
GSNOR Contributes to Demethylation and Expression of Transposable Elements and Stress-Responsive Genes

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Cultivation
2.2. Quantification of MTA, SAM, SAH, Cys GSH, and Hcys
2.3. Whole-Genome Bisulfite Sequencing and Data Analysis
2.4. RNA Sequencing
2.5. Acid Extraction of Histones
2.6. Quantification of Histone Methylation and Acetylation by LC-MS/MS
2.7. Accession Numbers
3. Results
3.1. GSNOR and SAHH1 Are Involved in Regulating Metabolite Levels of the Methylation Cycle
3.2. Loss of GSNOR1 and SAHH1 Functions Results in Altered Histone Methylation Levels
3.3. SAHH1 and GSNOR1 Functions Affect DNA Methylation
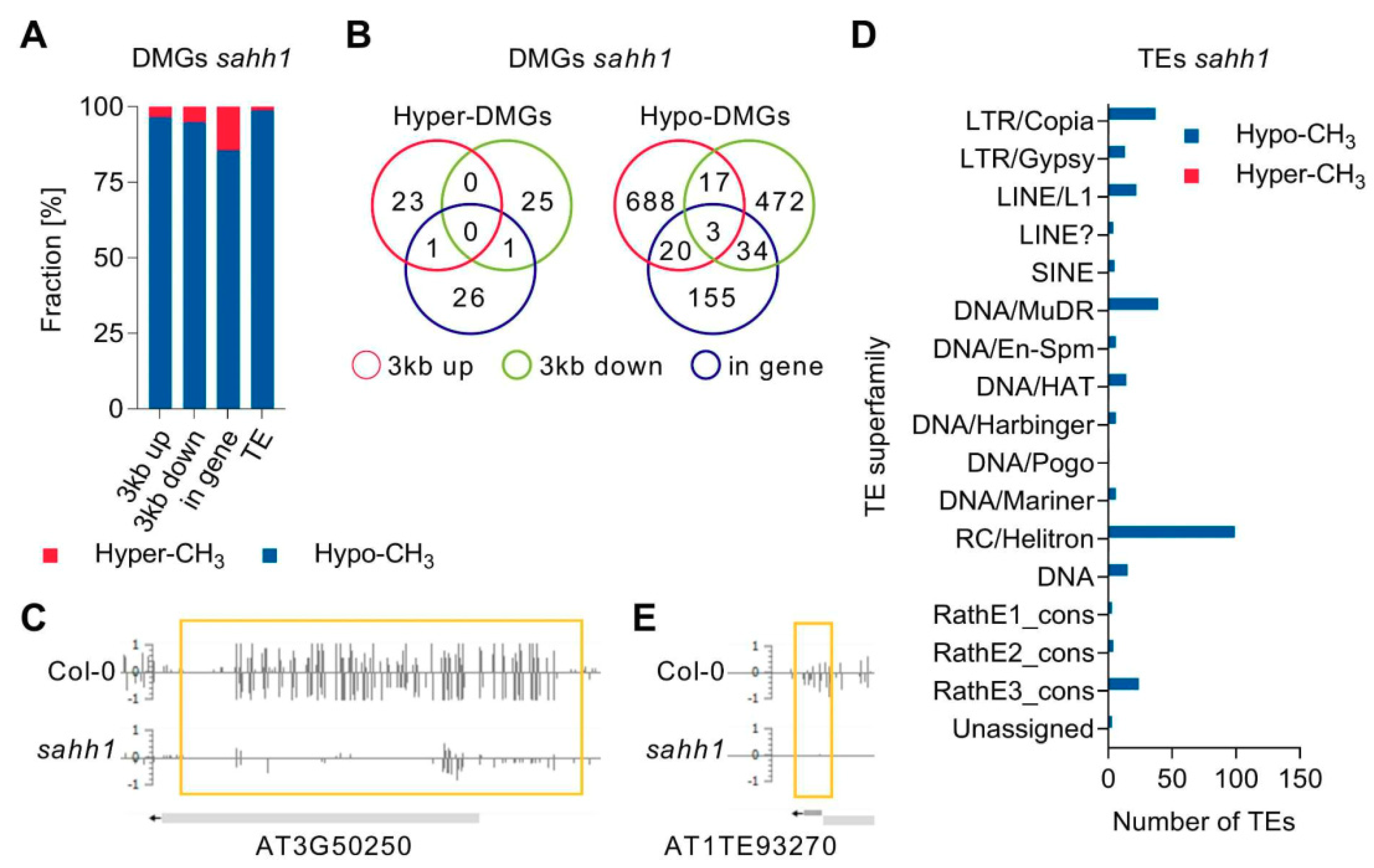
3.4. GSNOR1 and SAHH1 Regulate DNA Methylation of TEs and Genes
3.5. Transcriptomic Profiling of gsnor1-3 and sahh1 Plants
3.6. Integrative Analysis of WGBS and RNA-Seq Data
4. Discussion
4.1. Loss of GSNOR1 Function Results in an Increased Methylation Index
4.2. GSNOR1 Function Is Crucial for the Maintenance of Histone Methylation and DNA Methylation
4.3. Alteration in DNA Methylation Does Not Correlate with Gene Expression
4.4. GSNOR1 Regulates Demethylation and Expression of TEs and Stress-Responsive Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bethke, P.C.; Libourel, I.G.L.; Aoyama, N.; Chung, Y.-Y.; Still, D.W.; Jones, R.L. The Arabidopsis Aleurone Layer Responds to Nitric Oxide, Gibberellin, and Abscisic Acid and Is Sufficient and Necessary for Seed Dormancy. Plant Physiol. 2007, 143, 1173–1188. [Google Scholar] [CrossRef] [Green Version]
- Albertos, P.; Romero-Puertas, M.C.; Tatematsu, K.; Mateos, I.; Sanchez-Vicente, I.; Nambara, E.; Lorenzo, O. S-nitrosylation triggers ABI5 degradation to promote seed germination and seedling growth. Nat. Commun. 2015, 6, 8669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhu, J.K.; Lang, Z. Nitric oxide suppresses the inhibitory effect of abscisic acid on seed germination by S-nitrosylation of SnRK2 proteins. Plant Signal. Behav. 2015, 10, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa-Aragunde, N.; Graziano, M.; Lamattina, L. Nitric oxide plays a central role in determining lateral root development in tomato. Planta 2004, 218, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.Y.; Tian, Q.Y.; Li, L.H.; Zhang, W.H. Nitric oxide is involved in nitrate-induced inhibition of root elongation in Zea mays. Ann. Bot. 2007, 100, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Beligni, M.V.; Lamattina, L. Nitric oxide stimulates seed germination and de-etiolation, and inhibits hypocotyl elongation, three light-inducible responses in plants. Planta 2000, 210, 215–221. [Google Scholar] [CrossRef]
- Laxalt, A.M.; García-Mata, C.; Lamattina, L. The Dual Role of Nitric Oxide in Guard Cells: Promoting and Attenuating the ABA and Phospholipid-Derived Signals Leading to the Stomatal Closure. Front. Plant Sci. 2016, 7, 2007–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Neill, S.J.; Tang, Z.; Cai, W. Nitric Oxide Mediates Gravitropic Bending in Soybean Roots. Plant Physiol. 2005, 137, 663–670. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Tang, R.-H.; Hao, Y.; Stevens, R.D.; Cook, C.W.; Ahn, S.M.; Jing, L.; Yang, Z.; Chen, L.; Guo, F.; et al. Nitric Oxide Represses the Arabidopsis Floral Transition. Science 2004, 305, 1968–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seligman, K.; Saviani, E.E.; Oliveira, H.C.; Pinto-Maglio, C.A.F.; Salgado, I. Floral transition and nitric oxide emission during flower development in Arabidopsis thaliana is affected in nitrate reductase-deficient plants. Plant Cell Physiol. 2008, 49, 1112–1121. [Google Scholar] [CrossRef]
- Prado, A.M. Nitric oxide is involved in growth regulation and re-orientation of pollen tubes. Development 2004, 131, 2707–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishina, T.E.; Lamb, C.; Zeier, J. Expression of a nitric oxide degrading enzyme induces a senescence programme in Arabidopsis. Plant Cell Environ. 2007, 30, 39–52. [Google Scholar] [CrossRef]
- Trapet, P.; Kulik, A.; Lamotte, O.; Jeandroz, S.; Bourque, S.; Nicolas-Francès, V.; Rosnoblet, C.; Besson-Bard, A.; Wendehenne, D. NO signaling in plant immunity: A tale of messengers. Phytochemistry 2015, 112, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Lamattina, L.; Spoel, S.H.; Loake, G.J. Nitric oxide function in plant biology: A redox cue in deconvolution. New Phytol. 2014, 202, 1142–1156. [Google Scholar] [CrossRef]
- Fancy, N.N.; Bahlmann, A.-K.; Loake, G.J. Nitric oxide function in plant abiotic stress. Plant. Cell Environ. 2017, 40, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Buet, A.; Simontacchi, M. Nitric oxide and plant iron homeostasis. Ann. N. Y. Acad. Sci. 2015, 1340, 39–46. [Google Scholar] [CrossRef]
- Gross, F.; Durner, J.; Gaupels, F. Nitric oxide, antioxidants and prooxidants in plant defence responses. Front Plant Sci 2013, 4, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astier, J.; Gross, I.; Durner, J. Nitric oxide production in plants: An update. J. Exp. Bot. 2018, 69, 3401–3411. [Google Scholar] [CrossRef]
