic aspects of flavones: The anticancer properties of Scutellaria and its main active constituents wogonin, baicalein and baicalin. Cancer Treat. Rev. 2009, 35, 57–68. [Google Scholar] [CrossRef]
- Li, Q.; Yu, Z.; Xiao, D.; Wang, Y.; Zhao, L.; An, Y.; Gao, Y. Baicalein inhibits mitochondrial apoptosis induced by oxidative stress in cardiomyocytes by stabilizing MARCH5 expression. J. Cell. Mol. Med. 2020, 24, 2040–2051. [Google Scholar]
- Liu, B.Y.; Li, L.; Liu, G.L.; Ding, W.; Chang, W.G.; Xu, T.; Wang, J.X. Baicalein attenuates cardiac hypertrophy in mice via suppressing oxidative stress and activating autophagy in cardiomyocytes. Acta Pharmacol. Sin. 2020, 42, 701–714. [Google Scholar] [CrossRef]
- Cui, G.; Luk, S.C.W.; Li, R.A.; Chan, K.K.K.; Lei, S.W.; Wang, L.; Lee, S.M.Y. Cytoprotection of baicalein against oxidative stress-induced cardiomyocytes injury through the Nrf2/Keap1 pathway. J. Cardiovasc. Pharmacol. 2015, 65, 39–46. [Google Scholar]
- Wakabayashi, I. Inhibitory effects of baicalein and wogonin on lipopolysaccharide-induced nitric oxide production in macrophages. Pharmacol. Toxicol. 1999, 84, 288–291. [Google Scholar] [CrossRef]
- Cheng, P.Y.; Lee, Y.M.; Wu, Y.S.; Chang, T.W.; Jin, J.S.; Yen, M.H. Protective effect of baicalein against endotoxic shock in rats in vivo and in vitro. Biochem. Pharmacol. 2007, 73, 793–804. [Google Scholar] [CrossRef]
- Lee, Y.M.; Cheng, P.Y.; Chim, L.S.; Kung, C.W.; Ka, S.M.; Chung, M.T.; Sheu, J.R. Baicalein, an active component of Scutellaria baicalensis Georgi, improves cardiac contractile function in endotoxaemic rats via induction of heme oxygenase-1 and suppression of inflammatory responses. J. Ethnopharmacol. 2011, 135, 179–185. [Google Scholar] [CrossRef]
- Ai, W.; Zhang, Y.; Tang, Q.; Yan, L.; Bian, Z.; Liu, C.; Huang, H.; Bai, X.; Yin, L.; Li, H. Silibinin attenuates cardiac hypertrophy and fibrosis through blocking EGFR-dependent signaling. J. Cell. Biochem. 2010, 110, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Wu, Z.; Wu, B.; Liu, C.; Huang, R.; Yang, L.; Guo, R.; Wu, K.; Chen, J. Naringin protects cardiomyocytes against hyperglycemia-induced injuries in vitro and in vivo. J. Endocrinol. 2016, 230, 197–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.Y.; Jin, P.; He, Q.; Lu, L.H.; Ma, J.P.; Gao, W.L.; Bai, H.P.; Yang, J. Naringenin ameliorates hypoxia/reoxygenation-induced endoplasmic reticulum stress-mediated apoptosis in H9c2 myocardial cells: Involvement in ATF6, IRE1alpha and PERK signaling activation. Mol. Cell. Biochem. 2017, 424, 111–122. [Google Scholar] [CrossRef]
- Yu, L.M.; Dong, X.; Xue, X.D.; Zhang, J.; Li, Z.; Wu, H.J.; Yang, Z.L.; Yang, Y.; Wang, H.S. Naringenin improves mitochondrial function and reduces cardiac damage following ischemia-reperfusion injury: The role of the AMPK-SIRT3 signaling pathway. Food Funct. 2019, 10, 2752–2765. [Google Scholar] [CrossRef] [PubMed]
- Reyes, D.R.A.; Gomes, M.J.; Rosa, C.M.; Pagan, L.U.; Damatto, F.C.; Damatto, R.L.; Depra, I.; Campos, D.H.S.; Fernandez, A.A.H.; Martinez, P.F.; et al. N-Acetylcysteine Influence on Oxidative Stress and Cardiac Remodeling in Rats During Transition from Compensated Left Ventricular Hypertrophy to Heart Failure. Cell. Physiol. Biochem. 2017, 44, 2310–2321. [Google Scholar]
- Singal, P.K.; Kirshenbaum, L.A. A relative deficit in antioxidant reserve may contribute in cardiac failure. Can. J. Cardiol. 1990, 6, 47–49. [Google Scholar] [PubMed]
