
Study of the Antioxidant Effects of Coffee Phenolic Metabolites on C6 Glioma Cells Exposed to Diesel Exhaust Particles

 , ,
, ,  , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Determination of Intracellular Reactive Oxygen Species (ROS)
2.4. Determination of Cells Viability
2.5. Method Setup with Tert-Butylhydroperoxide (tBHP) and N-Acetylcysteine (NAC)
2.6. Assessment of Diesel Exhaust Particles (DEP) Effects
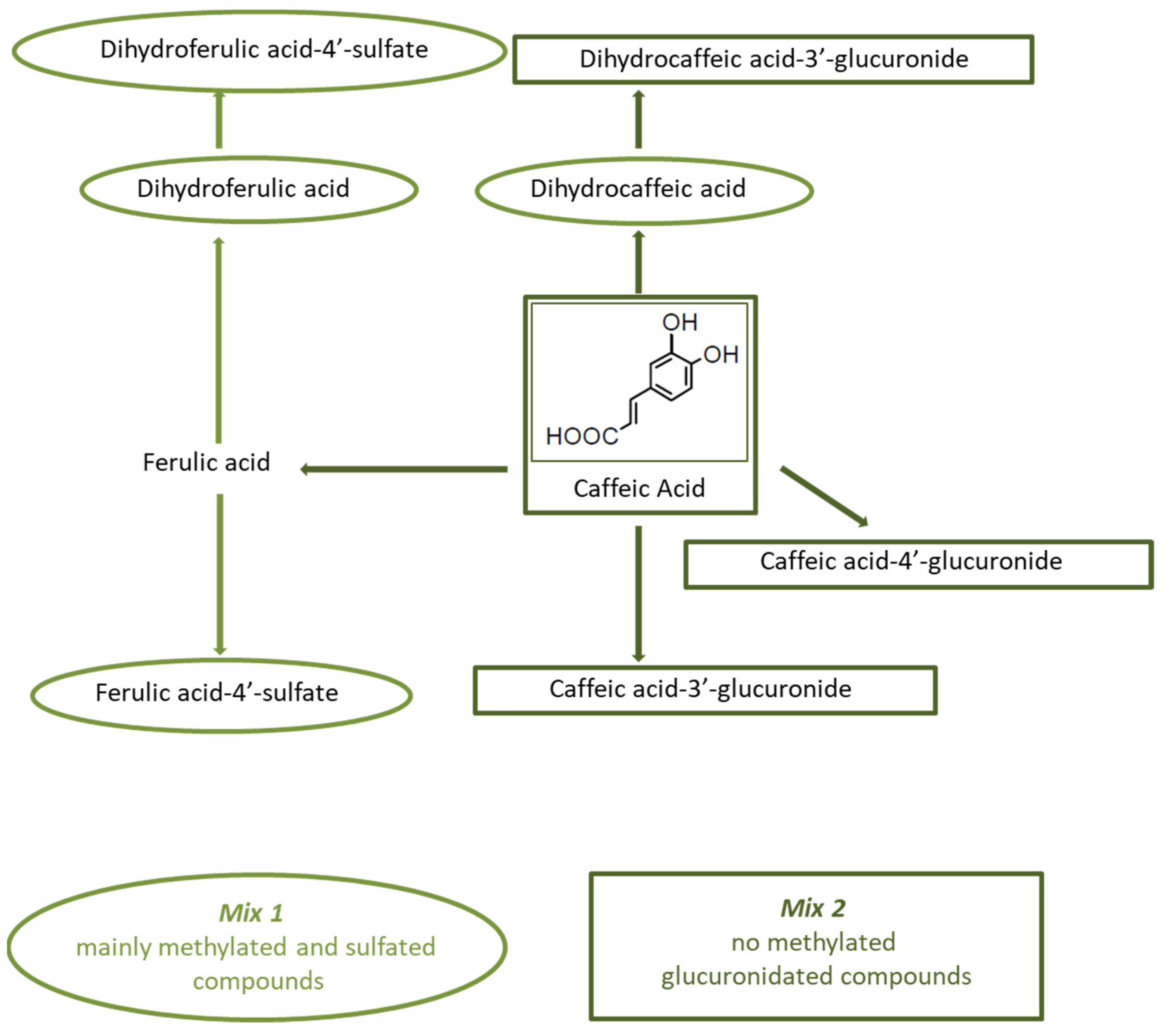
2.7. Coffee Phenolic Metabolites
2.8. SDS-PAGE Electrophoresis and Immunoblotting
2.9. Statistical Analysis
3. Results
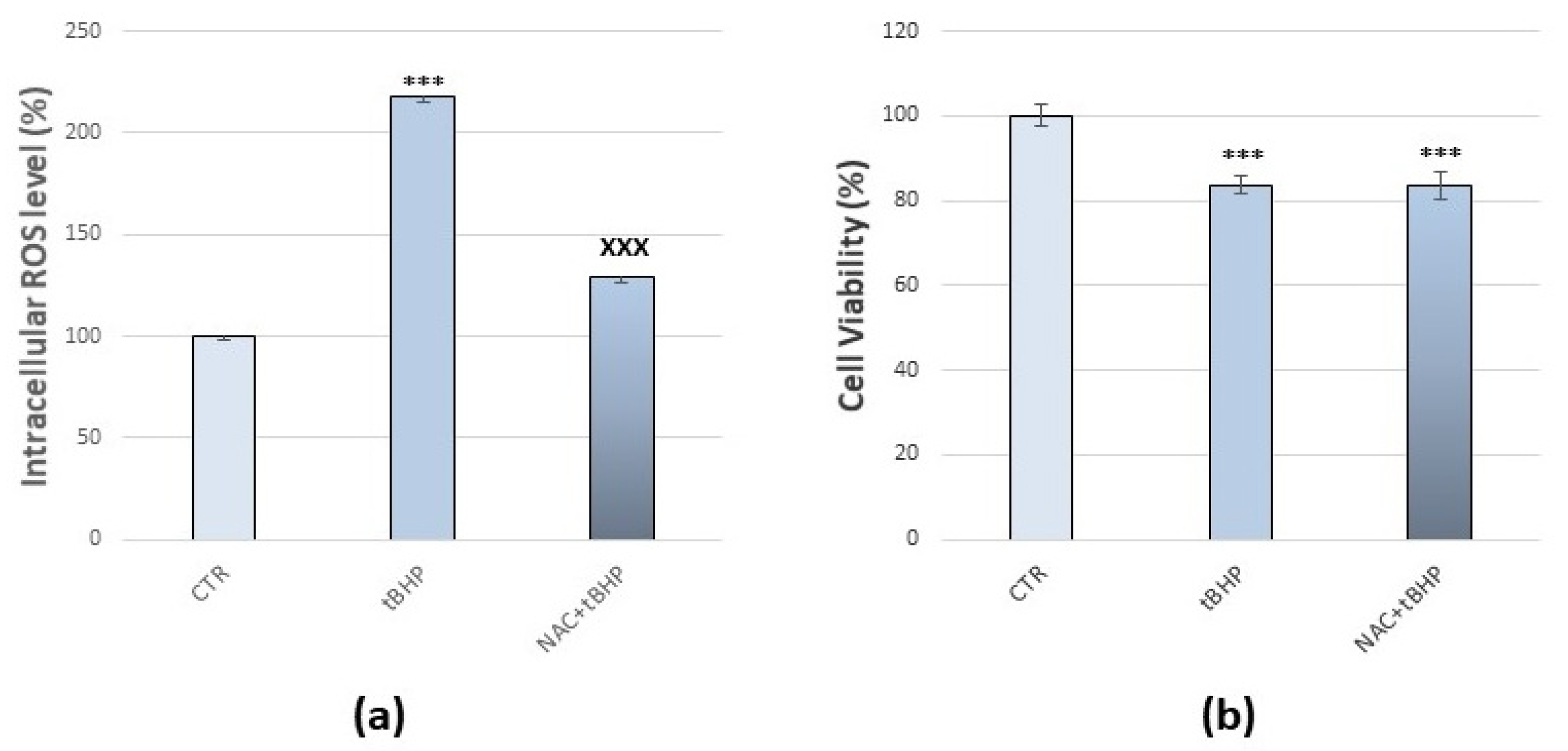
3.1. Method Setup with tBHP and NAC
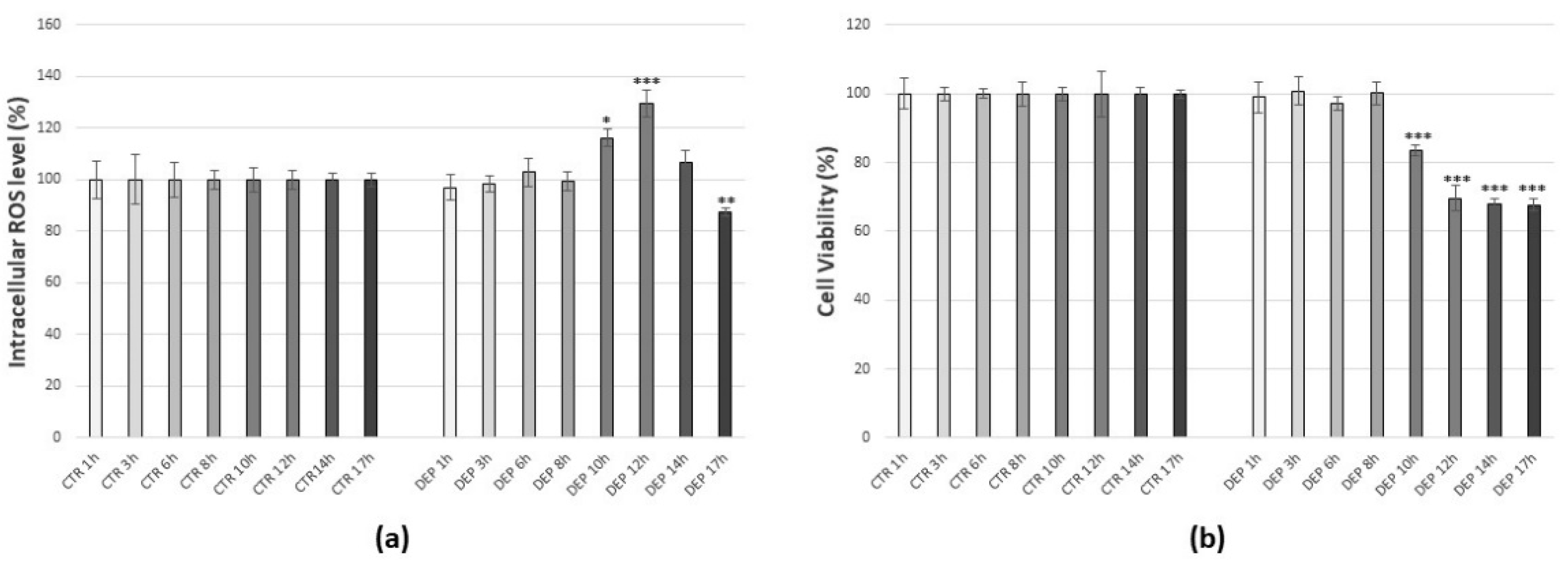
3.2. Effects of Diesel Exhaust Particles (DEPs)
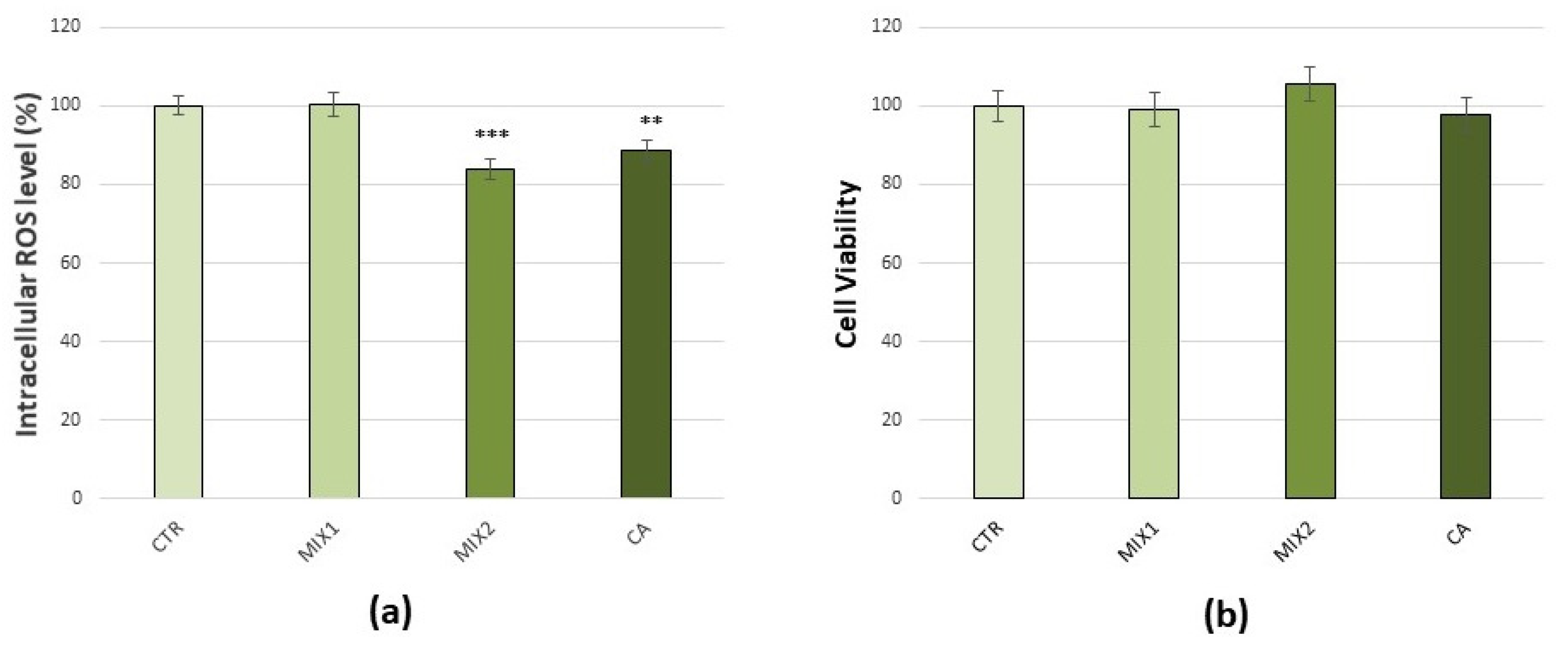
3.3. Effects of Coffee Phenolic Metabolites
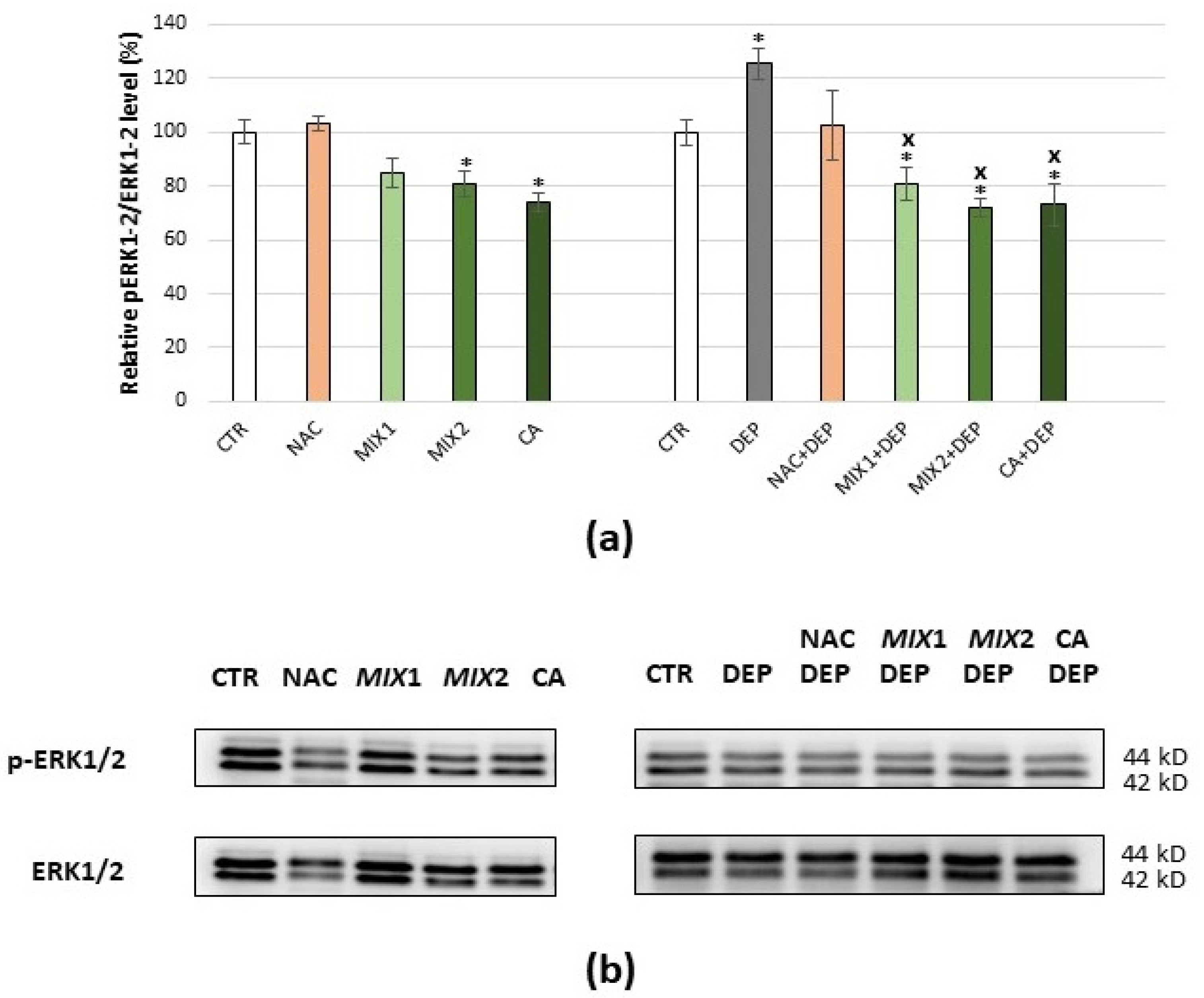
3.4. Effects of Coffee Phenolic Metabolites against DEP-Mediated Oxidative Stress
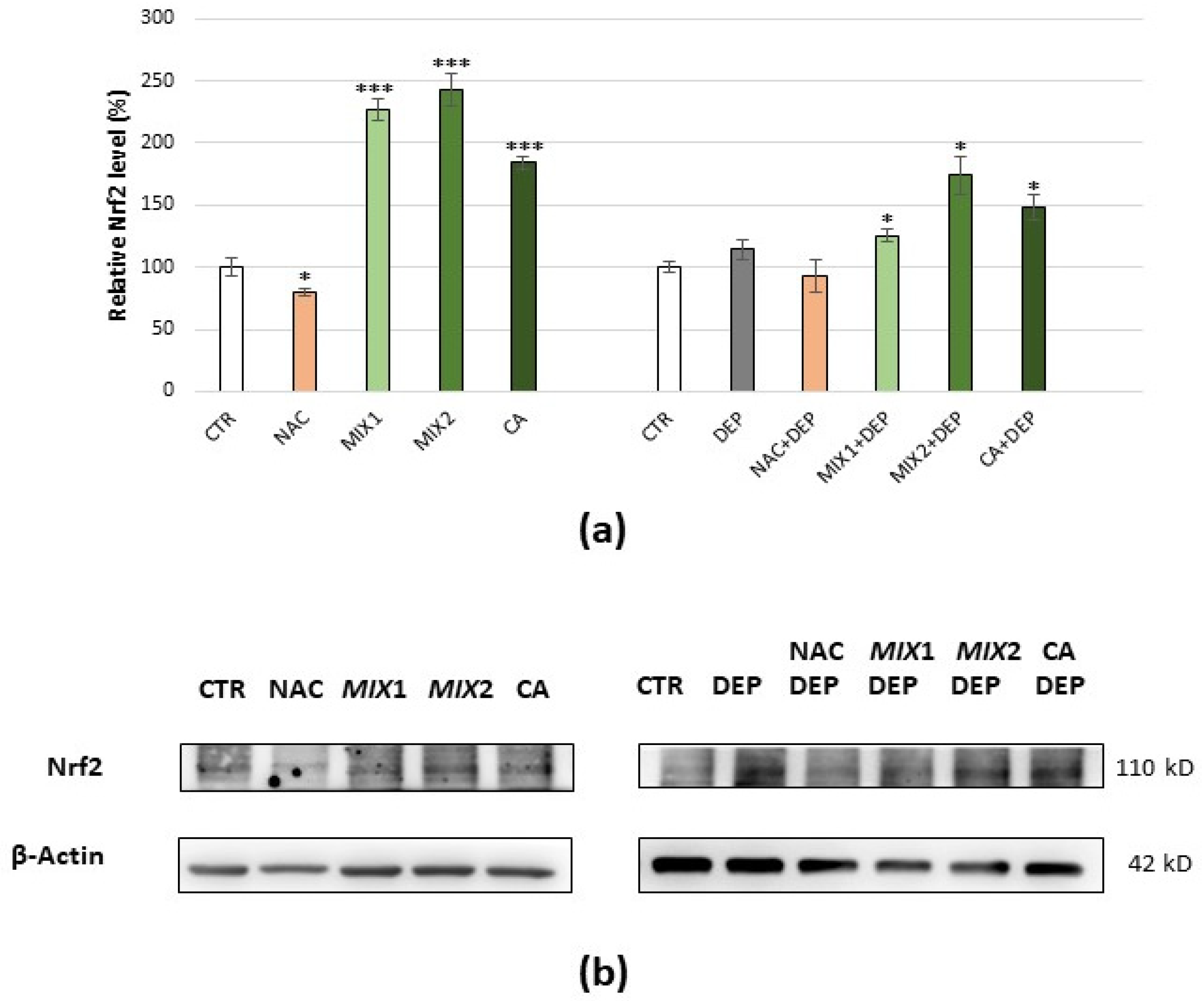
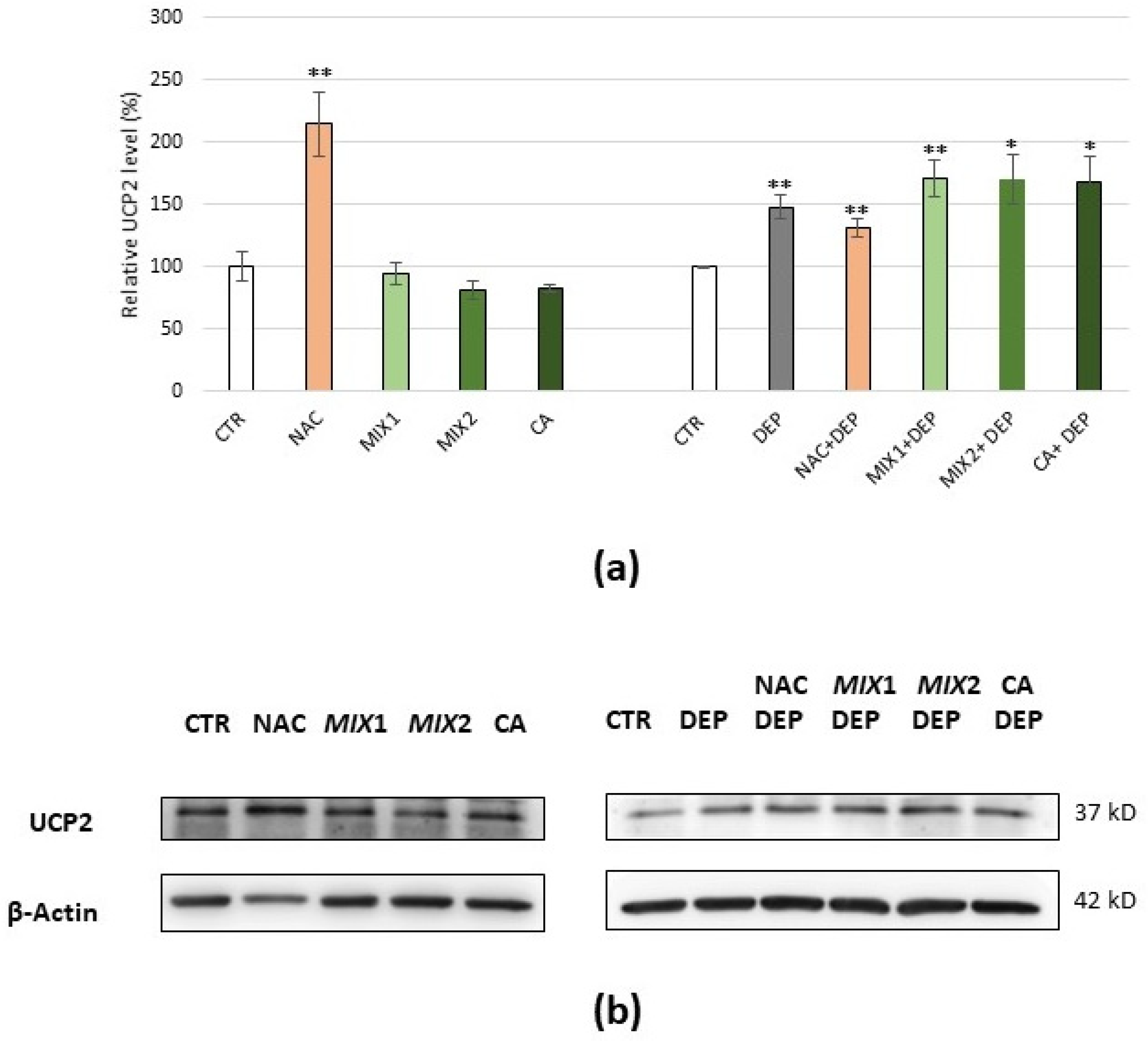
3.5. Analysis of Oxidative Stress-Related Proteins
4. Discussion
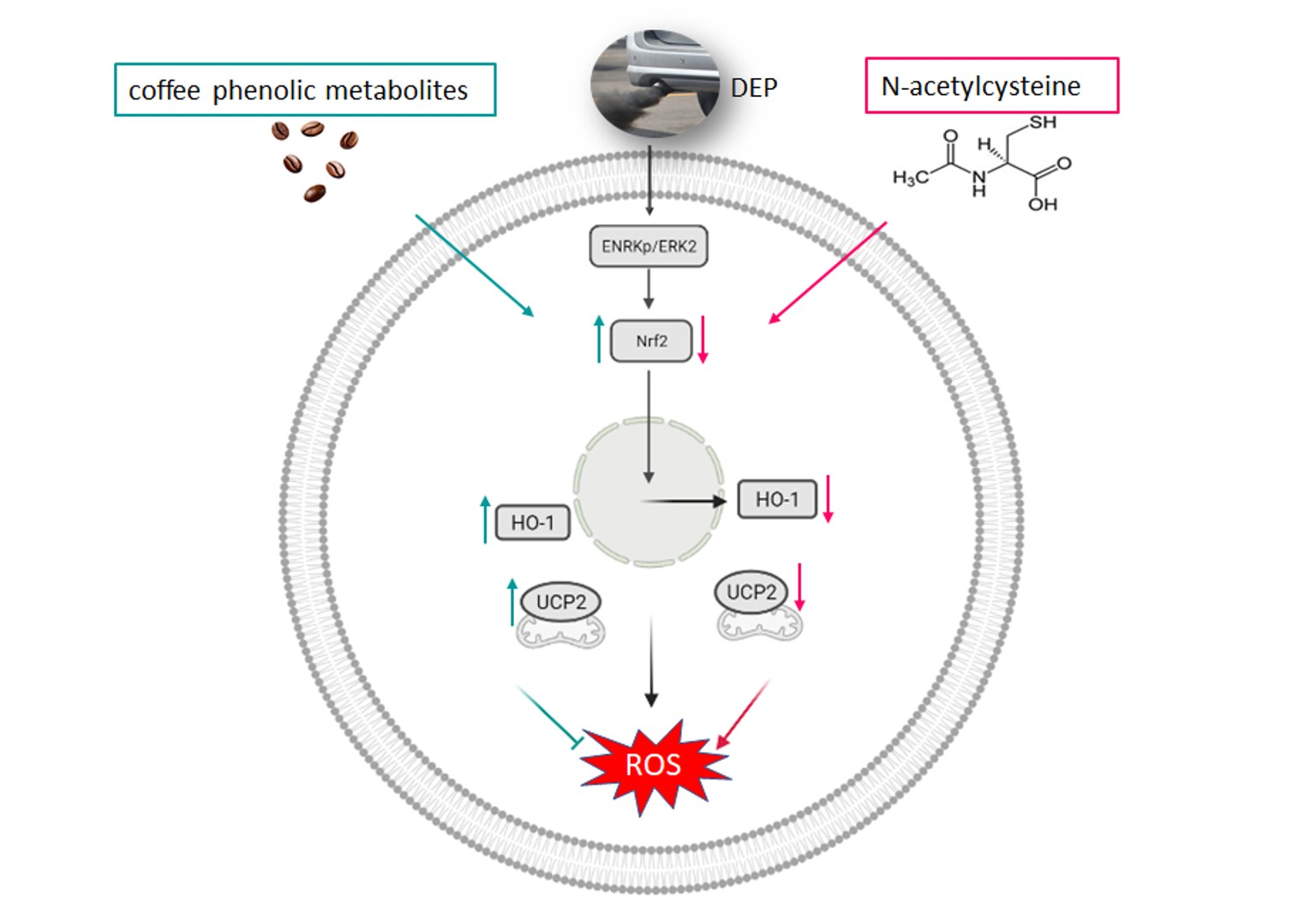
4.1. Antioxidant Strategies of Coffee Phenolic Metabolites against DEP-Induced Oxidative Stress
4.2. Proteins Involvement in Defense against Oxidative Stress
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Block, M.L.; Calderón-Garcidueñas, L. Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009, 32, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesterberg, T.W.; Long, C.M.; Lapin, C.A.; Hamade, A.K.; Valberg, P.A. Diesel exhaust particulate (DEP) and nanoparticle exposures: What do DEP human clinical studies tell us about potential human health hazards of nanoparticles. Inhal. Toxicol. 2010, 22, 679–694. [Google Scholar] [CrossRef]
- Ma, J.Y.; Ma, J.K. The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J. Environ. Sci. Health Part C Environ. Carcinog Ecotoxicol. Rev. 2002, 20, 117–147. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Wu, X.; Pei, Z.; Li, G.; Wang, T.; Qin, L.; Wilson, B.; Yang, J.; Hong, J.S.; Veronesi, B. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: The role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004, 18, 1618–1620. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Garciduenas, L.; Reed, W.; Maronpot, R.R.; Henriquez-Roldan, C.; Delgado-Chavez, R.; Calderon-Garciduenas, A.; Dragustinovis, I.; Franco-Lira, M.; Aragon-Flores, M.; Solt, A.C.; et al. Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol. Pathol. 2004, 32, 650–658. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Maronpot, R.R.; Torres-Jardon, R.; Henriquez-Roldan, C.; Schoonhoven, R.; Acuna-Ayala, H.; Villarreal-Calderon, A.; Nakamura, J.; Fernando, R.; Reed, W.; et al. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol. Pathol. 2003, 31, 524–538. [Google Scholar] [CrossRef] [Green Version]