- Wurm, C.J.; Lindermayr, C. Nitric oxide signaling in the plant nucleus: The function of nitric oxide in chromatin modulation and transcription. J. Exp. Bot. 2021, 72, 808–818. [Google Scholar] [CrossRef]
- Kolbert, Z.; Feigl, G.; Bordé, Á.; Molnár, Á.; Erdei, L. Protein tyrosine nitration in plants: Present knowledge, computational prediction and future perspectives. Plant Physiol. Biochem. 2017, 113, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, I.; Lindermayr, C. Nitric oxide-based protein modification: Formation and site-specificity of protein S-nitrosylation. Front. Plant Sci 2013, 4, 137. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Chen, L.; Zuo, J. Protein S-Nitrosylation in plants: Current progresses and challenges. J. Integr. Plant Biol. 2019, 61, 1206–1223. [Google Scholar] [CrossRef] [PubMed]
- Ageeva-Kieferle, A.; Rudolf, E.E.; Lindermayr, C. Redox-Dependent Chromatin Remodeling: A New Function of Nitric Oxide as Architect of Chromatin Structure in Plants. Front. Plant Sci. 2019, 10, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, I.; Ageeva, A.; König, E.-E.; Lindermayr, C. Chapter Two—S-Nitrosylation of Nuclear Proteins: New Pathways in Regulation of Gene Expression. In Nitric Oxide and Signaling in Plants; Wendehenne, D., Ed.; Academic Press: London, UK, 2016; Volume 77, pp. 15–39. [Google Scholar]
- Mengel, A.; Chaki, M.; Shekariesfahlan, A.; Lindermayr, C. Effect of nitric oxide on gene transcription—S-nitrosylation of nuclear proteins. Front. Plant Sci. 2013, 4, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León, J.; Costa, Á.; Castillo, M.C. Nitric oxide triggers a transient metabolic reprogramming in Arabidopsis. Sci. Rep. 2016, 6, 37945. [Google Scholar] [CrossRef]
- Freschi, L. Nitric oxide and phytohormone interactions: Current status and perspectives. Front Plant Sci. 2013, 4, 398. [Google Scholar] [CrossRef] [Green Version]
- Simontacchi, M.; García-Mata, C.; Bartoli, C.G.; Santa-María, G.E.; Lamattina, L. Nitric oxide as a key component in hormone-regulated processes. Plant Cell Rep. 2013, 32, 853–866. [Google Scholar] [CrossRef]
- Lindermayr, C. Crosstalk between reactive oxygen species and nitric oxide in plants: Key role of S-nitrosoglutathione reductase. Free Radic. Biol. Med. 2018, 122, 110–115. [Google Scholar] [CrossRef]
- Jahnová, J.; Luhová, L.; Petřivalský, M. S-Nitrosoglutathione Reductase—The Master Regulator of Protein S-Nitrosation in Plant NO Signaling. Plants 2019, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Begara-Morales, J.C.; Chaki, M.; Valderrama, R.; Sánchez-Calvo, B.; Mata-Pérez, C.; Padilla, M.N.; Corpas, F.J.; Barroso, J.B. Nitric oxide buffering and conditional nitric oxide release in stress response. J. Exp. Bot. 2018, 69, ery072. [Google Scholar] [CrossRef]
- Floryszak-Wieczorek, J.; Milczarek, G.; Arasimowicz, M.; Ciszewski, A. Do nitric oxide donors mimic endogenous NO-related response in plants? Planta 2006, 224, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, X.; Chen, L.; Sun, X.; Lu, C.; Zhang, L.; Wang, Y.; Zuo, J. Site-specific nitrosoproteomic identification of endogenously S-nitrosylated proteins in Arabidopsis. Plant Physiol. 2015, 167, 1731–1746. [Google Scholar] [CrossRef] [Green Version]
- Feechan, A.; Kwon, E.; Yun, B.W.; Wang, Y.; Pallas, J.A.; Loake, G.J. A central role for S-nitrosothiols in plant disease resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 8054–8059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, U.; Wie, C.; Fernandez, B.O.; Feelisch, M.; Vierling, E. Modulation of nitrosative stress by S-nitrosoglutathione reductase is critical for thermotolerance and plant growth in Arabidopsis. Plant Cell 2008, 20, 786–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Sun, S.; Wang, C.; Li, Y.; Liang, Y.; An, F.; Li, C.; Dong, H.; Yang, X.; Zhang, J.; et al. The Arabidopsis PARAQUAT RESISTANT2 gene encodes an S-nitrosoglutathione reductase that is a key regulator of cell death. Cell Res. 2009, 19, 1377–1387. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, H.; Ohtani, M.; Kurata, T.; Sakamoto, T.; Demura, T. Protein S-Nitrosylation Regulates Xylem Vessel Cell Differentiation in Arabidopsis. Plant Cell Physiol. 2018, 59, 17–29. [Google Scholar] [CrossRef]
- Yun, B.W.; Skelly, M.J.; Yin, M.; Yu, M.; Mun, B.G.; Lee, S.U.; Hussain, A.; Spoel, S.H.; Loake, G.J. Nitric oxide and S-nitrosoglutathione function additively during plant immunity. New Phytol. 2016, 211, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, I.; Holzmeister, C.; Wirtz, M.; Geerlof, A.; Fröhlich, T.; Römling, G.; Kuruthukulangarakoola, G.T.; Linster, E.; Hell, R.; Arnold, G.J.; et al. ROS-Mediated Inhibition of S-nitrosoglutathione Reductase Contributes to the Activation of Anti-oxidative Mechanisms. Front. Plant Sci. 2016, 7, 1669. [Google Scholar] [CrossRef] [Green Version]