- Dhaliwal, H.; Kirshenbaum, L.A.; Randhawa, A.K.; Singal, P.K. Correlation between antioxidant changes during hypoxia and recovery on reoxygenation. Am. J. Physiol. 1991, 261, H632–H638. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, L.A.; Singal, P.K. Antioxidant changes in heart hypertrophy: Significance during hypoxia-reoxygenation injury. Can. J. Physiol Pharmacol. 1992, 70, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Zinger, A.; Nevo, A.; Sekler, I.; Hershfinkel, M. Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J. Biol. Chem. 2010, 285, 26097–26106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, M.; Takebayashi, T.; Ohta, H.; Alcalde, R.E.; Itano, Y.; Matsumura, T. Zinc accumulation and metallothionein gene expression in the proliferating epidermis during wound healing in mouse skin. Histochem. Cell Biol. 1999, 112, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Lansdown, A.B.; Sampson, B.; Rowe, A. Sequential changes in trace metal, metallothionein and calmodulin concentrations in healing skin wounds. J. Anat. 1999, 195, 375–386. [Google Scholar] [CrossRef]
- Thomas, M.; Vidal, A.; Bhattacharya, S.K.; Ahokas, R.A.; Sun, Y.; Gerling, I.C.; Weber, K.T. Zinc dyshomeostasis in rats with aldosteronism. Response to spironolactone. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2361–H2366. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Rassaf, T.; Massion, P.B.; Kelm, M.; Balligand, J.L. Recent advances in the understanding of the role of nitric oxide in cardiovascular homeostasis. Pharmacol. Ther. 2005, 108, 225–256. [Google Scholar] [CrossRef] [PubMed]
- Zordoky, B.N.M.; Robertson, I.M.; Dyck, J.R.B. Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Biochim. Biophys. Acta 2015, 1852, 1155–1177. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.M.; Su, M.J.; Chen, J.K. Resveratrol protects myocardial ischemia-reperfusion injury through both NO-dependent and NO-independent mechanisms. Free Radic. Biol. Med. 2004, 36, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.S.; Wang, Z.B.; Ye, Z.; Lei, J.P.; Li, L.; Su, D.F.; Zheng, X. Resveratrol, an activator of SIRT1, upregulates AMPK and improves cardiac function in heart failure. Genet. Mol. Res. 2014, 13, 323–335. [Google Scholar] [CrossRef]
- Ahmet, I.; Tae, H.J.; Lakatta, E.G.; Talan, M. Long-term low dose dietary resveratrol supplement reduces cardiovascular structural and functional deterioration in chronic heart failure in rats. Can. J. Physiol. Pharmacol. 2017, 95, 268–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.S.; Jin, C.; Huang, X.; Liu, J.; Yan, W.S.; Huang, Q.; Kan, W. The mechanism of polydatin in shock treatment. Clin. Hemorheol. Microcirc. 2003, 29, 211–217. [Google Scholar] [PubMed]
- Zhao, Q.; Huang, H.X.; Jin, C.H. The regulation and its mechanism of polydatin on the β-adrenoreceptor in cardiac myocytes stimulated by lipopolysaccharide. Chin. Pharm. Bull. 2004, 20, 769–772. [Google Scholar]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Gupta, M.P. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 1–16. [Google Scholar]
- Huang, L.; Zhang, K.; Guo, Y.; Huang, F.; Yang, K.; Chen, L.; Yang, Q. Honokiol protects against doxorubicin cardiotoxicity via improving mitochondrial function in mouse hearts. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Du, G.H.; Qiu, Y.; Zhang, J.T. Protective effect of salvianolic acid a on ischemia-reperfusion induced injury in isolated rat heart. Yao Xue Xue Bao 1995, 30, 731–735. [Google Scholar]