- Wellenius, G.A.; Schwartz, J.; Mittleman, M.A. Air pollution and hospital admissions for ischemic and hemorrhagic stroke among medicare beneficiaries. Stroke 2005, 36, 2549–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-Garciduenas, L.; Franco-Lira, M.; Torres-Jardon, R.; Henriquez-Roldan, C.; Barragan-Mejia, G.; Valencia-Salazar, G.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Villarreal-Calderon, R.; Reed, W. Pediatric respiratory and systemic effects of chronic air pollution exposure: Nose, lung, heart, and brain pathology. Toxicol. Pathol. 2007, 35, 154–162. [Google Scholar] [CrossRef]
- Peters, A.; Veronesi, B.; Calderon-Garciduenas, L.; Gehr, P.; Chen, L.C.; Geiser, M.; Reed, W.; Rothen-Rutishauser, B.; Schurch, S.; Schulz, H. Translocation and potential neurological effects of fine and ultrafine particles a critical update. Part. Fibre Toxicol. 2006, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Anika, M.S.; Hartz, B.B.; Block, M.L.; Hong, J.S.; Miller, D.S. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoproteinup-regulation at the blood-brain barrier. FASEB J. 2008, 22, 2723–2733. [Google Scholar] [CrossRef] [Green Version]
- Møller, P.; Jacobsen, N.R.; Folkmann, J.K.; Danielsen, P.H.; Mikkelsen, L.; Hemmingsen, J.G.; Vesterdal, L.K.; Forchhammer, L.; Wallin, H.; Loft, S. Role of oxidative damage in toxicity of particulates. Free Radic. Res. 2010, 44, 1–46. [Google Scholar] [CrossRef]
- Totlandsdal, A.I.; Herseth, J.I.; Bølling, A.K.; Kubátová, A.; Braun, A.; Cochran, R.E.; Refsnes, M.; Ovrevik, J.; Låg, M. Differential effects of the particle core and organic extract of diesel exhaust particles. Toxicol. Lett. 2012, 208, 262–268. [Google Scholar] [CrossRef]