- Chaki, M.; Shekariesfahlan, A.; Ageeva, A.; Mengel, A.; von Toerne, C.; Durner, J.; Lindermayr, C. Identification of nuclear target proteins for S-nitrosylation in pathogen-treated Arabidopsis thaliana cell cultures. Plant Sci. 2015, 238, 115–126. [Google Scholar] [CrossRef]
- Imran, Q.M.; Hussain, A.; Lee, S.U.; Mun, B.G.; Falak, N.; Loake, G.J.; Yun, B.W. Transcriptome profile of NO-induced Arabidopsis transcription factor genes suggests their putative regulatory role in multiple biological processes. Sci. Rep. 2018, 8, 771. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Mengel, A.; Ageeva, A.; Georgii, E.; Bernhardt, J.; Wu, K.; Durner, J.; Lindermayr, C. Nitric Oxide Modulates Histone Acetylation at Stress Genes by Inhibition of Histone Deacetylases. Plant Physiol. 2017, 173, 1434–1452. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Torres-Garcia, J.; Latrasse, D.; Benhamed, M.; Schilderink, S.; Zhou, W.; Kulikova, O.; Hirt, H.; Bisseling, T. Plant-specific histone deacetylases HDT1/2 regulate GIBBERELLIN 2-OXIDASE2 expression to control arabidopsis root meristem cell number. Plant Cell 2017, 29, 2183–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.; Zhuang, T.; Yin, W.; Miao, Y.; Wang, B.; Zhang, Y.; Lin, X.; Xu, C.; von Wettstein, D.; Rustgi, S.; et al. DNA Methylation Changes Induced in Rice by Exposure to High Concentrations of the Nitric Oxide Modulator, Sodium Nitroprusside. Plant Mol. Biol. Report. 2015, 33, 1428–1440. [Google Scholar] [CrossRef]
- Su, X.; Wellen, K.E.; Rabinowitz, J.D. Metabolic control of methylation and acetylation. Curr. Opin. Chem. Biol. 2016, 30, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelova, V.; Ambach, L.; Rébeillé, F.; Stove, C.; Van Der Straeten, D. Folates in Plants: Research Advances and Progress in Crop Biofortification. Front. Chem. 2017, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Sauter, M.; Moffatt, B.; Saechao, M.C.; Hell, R.; Wirtz, M. Methionine salvage and S-adenosylmethionine: Essential links between sulfur, ethylene and polyamine biosynthesis. Biochem. J. 2013, 451, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Ma, L.; Pang, H.; Wang, P.; Liu, L.; Cheng, Y.; Cheng, J.; Guo, Y.; Li, Q. METHIONINE SYNTHASE1 is involved in chromatin silencing by maintaining dna and histone methylation. Plant Physiol. 2019, 181, 249–261. [Google Scholar] [CrossRef]
- Huang, X.-Y.; Li, M.; Luo, R.; Zhao, F.-J.; Salt, D.E. Epigenetic regulation of sulfur homeostasis in plants. J. Exp. Bot. 2019, 70, 4171–4182. [Google Scholar] [CrossRef] [PubMed]
- Rocha, P.S.; Sheikh, M.; Melchiorre, R.; Fagard, M.; Boutet, S.; Loach, R.; Moffatt, B.; Wagner, C.; Vaucheret, H.; Furner, I. The Arabidopsis HOMOLOGY-DEPENDENT GENE SILENCING1 gene codes for an S-adenosyl-L-homocysteine hydrolase required for DNA methylation-dependent gene silencing. Plant Cell 2005, 17, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Baubec, T.; Dinh, H.Q.; Pecinka, A.; Rakic, B.; Rozhon, W.; Wohlrab, B.; von Haeseler, A.; Scheid, O.M. Cooperation of multiple chromatin modifications can generate unanticipated stability of epigenetic States in Arabidopsis. Plant Cell 2010, 22, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Li, C.H.; Yu, N.; Jiang, S.M.; Shangguan, X.X.; Wang, L.J.; Chen, X.Y. Down-regulation of S-adenosyl-L-homocysteine hydrolase reveals a role of cytokinin in promoting transmethylation reactions. Planta 2008, 228, 125–136. [Google Scholar] [CrossRef]
- Jordan, N.D.; West, J.P.; Bottley, A.; Sheikh, M.; Furner, I. Transcript profiling of the hypomethylated hog1 mutant of Arabidopsis. Plant Mol. Biol. 2007, 65, 571–586. [Google Scholar] [CrossRef]
- Ouyang, B.; Fei, Z.; Joung, J.G.; Kolenovsky, A.; Koh, C.; Nowak, J.; Caplan, A.; Keller, W.A.; Cui, Y.; Cutler, A.J.; et al. Transcriptome profiling and methyl homeostasis of an Arabidopsis mutant deficient in S-adenosylhomocysteine hydrolase1 (SAHH1). Plant Mol. Biol. 2012, 79, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Masuta, C.; Uehara, K.; Kataoka, J.; Koiwai, A.; Noma, M. Morphological changes and hypomethylation of DNA in transgenic tobacco expressing antisense RNA of the S-adenosyl-L-homocysteine hydrolase gene. Plant Mol. Biol. 1997, 35, 981–986. [Google Scholar] [CrossRef]
- Fojtová, M.; Kovarík, A.; Votruba, I.; Holý, A. Evaluation of the impact of S-adenosylhomocysteine metabolic pools on cytosine methylation of the tobacco genome. Eur. J. Biochem. 1998, 252, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovavĭk, A.; Koukalová, B.; Holý, A.; Bezděk, M. Sequence-specific hypomethylation of the tobacco genome induced with dihydroxypropyladenine, ethionine and 5-azacytidine. FEBS Lett. 1994, 353, 309–311. [Google Scholar] [CrossRef] [Green Version]