- Wang, S.B.; Tian, S.; Yang, F.; Yang, H.G.; Yang, X.Y.; Du, G.H. Cardioprotective effect of salvianolic acid A on isoproterenol-induced myocardial infarction in rats. Eur. J. Pharmacol. 2009, 615, 125–132. [Google Scholar]
- Wang, X.; Guo, D.; Li, W.; Zhang, Q.; Jiang, Y.; Wang, Q.; Li, C.; Qiu, Q.; Wang, Y. Danshen (Salvia miltiorrhiza) restricts MD2/TLR4-MyD88 complex formation and signalling in acute myocardial infarction-induced heart failure. J. Cell. Mol. Med. 2020, 24, 10677–10692. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, K.; Zhu, F.; Wu, Z.; Chu, X.; Zhang, X.; Zhang, Y.; Zhang, J.; Chu, L. Salvia miltiorrhiza (Danshen) inhibits L-type calcium current and attenuates calcium transient and contractility in rat ventricular myocytes. J. Ethnopharmacol. 2014, 158, 397–403. [Google Scholar] [CrossRef] [PubMed]
- McLennan, P.L.; Owen, A.J.; Slee, E.L.; Theiss, M.L. Myocardial function, ischaemia and n-3 polyunsaturated fatty acids: A membrane basis. J. Cardiovasc. Med. 2007, 8, S15–S18. [Google Scholar] [CrossRef]
- Ganguly, R.; Hasanally, D.; Stamenkovic, A.; Maddaford, T.G.; Chaudhary, R.; Pierce, G.N.; Ravandi, A. Alpha linolenic acid decreases apoptosis and oxidized phospholipids in cardiomyocytes during ischemia/reperfusion. Mol. Cell. Biochem. 2018, 437, 163–175. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, X.; Wang, H.; Chen, Z. Beta carotene protects H9c2 cardiomyocytes from advanced glycation end product-induced endoplasmic reticulum stress, apoptosis, and autophagy via the PI3K/Akt/mTOR signaling pathway. Ann. Transl. Med. 2020, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Zhang, Z.; Li, W. Protective effect of chlorogenic acid preconditioning on myocardial ischemia-reperfusion injury in rats. Chin. J. Mod. Appl. Pharm. 2019, 36, 682–685. [Google Scholar]
- He, W.F.; Xue, D.Q.; Yao, L.G.; Li, J.Y.; Li, J.; Guo, Y.W. Hainanerectamines A-C, alkaloids from the Hainan sponge Hyrtios erecta. Mar. Drugs 2014, 12, 3982–3993. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Su, C.P.; Wang, Q.; Wu, F.J.; Bai, R.; Zhang, H.M.; Liu, J.Y.; Lu, W.J.; Wang, W.; Lan, F.; et al. Chlorogenic acid: A potent molecule that protects cardiomyocytes from TNF-alpha-induced injury via inhibiting NF-kappaB and JNK signals. J. Cell. Mol. Med. 2019, 23, 4666–4678. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Wan, C.X.; Huang, S.; Wang, H.; Fan, D.; Wu, H.M.; Wu, Q.; Ma, Z.; Deng, W.; Tang, Q.Z. Oridonin protects against cardiac hypertrophy by promoting P21-related autophagy. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhang, M.; Zhou, F.; Cao, W.; Bi, L.; Xie, Y.; Yang, Q.; Wang, S. Cinnamaldehyde ameliorates LPS-induced cardiac dysfunction via TLR4-NOX4 pathway: The regulation of autophagy and ROS production. J. Mol. Cell. Cardiol. 2016, 101, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Geng, Q.; Huang, H.; Yao, H.; Du, T.; Chen, L.; Wu, Z.; Miao, X.; Shi, P. Antioxidative and cardioprotective effects of Schisandra chinensis bee pollen extract on isoprenaline-induced myocardial infarction in rats. Molecules 2019, 24, 1090. [Google Scholar] [CrossRef] [Green Version]