- Cheung, K.L.; Ntziachristos, L.; Tzamkiozis, T.; Schauer, J.J.; Samaras, Z.; Moore, K.F.; Sioutas, C. Emissions of particulate trace elements, metals and organic species from gasoline, diesel, and biodiesel passenger vehicles and their relation to oxidative potential. Aerosol. Sci. Technol. 2010, 44, 500–513. [Google Scholar] [CrossRef]
- Ristovski, Z.D.; Miljevic, B.; Surawski, M.C.; Morawska, L.; Fong, K.M.; Goh, F.; Yang, I.A. Respiratory health effects of diesel particulate matter. Respirology 2012, 17, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, F.; Milani, C.; Botto, L.; Lonati, E.; Bulbarelli, A.; Palestini, P. Involvement of MEK-ERK1–2 pathway in the antioxidant response in C6 glioma cells after diesel exhaust particles exposure. Toxicol. Lett. 2016, 250–251, 57–65. [Google Scholar] [CrossRef]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the Nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.H.; Jang, J.H.; Na, H.K.; Cha, Y.N.; Surh, Y.J. Carbon monoxide produced by heme oxygenase-1 in response to nitrosative stress induces expression of glutamatecysteine ligase in PC12 cells via activation of phosphatidylinositol 3-kinase and Nrf2 signaling. J. Biol. Chem. 2007, 282, 28577–28586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iles, K.E.; Dickinson, D.A.; Wigley, A.F.; Welty, N.E.; Blank, V.; Forman, H.J. HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radic. Biol. Med. 2005, 39, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Alam, J.; Venkatesan, M.I.; Eiguren-Fernandez, A.; Schmitz, D.; Di Stefano, E.; Slaughter, N.; Killeen, E.; Wang, X.; Huang, A.; et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: Protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 2004, 173, 3467–3481. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [Google Scholar] [CrossRef]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Horvath, T.L. Uncoupling protein-2 regulates lifespan in mice. Am. J. Physiol.Endocrinol. Metab. 2009, 296, E621–E627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim-Han, J.S.; Dugan, L.L. Mitochondrial Uncoupling Proteins in the Central Nervous System. Antioxid. Redox Signal. 2005, 7, 1173–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, C.H.; Joo, S.H. Downregulation of Reactive Oxygen Species in Apoptosis. J. Cancer Prev. 2016, 21, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youdim, K.A.; Joseph, J.A. A possible emerging role of phytochemicals in improving age-related neurological dysfunctions: A multiplicity of effects. Free Radic. Biol. Med. 2001, 30, 583–594. [Google Scholar] [CrossRef]
- Youdim, K.A.; Spencer, J.P.E.; Schroeter, H.; Rice-Evans, C. Dietary flavonoids as potential neuroprotectants. Biol. Chem. 2002, 383, 503–519. [Google Scholar] [CrossRef]
- Potì, F.; Santi, D.; Spaggiari, G.; Zimetti, F.; Zanotti, I. Polyphenol Health Effects on Cardiovascular and Neurodegenerative Disorders: A Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 351. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [Green Version]
- Bento-Silva, A.; Koistinen, V.M.; Mena, P.; Bronze, M.R.; Hanhineva, K.; Sahlstrøm, S.; Kitrytė, V.; Moco, S.; Aura, A.M. Factors affecting intake, metabolism and health benefits of phenolic acids: Do we understand individual variability? Eur. J. Nutr. 2020, 59, 1275–1293. [Google Scholar] [CrossRef] [Green Version]
- Ziauddeen, N.; Rosi, A.; Del Rio, D.; Amoutzopoulos, B.; Nicholson, S.; Page, P.; Scazzina, F.; Brighenti, F.; Ray, S.; Mena, P. Dietary intake of (poly)phenols in children and adults: Cross-sectional analysis of UK National Diet and Nutrition Survey Rolling Programme (2008–2014). Eur. J. Nutr. 2019, 58, 3183–3198. [Google Scholar] [CrossRef]
- Zamora-Ros, R.; Rothwell, J.A.; Scalbert, A.; Knaze, V.; Romieu, I.; Slimani, N.; Fagherazzi, G.; Perquier, F.; Touillaud, M.; Molina-Montes, E.; et al. Dietary intakes and food sources of phenolic acids in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Br. J. Nutr. 2013, 110, 1500–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, I.A.; Mena, P.; Calani, L.; Cid, C.; Del Rio, D.; Lean, M.E.J.; Crozier, A. Variations in caffeine and chlorogenic acid contents of coffees: What are we drinking? Food Funct. 2014, 5, 1718–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, M.N.; Johnston, K.L.; Knight, S.; Kuhnert, N. Hierarchical scheme for LC-MSn identification of chlorogenic acids. J. Agric. Food Chem. 2003, 51, 2900–2911. [Google Scholar] [CrossRef]
- Morton, K.; Knight, K.; Kalman, D.; Hewlings, S. A Prospective Randomized, Double-Blind, Two-Period Crossover Pharmacokinetic Trial Comparing Green Coffee Bean Extract—A Botanically Sourced Caffeine—With a Synthetic USP Control. Clin. Pharmacol. Drug Dev. 2018, 7, 871–879. [Google Scholar] [CrossRef]