- Fulnecek, J.; Matyasek, R.; Votruba, I.; Holy, A.; Krizova, K.; Kovarik, A. Inhibition of SAH-hydrolase activity during seed germination leads to deregulation of flowering genes and altered flower morphology in tobacco. Mol. Genet. Genom. 2011, 285, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Wang, L.; Wang, J.; Zhao, X.; Cheng, J.; Yu, W.; Jin, D.; Li, Q.; Gong, Z. METHIONINE ADENOSYLTRANSFERASE 4 mediates DNA and histone methylation. Plant Physiol. 2018, 177, 652–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, B.; Vera, P. Folate Metabolism Interferes with Plant Immunity through 1C Methionine Synthase-Directed Genome-wide DNA Methylation Enhancement. Mol. Plant 2019, 12, 1227–1242. [Google Scholar] [CrossRef]
- Wu, X.; Li, F.; Kolenovsky, A.; Caplan, A.; Cui, Y.; Cutler, A.; Tsang, E.W.T. A mutant deficient in S-adenosylhomocysteine hydrolase in Arabidopsis shows defects in root-hair development. Botany 2009, 87, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Morel, J.-B.; Mourrain, P.; Béclin, C.; Vaucheret, H. DNA methylation and chromatin structure affect transcriptional and post-transcriptional transgene silencing in Arabidopsis. Curr. Biol. 2000, 10, 1591–1594. [Google Scholar] [CrossRef] [Green Version]
- Murashige, T.; Skoog, F. A Revised Medium for Rapid Growth and Bio Assays with Tobacco Tissue Cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Bürstenbinder, K.; Rzewuski, G.; Wirtz, M.; Hell, R.; Sauter, M. The role of methionine recycling for ethylene synthesis in Arabidopsis. Plant J. 2007, 49, 238–249. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef]
- Hagmann, J.; Becker, C.; Müller, J.; Stegle, O.; Meyer, R.C.; Wang, G.; Schneeberger, K.; Fitz, J.; Altmann, T.; Bergelson, J.; et al. Century-scale Methylome Stability in a Recently Diverged Arabidopsis thaliana Lineage. PLoS Genet. 2015, 11, e1004920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, C.L.; Alonso, C.; Becker, C.; Bossdorf, O.; Bucher, E.; Colomé-Tatché, M.; Durka, W.; Engelhardt, J.; Gaspar, B.; Gogol-Döring, A.; et al. Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 2017, 20, 1576–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.T.; Germain, H.; Wiermer, M.; Bi, D.; Xu, F.; Garcia, A.V.; Wirthmueller, L.; Despres, C.; Parker, J.E.; Zhang, Y.; et al. Nuclear pore complex component MOS7/Nup88 is required for innate immunity and nuclear accumulation of defense regulators in Arabidopsis. Plant Cell 2009, 21, 2503–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feller, C.; Forné, I.; Imhof, A.; Becker, P.B. Global and specific responses of the histone acetylome to systematic perturbation. Mol. Cell 2015, 57, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Peri, S.; Steen, H.; Pandey, A. GPMAW—A software tool for analyzing proteins and peptides. Trends Biochem. Sci. 2001, 26, 687–689. [Google Scholar] [CrossRef]
- Lindermayr, C.; Saalbach, G.; Durner, J. Proteomic identification of S-nitrosylated proteins in Arabidopsis. Plant Physiol. 2005, 137, 921–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, A.; Rossignol, M.; Peltier, J.B. Proteomics investigation of endogenous S-nitrosylation in Arabidopsis. Biochem. Biophys. Res. Commun. 2011, 416, 331–336. [Google Scholar] [CrossRef]
- Maldonado-Alconada, A.M.; Echevarría-Zomeño, S.; Lindermayr, C.; Redondo-López, I.; Durner, J.; Jorrín-Novo, J.V. Proteomic analysis of Arabidopsis protein S-nitrosylation in response to inoculation with Pseudomonas syringae. Acta Physiol. Plant. 2011, 33, 1493–1514. [Google Scholar] [CrossRef]
- Puyaubert, J.; Fares, A.; Reze, N.; Peltier, J.B.; Baudouin, E.; Rézé, N.; Peltier, J.B.; Baudouin, E.; Reze, N.; Peltier, J.B.; et al. Identification of endogenously S-nitrosylated proteins in Arabidopsis plantlets: Effect of cold stress on cysteine nitrosylation level. Plant Sci. 2014, 215, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Puyaubert, J.; Baudouin, E. New clues for a cold case: Nitric oxide response to low temperature. Plant Cell Env. 2014, 37, 2623–2630. [Google Scholar] [CrossRef]
- Song, X.; Cao, X. Context and Complexity: Analyzing Methylation in Trinucleotide Sequences. Trends Plant Sci. 2017, 22, 351–353. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zhang, S.; Li, K.; Thimmapuram, J.; Xie, S. ViewBS: A powerful toolkit for visualization of high-throughput bisulfite sequencing data. Bioinformatics 2018, 34, 708–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, D.R.; Cornatzer, W.E.; Duerre, J.A. Relationship between tissue levels of S-adenosylmethionine, S-adenosylhomocysteine, and transmethylation reactions. Can. J. Biochem. 1979, 57, 56–64. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Romá-Mateo, C.; Pérez-Machado, G.; Peiró-Chova, L.; Pallardó, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef]