- Denisow, B.; Denisow-Pietrzyk, M. Biological and therapeutic properties of bee pollen: A review. J. Sci. Food Agric. 2016, 96, 4303–4309. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, M. Microbiology of pollen and bee bread: The yeasts. Apidologie 1979, 10, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Morais, M.; Moreira, L.; Feás, X.; Estevinho, L.M. Honeybee-collected pollen from five Portuguese Natural Parks: Palynological origin, phenolic content, antioxidant properties and antimicrobial activity. Food Chem. Toxicol. 2011, 49, 1096–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Shen, Z.; Geng, Q.; Wu, Z.; Shi, P.; Miao, X. Protective effect of Schisandra chinensis been pollen extract on liver and kidney injury induced by cisplatin in rats. Biomed. Pharmacother. 2017, 95, 1765–1776. [Google Scholar] [CrossRef]
- Ryan, E.A.; Pick, M.E.; Marceau, C. Use of alternative medicines in diabetes mellitus. Diabet. Med. 2001, 18, 242–245. [Google Scholar] [CrossRef]
- Tessema, B.; Mulu, A.; Kassu, A.; Yismaw, G. An in vitro assessment of the antibacterial effect of garlic (Allium sativum) on bacterial isolates from wound infections. Ethiop. Med. J. 2006, 44, 385–389. [Google Scholar] [PubMed]
- Silagy, C.; Neil, A. Garlic as a lipid lowering agent–a meta-analysis. J. R. Coll. Physicians Lond. 1994, 28, 39–45. [Google Scholar]
- Wang, H.C.; Pao, J.; Lin, S.Y.; Sheen, L.Y. Molecular mechanisms of garlic-derived allyl sulfides in the inhibition of skin cancer progression. Ann. N. Y. Acad. Sci. 2012, 1271, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Adaki, S.; Adaki, R.; Shah, K.; Karagir, A. Garlic: Review of literature. Indian J. Cancer 2014, 51, 577–581. [Google Scholar] [CrossRef]
- Charron, C.S.; Dawson, H.D.; Novotny, J.A. Garlic influences gene expression in vivo and in vitro. J. Nutr. 2016, 146, 444S–449S. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Li, S.; Tang, X.; Li, Z.; Zhang, J.; Xue, X.; Han, J.; Liu, Y.; Zhang, Y.; Zhang, Y.; et al. Diallyl trisulfide ameliorates myocardial ischemia-reperfusion injury by reducing oxidative stress and endoplasmic reticulum stress-mediated apoptosis in type 1 diabetic rats: Role of SIRT1 activation. Apoptosis 2017, 22, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Militaru, C.; Donoiu, I.; Craciun, A.; Scorei, I.D.; Bulearca, A.M.; Scorei, R.I. Oral resveratrol and calcium fructoborate supplementation in subjects with stable angina pectoris: Effects on lipid profiles, inflammation markers, and quality of life. Nutrition 2013, 29, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, Y.S.; Wu, J.C.; Huang, Q.; Shahidi, F.; Wang, Y.J.; Ho, C.T.; Pan, M.H. Metabolic and colonic microbiota transformation may enhance the bioactivities of dietary polyphenols. J. Funct. Foods 2014, 7, 3–25. [Google Scholar] [CrossRef]
- Mills, C.E.; Flury, A.; Marmet, C.; Poquet, L.; Rimoldi, S.F.; Sartori, C.; Rexhaj, E.; Brenner, R.; Allemann, Y.; Zimmermann, D.; et al. Mediation of coffee-induced improvements in human vascular function by chlorogenic acids and its metabolites: Two randomized, controlled, crossover intervention trials. Clin. Nutr. 2017, 36, 1520–1529. [Google Scholar] [CrossRef] [Green Version]
- Samavat, H.; Newman, A.R.; Wang, R.; Yuan, J.M.; Wu, A.H.; Kurzer, M.S. Effects of green tea catechin extract on serum lipids in postmenopausal women: A randomized, placebo-controlled clinical trial. Am. J. Clin. Nutr. 2016, 104, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Qiu, B.; Jia, M.; Liu, W.; Guo, X.F.; Li, N.; Xu, Z.X.; Du, F.L.; Xu, T.; Li, D. Effects of α-linolenic acid intake on blood lipid profiles: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2020, 9, 1–17. [Google Scholar] [CrossRef]
- Feliciano, R.P.; Pritzel, S.; Heiss, C.; Rodriguez-Mateos, A. Flavonoid intake and cardiovascular disease risk. Curr. Opin. Food Sci. 2015, 2, 92–99. [Google Scholar] [CrossRef]