- Clifford, M.N. Chlorogenic acids and other cinnamates—nature, occurrence and dietary burden. J. Sci. Food Agric. 1999, 79, 362–372. [Google Scholar] [CrossRef]
- Stalmach, A.; Mullen, W.; Nagai, C.; Crozier, A. On-line HPLC analysis of the antioxidant activity of phenolic compounds in brewed paper-filtered coffee. Brasil. J. Plant. Physiol. 2006, 18, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Stalmach, A.; Mullen, W.; Barron, D.; Uchida, K.; Yokota, T.; Cavin, C.; Steiling, H.; Williamson, G.; Crozier, A. Metabolite profiling of hydroxycinnamate derivatives in plasma and urine after the ingestion of coffee by humans: Identification of biomarkers of coffee consumption. Drug Metab. Dispos. 2009, 37, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalmach, A.; Steiling, H.; Williamson, G.; Crozier, A. Bioavailability of chlorogenic acids following acute ingestion of coffee by humans with an ileostomy. Arch. Biochem. Biophys. 2010, 501, 98–105. [Google Scholar] [CrossRef]
- Crozier, A.; Jaganath, I.B.; Clifford, M.N. Dietary phenolics: Chemistry, bioavailability and effects on health. Nat. Prod. Rep. 2009, 26, 1001–1043. [Google Scholar] [CrossRef]
- Donovan, J.L.; Manach, C.; Faulks, R.M.; Kroon, P.A. Absorption and metabolism of dietary secondary metabolites. In Plant Secondary Metabolites: Occurrence, Structure and Role in the Human Diet; Crozier, A., Clifford, M.N., Ashihara, H., Eds.; Blackwell Publishing: Oxford, UK, 2006; pp. 303–351. [Google Scholar]