- Detich, N.; Hamm, S.; Just, G.; Knox, J.D.; Szyf, M. The methyl donor S-Adenosylmethionine inhibits active demethylation of DNA. A candidate novel mechanism for the pharmacological effects of S-Adenosylmethionine. J. Biol. Chem. 2003, 278, 20812–20820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascale, R.M.; Simile, M.M.; Satta, G.; Seddaiu, M.A.; Daino, L.; Pinna, G.; Gaspa, M.A.L.V.; Feo, F. Comparative effects of L-methionine, S-adenosyl-L-methionine and 5’-methylthioadenosine on the growth of preneoplastic lesions and DNA methylation in rat liver during the early stages of hepatocarcinogenesis. Anticancer Res. 1991, 11, 1617–1624. [Google Scholar]
- Ara, A.I.; Xia, M.; Ramani, K.; Mato, J.M.; Lu, S.C. S-Adenosylmethionine Inhibits Lipopolysaccharide-Induced Gene Expression via Modulation of Histone Methylation. Hepatology 2008, 47, 1655–1666. [Google Scholar] [CrossRef] [Green Version]
- Speckmann, B.; Schulz, S.; Hiller, F.; Hesse, D.; Schumacher, F.; Kleuser, B.; Geisel, J.; Obeid, R.; Grune, T.; Kipp, A.P. Selenium increases hepatic DNA methylation and modulates one-carbon metabolism in the liver of mice. J. Nutr. Biochem. 2017, 48, 112–119. [Google Scholar] [CrossRef]
- Luka, Z.; Capdevila, A.; Mato, J.M.; Wagner, C. A Glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res. 2006, 15, 393–397. [Google Scholar] [CrossRef]
- Lindermayr, C.; Rudolf, E.E.; Durner, J.; Groth, M. Interactions between metabolism and chromatin in plant models. Mol. Metab. 2020, 38, 100951. [Google Scholar] [CrossRef]
- Hu, J.; Yang, H.; Mu, J.; Lu, T.; Peng, J.; Deng, X.; Kong, Z.; Bao, S.; Cao, X.; Zuo, J. Nitric Oxide Regulates Protein Methylation during Stress Responses in Plants. Mol. Cell 2017, 67, 702–710. [Google Scholar] [CrossRef]
- Li, X.; Harris, C.J.; Zhong, Z.; Chen, W.; Liu, R.; Jia, B.; Wang, Z.; Li, S.; Jacobsen, S.E.; Du, J. Mechanistic insights into plant SUVH family H3K9 methyltransferases and their binding to context-biased non-CG DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, E8793–E8802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Wang, Z.; Li, S.; Yu, B.; Liu, J.Y.; Chen, X. Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev. 2009, 23, 2850–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Wang, H.L.V.; Lee, J.; Dinwiddie, B.L.; Belostotsky, D.A.; Chekanova, J.A. The Role of the Arabidopsis Exosome in siRNA-Independent Silencing of Heterochromatic Loci. PLoS Genet. 2013, 9, e1003411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.W.; Shao, C.R.; Zhang, C.J.; Zhou, J.X.; Zhang, S.W.; Li, L.; Chen, S.; Huang, H.W.; Cai, T.; He, X.J. The SET Domain Proteins SUVH2 and SUVH9 Are Required for Pol V Occupancy at RNA-Directed DNA Methylation Loci. PLoS Genet. 2014, 10, e1003948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyf, M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 243–263. [Google Scholar] [CrossRef] [Green Version]
- Bovee, R.; Pham, V.; Fernandez, J.; Tretyakova, N.; Thomas, D.D. Nitric oxide is an epigenetic regulator of gene expression by directly controlling DNA methylation patterns. Free Radic. Biol. Med. 2018, 120, S114. [Google Scholar] [CrossRef]
- Li, Y.; Kumar, S.; Qian, W. Active DNA demethylation: Mechanism and role in plant development. Plant Cell Rep. 2018, 37, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Crack, J.C.; Green, J.; Thomson, A.J.; Brun, N.E.L. Iron-sulfur clusters as biological sensors: The chemistry of reactions with molecular oxygen and nitric oxide. Acc. Chem. Res. 2014, 47, 3196–3205. [Google Scholar] [CrossRef]
- Navarre, D.A.; Wendehenne, D.; Durner, J.; Noad, R.; Klessig, D.F. Nitric oxide modulates the activity of tobacco aconitase. Plant Physiol. 2000, 122, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Q.; Yuan, W.; Cao, Z.; Qi, B.; Kumar, S.; Li, Y.; Qian, W. The cytosolic Fe-S cluster assembly component MET18 is required for the full enzymatic activity of ROS1 in active DNA demethylation. Sci. Rep. 2016, 6, 26443. [Google Scholar] [CrossRef] [Green Version]
- Hmadcha, B.A.; Bedoya, F.J.; Sobrino, F.; Pintado, E. Methylation-dependent Gene Silencing Induced by Interleukin 1β Via Nitric Oxide Production. J. Exp. Med. 1999, 190, 1595–1603. [Google Scholar] [CrossRef] [Green Version]
- Hewezi, T.; Lane, T.; Piya, S.; Rambani, A.; Rice, J.H.; Staton, M. Cyst Nematode Parasitism Induces Dynamic Changes in the Root Epigenome. Plant Physiol. 2017, 174, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Takuno, S.; Gaut, B.S. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. USA 2013, 110, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Lee, U.S.; Wagner, D. Tug of war: Adding and removing histone lysine methylation in Arabidopsis. Curr. Opin. Plant Biol. 2016, 34, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Begara-Morales, J.C.; Sánchez-Calvo, B.; Luque, F.; Leyva-Pérez, M.O.; Leterrier, M.; Corpas, F.J.; Barroso, J.B. Differential transcriptomic analysis by RNA-seq of GSNO-responsive genes between arabidopsis roots and leaves. Plant Cell Physiol. 2014, 55, 1080–1095. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, H. Writing and Reading Histone H3 Lysine 9 Methylation in Arabidopsis. Front. Plant Sci. 2020, 11, 452. [Google Scholar] [CrossRef]