{kind=link}
| Cardiac Disease | Antioxidant | Source | Effects/Mechanisms | References |
|---|---|---|---|---|
| Arrhythmias | Acacetin | Honey | Anti-AF, ↓IKur | [177] |
| α-Linolenic acid | Seed, nut, and their oil | Anti-VF, Anti-HF | [223] | |
| Resveratrol | Red wine, blueberry | Anti-arrhythmias | [212] | |
| Salvianolic acid A | Danshen | Anti-VF | [221] | |
| HF/Contractile dysfunction | Polydatin | Hu zhang (polygonum cuspidatum) | ↓ICa, ↑RyR activity, ↑Myofilament Ca2+ sensitivity | [186,215,216] |
| Quercetin | Oak, blueberry | ↓ICa, ↑Ca2+ transient | [112] | |
| Luteolin | Celery, parsley | ↑Contraction, ↑SERCA | [165] | |
| NAC | Onion | ↓HF | [202] | |
| Resveratrol | Red wine, blueberry | ↑Contraction | [214] | |
| Salvianolic acid A | Danshen | ↓ICa, Ca2+ transient, contraction | [222] | |
| Hypertrophy | Rutin | Tea, buckwheat, tobacco | ↓Intracellular Ca2+ | [183] |
| NAC | Onion | ↓MAPK | [202] | |
| α-Linolenic acid | Seeds, nuts, and their oils | ? | [223] | |
| Silymarin | Milk thistle | ↓EGFR | [194] | |
| Honokiol | Magnolia tree bark | ↑Mitochondrial sirtuin 3 | [217] | |
| Isorhamnetin | Hippophae rhamnoides L. | ↓PI3K-Akt | [184] | |
| Apigenin | Chamomile | ? | [190] | |
| Oridonin | Rabdosia rubescens | ? | [229] | |
| MI I-R injury | Resveratrol Polydatin | Red wine, blueberry Hu-zhang | ↑AMPK-sirtuin 1, ↓apoptosis | [184,213] |
| Luteolin | Celery, parsley | ↑Akt, ↓apoptosis | [163] | |
| β-carotene | Carrots, spinach, tomatoes | ↑p-Akt, ↓apoptosis | [225] | |
| EGCG/catechin | Green tea | ↓p-STAT-1, ↓apoptosis | [161,162] | |
| Kaempferol | Honey | ↓Inflammation | [179] | |
| Quercetin | Oak, blueberry | ↓Inflammation | [166,167] | |
| Isorhamnetin Rhamnetin | Hippophae rhamnoides L. Spiraea | ↓ROS, ↓ERK ↓ROS | [185,187,188] | |
| Apigenin | Chamomile | ↑PI3K-Akt | [189] | |
| Chlorogenic acid | Eucommia ulmoides | ↓MEK/ERK | [209] | |
| Protein B | [210] | |||
| Rutin | Tea, buckwheat, tobacco | ↓p-Akt | [180,181,182] | |
| ↑Sirtuin 1 | [181,182] | |||
| DATS, DADS, DAS | Garlic | ↑Nrf2/HO-1 | [242] | |
| Baicalin | Scutellaria baicalensis | ↑Autophagy, ↑MARCH5 ↑HO-1 | [192,193,194,197] | |
| Naringin | Citrus | ↑ATF6-IRE1α-ERK ↑AMPK-sirtuin 3 | [196,197] | |
| Zinc | Meat, oysters | ↓Oxidation | [203,204,205,206,207,208,209] | |
| Salvianolic acid A | Danshen | ↓MD2-TLR4-MyD88, ↓TRAF6-NF-κB | [221] | |
| SCBPE | Bee pollen | ↑Nrf2, HO-1, and Bcl2 | [231] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, S.-H.; Kim, J.-C.; Eslenur, N.; Trinh, T.N.; Do, L.N.H. Modulations of Cardiac Functions and Pathogenesis by Reactive Oxygen Species and Natural Antioxidants. Antioxidants 2021, 10, 760. https://doi.org/10.3390/antiox10050760
Woo S-H, Kim J-C, Eslenur N, Trinh TN, Do LNH. Modulations of Cardiac Functions and Pathogenesis by Reactive Oxygen Species and Natural Antioxidants. Antioxidants. 2021; 10(5):760. https://doi.org/10.3390/antiox10050760
Chicago/Turabian StyleWoo, Sun-Hee, Joon-Chul Kim, Nipa Eslenur, Tran Nguyet Trinh, and Long Nguyen Hoàng Do. 2021. "Modulations of Cardiac Functions and Pathogenesis by Reactive Oxygen Species and Natural Antioxidants" Antioxidants 10, no. 5: 760. https://doi.org/10.3390/antiox10050760
APA StyleWoo, S. -H., Kim, J. -C., Eslenur, N., Trinh, T. N., & Do, L. N. H. (2021). Modulations of Cardiac Functions and Pathogenesis by Reactive Oxygen Species and Natural Antioxidants. Antioxidants, 10(5), 760. https://doi.org/10.3390/antiox10050760