- Chen, L.T.; Wu, K. Role of histone deacetylases HDA6 and HDA19 in ABA and abiotic stress response. Plant Signal. Behav. 2010, 5, 1318–1320. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Hale, C.J.; Feng, S.; Caro, E.; Jacob, Y.; Michaels, S.D.; Jacobsen, S.E. DNA methyltransferases are required to induce heterochromatic re-replication in Arabidopsis. PLoS Genet. 2012, 8, e1002808. [Google Scholar] [CrossRef]
- Negi, P.; Rai, A.N.; Suprasanna, P. Moving through the stressed genome: Emerging regulatory roles for transposons in plant stress response. Front. Plant Sci. 2016, 7, 1448. [Google Scholar] [CrossRef] [Green Version]
- Bouvet, G.F.; Jacobi, V.; Plourde, K.V.; Bernier, L. Stress-induced mobility of OPHIO1 and OPHIO2, DNA transposons of the Dutch elm disease fungi. Fungal Genet. Biol. 2008, 45, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Johns, M.A.; Mottinger, J.; Freeling, M. A low copy number, copia-like transposon in maize. EMBO J. 1985, 4, 1093–1101. [Google Scholar] [CrossRef]
- Hirochika, H. Activation of tobacco retrotransposons during tissue culture. EMBO J. 1993, 12, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Hirochika, H.; Sugimoto, K.; Otsuki, Y.; Tsugawa, H.; Kanda, M. Retrotransposons of rice involved in mutations induced by tissue culture. Proc. Natl. Acad. Sci. USA 1996, 93, 7783–7788. [Google Scholar] [CrossRef] [Green Version]
- Turcich, M.P.; Bokhari-Riza, A.; Hamilton, D.A.; He, C.; Messier, W.; Stewart, C.-B.; Mascarenhas, J.P. PREM-2, a copia-type retroelement in maize is expressed preferentially in early microspores. Sex. Plant Reprod. 1996, 9, 65–74. [Google Scholar] [CrossRef]
- Grandbastien, M.-A.; Lucas, H.; Morel, J.-B.; Mhiri, C.; Vernhettes, S.; Casacuberta, J.M. The expression of the tobacco Tnt1 retrotransposon is linked to plant defense responses. Genetica 1997, 100, 241–252. [Google Scholar] [CrossRef]
- Grandbastien, M.-A.; Audeon, C.; Bonnivard, E.; Casacuberta, J.M.; Chalhoub, B.; Costa, A.-P.P.; Le, Q.H.; Melayah, D.; Petit, M.; Poncet, C.; et al. Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogenet. Genome Res. 2005, 110, 229–241. [Google Scholar] [CrossRef]
- Bui, Q.T.; Grandbastien, M.-A. LTR Retrotransposons as Controlling Elements of Genome Response to Stress? In Plant Transposable Elements—Impact on Genome Structure and Function; Grandbastien, M.-A., Casacuberta, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 24, pp. 273–296. [Google Scholar]
- McClintock, B. The significance of responses of the genome to challenge. Science 1984, 226, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Fouché, S.; Badet, T.; Oggenfuss, U.; Plissonneau, C.; Francisco, C.S.; Croll, D. Stress-Driven Transposable Element De-repression Dynamics and Virulence Evolution in a Fungal Pathogen. Mol. Biol. Evol. 2020, 37, 221–239. [Google Scholar] [CrossRef]
- Shapiro, J.A. A 21st century view of evolution: Genome system architecture, repetitive DNA, and natural genetic engineering. Gene 2005, 345, 91–100. [Google Scholar] [CrossRef]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Rusterucci, C.; Espunya, M.C.; Diaz, M.; Chabannes, M.; Martinez, M.C. S-Nitrosoglutathione Reductase Affords Protection against Pathogens in Arabidopsis, Both Locally and Systemically. Plant Physiol. 2007, 143, 1282–1292. [Google Scholar] [CrossRef] [Green Version]
- Kroumanová, K.; Kocourková, D.; Daněk, M.; Lamparová, L.; Pospíchalová, R.; Malínská, K.; Krčková, Z.; Burketová, L.; Valentová, O.; Martinec, J.; et al. Characterisation of arabidopsis flotillins in response to stresses. Biol. Plant. 2019, 63, 144–152. [Google Scholar] [CrossRef]
- Daněk, M.; Valentová, O.; Martinec, J. Flotillins, Erlins, and HIRs: From Animal Base Camp to Plant New Horizons. Crit. Rev. Plant Sci. 2016, 35, 191–214. [Google Scholar] [CrossRef]
- Heitz, T.; Widemann, E.; Lugan, R.; Miesch, L.; Ullmann, P.; Désaubry, L.; Holder, E.; Grausem, B.; Kandel, S.; Miesch, M.; et al. Cytochromes P450 CYP94C1 and CYP94B3 catalyze two successive oxidation steps of plant hormone jasmonoyl-isoleucine for catabolic turnover. J. Biol. Chem. 2012, 287, 6296–6306. [Google Scholar] [CrossRef] [Green Version]
- Aubert, Y.; Widemann, E.; Miesch, L.; Pinot, F.; Heitz, T. CYP94-mediated jasmonoyl-isoleucine hormone oxidation shapes jasmonate profiles and attenuates defence responses to Botrytis cinerea infection. J. Exp. Bot. 2015, 66, 3879–3892. [Google Scholar] [CrossRef] [Green Version]
- Kandel, S.; Sauveplane, V.; Compagnon, V.; Franke, R.; Millet, Y.; Schreiber, L.; Werck-Reichhart, D.; Pinot, F. Characterization of a methyl jasmonate and wounding-responsive cytochrome P450 of Arabidopsis thaliana catalyzing dicarboxylic fatty acid formation in vitro. FEBS J. 2007, 274, 5116–5127. [Google Scholar] [CrossRef]
- Wünsche, H.; Baldwin, I.T.; Wu, J. S-Nitrosoglutathione reductase (GSNOR) mediates the biosynthesis of jasmonic acid and ethylene induced by feeding of the insect herbivore Manduca sexta and is important for jasmonate-elicited responses in Nicotiana attenuata. J. Exp. Bot. 2011, 62, 4605–4616. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motif | Sequence of Peptide | Mean % Abundance ± SD | ||
|---|---|---|---|---|
| sahh1 | Col-0/wt | gsnor1-3 | ||
| H3.K4_noPTM | TKQTAR | 42.81 ± 1.81 | 43.64 ± 3.12 | 42.70 ± 3.94 |
| H3.K4me1 | TKme1QTAR | 56.71 ± 1.75 | 55.71 ± 2.56 | 56.43 ± 3.63 |
| H3.K4me2 | TKme2QTAR | 0.21 ± 0.05 | 0.29 ± 0.24 | 0.40 ± 0.16 |
| H3.K4me3 | TKme3QTAR | 0.27 ± 0.08 | 0.36 ± 0.3 | 0.47 ± 0.19 |
| H3.K9_K14_noPTM | KSTGGKAPR | 42.62 ± 0.65 ** | 39.68 ± 0.26 | 37.09 ± 0.78 ** |
| H3.K9ac | KacSTGGKAPR | 1.97 ± 0.31 | 1.55 ± 0.18 | 1.58 ± 0.10 |
| H3.K14ac | KSTGGKacAPR | 27.07 ± 0.79 | 27.02 ± 0.19 | 27.46 ± 0.56 |
| H3.K9ac_K14ac | KacSTGGKacAPR | 3.02 ± 0.12 | 3.13 ± 0.13 | 2.85 ± 0.20 |
| H3.K9me1_K14ac | Kme1STGGKacAPR | 1.17 ± 0.06 | 1.13 ± 0.08 | 1.24 ± 0.14 |
| H3.K9me2_K14ac | Kme2STGGKacAPR | 0.16 ± 0.03 | 0.16 ± 0.06 | 0.22 ± 0.06 |
| H3.K9me3_K14ac | Kme3STGGKacAPR | 0.02 ± 0.00 | 0.03 ± 0.02 | 0.03 ± 0.01 |
| H3.K9me1 | Kme1STGGKAPR | 20.01 ± 0.34 | 21.31 ± 0.61 | 22.14 ± 0.50 |
| H3.K9me2 | Kme2STGGKAPR | 3.88 ± 0.09 *** | 5.83 ± 0.38 | 7.17 ± 0.41 ** |
| H3.K9me3 | Kme3STGGKAPR | 0.10 ± 0.02 | 0.17 ± 0.03 | 0.22 ± 0.02 |
| H3.K18_K23_noPTM | KQLATKAAR | 60.62 ± 0.93 * | 62.46 ± 0.11 | 62.72 ± 0.83 |
| H3.K18ac | KacQLATKAAR | 24.33 ± 0.30 ** | 22.82 ± 0.25 | 22.17 ± 0.57 |
| H3.K23ac | KQLATKacAAR | 6.90 ± 0.35 | 6.82 ± 0.08 | 7.14 ± 0.22 |
| H3.K18ac_K23ac | KacQLATKacAAR | 8.15 ± 0.40 | 7.90 ± 0.09 | 7.97 ± 0.25 |
| H3.1.K27_K36_K37_noPTM | KSAPATGGVKKPHR | 10.19 ± 1.33 | 9.55 ± 1.09 | 9.22 ± 0.83 |
| H3.1.K27ac | KacSAPATGGVKKPHR | 0.18 ± 0.01 | 0.17 ± 0.02 | 0.17 ± 0.01 |
| H3.1.K36ac | KSAPATGGVKacKPHR | 0.15 ± 0.01 | 0.14 ± 0.01 | 0.13 ± 0.01 |
| H3.1.K27ac_K36me2 | KacSAPATGGVKme2KPHR | 0.08 ± 0.01 | 0.07 ± 0.01 | 0.09 ± 0.03 |
| H3.1.K27ac_K36me3 | KacSAPATGGVKme3KPHR | 0.78 ± 0.14 | 0.76 ± 0.09 | 0.63 ± 0.03 |
| H3.1.K27me2_K36ac | Kme2SAPATGGVKacKPHR | 0.16 ± 0.02 | 0.16 ± 0.02 | 0.14 ± 0.02 |
| H3.1.K27me3_K36ac | Kme3SAPATGGVKacKPHR | 0.08 ± 0.04 | 0.07 ± 0.01 | 0.08 ± 0.04 |
| H3.1.K27me1 | Kme1SAPATGGVKKPHR | 49.45 ± 3.87 | 49.40 ± 3.38 | 43.24 ± 0.26 |
| H3.1.K27me2 | Kme2SAPATGGVKKPHR | 18.68 ± 1.87 | 19.48 ± 1.46 | 24.08 ± 0.77 * |
| H3.1.K27me3 | Kme3SAPATGGVKKPHR | 6.53 ± 1.63 | 6.68 ± 1.28 | 7.74 ± 0.24 |
| H3.1.K36me1 | KSAPATGGVKme1KPHR | 2.27 ± 0.36 | 2.32 ± 0.29 | 2.85 ± 0.78 |
| H3.1.K36me2 | KSAPATGGVKme2KPHR | 1.49 ± 0.05 | 1.41 ± 0.08 | 1.63 ± 0.26 |
| H3.1.K36me3 | KSAPATGGVKme3KPHR | 9.97 ± 1.76 | 9.78 ± 1.43 | 10.22 ± 0.59 |
| CG | CHG | CHH | |
|---|---|---|---|
| Col-0/wt | 22.90 ± 2.45% | 6.38 ± 1.28% | 1.54 ± 0.27% |
| gsnor1-3 | 20.32 ± 0.19% | 5.59 ± 0.07% | 1.38 ± 0.03% |
| sahh1 | 19.43 ± 1.18% | 3.11 ± 0.26% | 0.95 ± 0.10% |
| Overlap of Significantly Downregulated Genes with DMGs | ||||||||
| Chr | Start | bp | CH3 | Feature | Gene ID | log2FC | padj. | Description |
| Chr5 | 8751681 | 42 | + | 3kb down | AT5G25250 | −3.37 | 2.0 × 10−266 | Flotillin-like protein 1 (UV-stress) |
| Chr5 | 9309455 | 206 | − | 3kb up | AT5G26690 | −3.35 | 2.3 × 10−23 | Heavy metal-associated isoprenylated plant protein 2 (stress response; but not much data available) |
| Chr2 | 11812888 | 185 | + | 3kb down | AT2G27690 | −2.94 | 7.6 × 10−36 | Cytochrome P450 94C1 (jasmonoyl-L-isoleucine; wounding) |
| Chr5 | 8751681 | 42 | + | 3kb up | AT5G25260 | −2.80 | 2.0 × 10−64 | Flotillin-like protein 2 (UV-stress) |
| Chr2 | 9741371 | 43 | + | in gene | AT2G22880 | −2.24 | 3.8 × 10−12 | At2g22880 (Hypoxia, UV-stress, salt, wounding) |
| Chr2 | 15110344 | 63 | + | 3kb up | AT2G35980 | −2.07 | 1.7 × 10−8 | NDR1/HIN1-like protein 10 (Hypoxia, salt, biotic stress) |
| Chr2 | 18325130 | 77 | + | 3kb up | AT2G44380 | −2.05 | 1.8 × 10−8 | At2g44380 (biotic stress) |
| Chr1 | 24395763 | 100 | + | 3kb up | AT1G65610 | −1.82 | 4.4 × 10−6 | Endoglucanase 7 (biotic stress) |
| Chr5 | 5767502 | 32 | - | 3kb up | AT5G17490 | −1.56 | 6.8 × 10−10 | DELLA protein RGL3 (wounding, cold) |
| Chr3 | 22556563 | 37 | + | 3kb down | AT3G60966 | −1.45 | 1.8 × 10−4 | RING/U-box superfamily protein |
| Chr2 | 18325130 | 77 | + | 3kb up | AT2G44400 | −1.39 | 1.1 × 10−3 | Cysteine/Histidine-rich C1 domain family protein |
| Chr1 | 27068879 | 85 | + | 3kb down | AT1G71910 | −1.34 | 7.2 × 10−6 | At1g71910 |
| Chr2 | 3304271 | 210 | − | in gene | AT2G07774 | −1.31 | 4.1 × 10−5 | unknown protein |
| Chr5 | 18136940 | 44 | + | 3kb up | AT5G44920 | −1.21 | 2.5 × 10−3 | TIR domain-containing protein |
| Chr5 | 18136984 | 64 | + | 3kb up | AT5G44920 | −1.21 | 2.5 × 10−3 | TIR domain-containing protein |
| Chr3 | 3063382 | 181 | − | in gene | AT3G09960 | −1.21 | 4.7 × 10−3 | Calcineurin-like metallo-phosphoesterase superfamily protein |
| Chr5 | 18779966 | 240 | + | in gene | AT5G46295 | −1.19 | 2.3 × 10−4 | Transmembrane protein |
| Chr5 | 18780206 | 180 | + | 3kb up | AT5G46295 | −1.19 | 2.3 × 10−4 | Transmembrane protein |
| Chr2 | 12426536 | 39 | − | 3kb down | AT2G28940 | −1.17 | 1.7 × 10−17 | At2g28940 |
| Chr1 | 4123656 | 44 | − | 3kb up | AT1G12160 | −1.13 | 2.0 × 10−4 | Flavin-containing monooxygenase FMO GS-OX-like 1 |
| Chr1 | 21823145 | 288 | − | 3kb down | AT1G59124 | −1.08 | 2.9 × 10−19 | Probable disease resistance protein RF45 |
| Overlap of Significantly Upregulated Genes with DMGs | ||||||||
| Chr | Start | bp | CH3 | Feature | Gene Id | log2FC | padj. | Description |
| Chr5 | 7376314 | 54 | − | 3kb up | AT5G22300 | 1.82 | 1.3 × 10−39 | Bifunctional nitrilase/nitrile hydratase NIT4 (UV stress, biotic stress) |
| Chr3 | 9173846 | 95 | − | 3kb up | AT3G25190 | 1.30 | 2.7 × 10−4 | Vacuolar iron transporter homolog 2.1 (biotic stress) |
| Chr5 | 17145940 | 99 | + | 3kb up | AT5G42760 | 1.29 | 1.8 × 10−8 | Leucine carboxyl methyltransferase |
| Chr4 | 13766210 | 58 | + | 3kb up | AT4G27570 | 1.02 | 1.1 × 10−2 | UDP-glycosyltransferase 79B3 (jasmonate, cold) |
| Chr5 | 9637396 | 186 | + | 3kb up | AT5G27330 | 1.02 | 2.3 × 10−11 | Prefoldin chaperone subunit family protein |
| Overlap of Significantly Downregulated Genes with DMGs | ||||||||
| Chr | Start | bp | CH3 | Feature | Gene ID | log2FC | padj. | Description |
| Chr5 | 19178939 | 108 | − | 3kb up | AT5G47230 | −1.94 | 1.3 × 10−25 | Ethylene responsive element binding factor 5 ERF5 |
| Chr5 | 5907343 | 107 | − | 3kb up | AT5G17860 | −1.88 | 1.3 × 10−8 | Cation/calcium exchanger 1 |
| Chr2 | 18497356 | 377 | − | 3kb down | AT2G44840 | −1.79 | 9.7 × 10−16 | Ethylene-responsive transcription factor 13 ERF13 |
| Chr1 | 13837861 | 133 | − | 3kb up | AT1G36622 | −1.51 | 4.6 × 10−5 | Transmembrane protein |
| Chr1 | 13837994 | 23 | − | 3kb up | AT1G36622 | −1.51 | 4.6 × 10−5 | Transmembrane protein |
| Chr5 | 7261113 | 306 | − | 3kb up | AT5G21960 | −1.26 | 2.2 × 10−3 | Ethylene-responsive transcription factor ERF016 |
| Chr5 | 16023667 | 82 | − | 3kb up | AT5G40010 | −1.24 | 8.2 × 10−4 | AAA-ATPase ASD, mt |
| Chr1 | 26140005 | 248 | − | 3kb up | AT1G69530 | −1.08 | 1.7 × 10−128 | Expansin |
| Chr4 | 14031509 | 89 | − | 3kb down | AT4G28350 | −1.07 | 8.3 × 10−10 | Probable L-type lectin-domain containing receptor kinase VII.2 |
| Overlap of Significantly Upregulated Genes with DMGs | ||||||||
| Chr | start | bp | CH3 | Feature | Gene ID | log2FC | padj. | Description |
| Chr5 | 9206475 | 54 | − | in gene | AT5G26270 | 5.31 | 3.0 × 10−53 | unknown protein |
| Chr3 | 20260251 | 114 | − | in gene | AT3G54730 | 3.69 | 1.3 × 10−15 | Putative transmembrane protein At3g54730 |
| Chr3 | 20260365 | 94 | − | in gene | AT3G54730 | 3.69 | 1.3 × 10−15 | Putative transmembrane protein At3g54730 |
| Chr3 | 20260459 | 7 | + | 3kb down | AT3G54730 | 3.69 | 1.3 × 10−15 | Putative transmembrane protein At3g54730 |
| Chr5 | 18208166 | 230 | − | in gene | AT5G45095 | 3.12 | 7.8 × 10−11 | Putative uncharacterized protein |
| Chr1 | 12851246 | 141 | − | 3kb up | AT1G35140 | 2.38 | 1.2 × 10−257 | Protein EXORDIUM-like 1 |
| Chr4 | 6431517 | 56 | + | in gene | AT4G10380 | 1.44 | 6.9 × 10−10 | At4g10380 |
| Chr2 | 12887310 | 93 | − | 3kb down | AT2G30210 | 1.44 | 9.8 × 10−4 | Laccase-3 |
| Chr2 | 13160854 | 47 | − | 3kb up | AT2G30930 | 1.43 | 2.3 × 10−99 | Expressed protein |
| Chr1 | 3980123 | 55 | − | in gene | AT1G11785 | 1.31 | 2.6 × 10−3 | Putative uncharacterized protein |
| Chr3 | 9173846 | 95 | − | 3kb up | AT3G25190 | 1.29 | 6.6 × 10−4 | Vacuolar iron transporter homolog 2.1 |
| Chr3 | 21509510 | 77 | − | 3kb up | AT3G58070 | 1.14 | 2.0 × 10−3 | GIS |
| Chr3 | 20206910 | 10 | − | 3kb up | AT3G54580 | 1.01 | 4.4 × 10−2 | Proline-rich extensin-like family protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudolf, E.E.; Hüther, P.; Forné, I.; Georgii, E.; Han, Y.; Hell, R.; Wirtz, M.; Imhof, A.; Becker, C.; Durner, J.; et al. GSNOR Contributes to Demethylation and Expression of Transposable Elements and Stress-Responsive Genes. Antioxidants 2021, 10, 1128. https://doi.org/10.3390/antiox10071128
Rudolf EE, Hüther P, Forné I, Georgii E, Han Y, Hell R, Wirtz M, Imhof A, Becker C, Durner J, et al. GSNOR Contributes to Demethylation and Expression of Transposable Elements and Stress-Responsive Genes. Antioxidants. 2021; 10(7):1128. https://doi.org/10.3390/antiox10071128
Chicago/Turabian StyleRudolf, Eva Esther, Patrick Hüther, Ignasi Forné, Elisabeth Georgii, Yongtao Han, Rüdiger Hell, Markus Wirtz, Axel Imhof, Claude Becker, Jörg Durner, and et al. 2021. "GSNOR Contributes to Demethylation and Expression of Transposable Elements and Stress-Responsive Genes" Antioxidants 10, no. 7: 1128. https://doi.org/10.3390/antiox10071128
APA StyleRudolf, E. E., Hüther, P., Forné, I., Georgii, E., Han, Y., Hell, R., Wirtz, M., Imhof, A., Becker, C., Durner, J., & Lindermayr, C. (2021). GSNOR Contributes to Demethylation and Expression of Transposable Elements and Stress-Responsive Genes. Antioxidants, 10(7), 1128. https://doi.org/10.3390/antiox10071128