- Carregosa, D.; Carecho, R.; Figueira, I.; Santos, C.N. Low-Molecular Weight Metabolites from Polyphenols as Effectors for Attenuating Neuroinflammation. J. Agric. Food Chem. 2020, 68, 1790–1807. [Google Scholar] [CrossRef] [Green Version]
- Jäger, A.K.; Saaby, L. Flavonoids and the CNS. Molecules 2011, 16, 1471–1485. [Google Scholar] [CrossRef] [Green Version]
- Kuresh, A.Y.; Dobbie, M.S.; Kuhnle, G.; Proteggente, A.R.; Abbott, N.J.; Rice-Evans, C. Interaction between flavonoids and the blood–brain barrier: In vitro studies. J. Neurochem. 2003, 85, 180–192. [Google Scholar] [CrossRef]
- Shaoa, J.; Li, X.; Lu, X.; Jiang, C.; Hud, Y.; Li, Q.; You, Y.; Fu, Z. Enhanced growth inhibition effect of Resveratrol incorporated into biodegradable nanoparticles against glioma cells is mediated by the induction of intracellular reactive oxygen species levels. Colloids Surf. B Biointerfaces 2009, 72, 40–47. [Google Scholar] [CrossRef]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 1989, 119, 203–210. [Google Scholar] [CrossRef]
- Ghosh, M.; Manna, P.; Sil, P.C. Protective role of a coumarin-derived schiff base scaffold against tertiary butyl hydroperoxide (TBHP)-induced oxidative impairment and cell death via MAPKs, NF-κB and mitochondria-dependent pathways. Free Radic. Res. 2011, 45, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 2004, 117, 2417–2426. [Google Scholar] [CrossRef] [Green Version]
- Alía, M.; Ramos, S.; Mateos, R.; Bravo, L.; Goya, L. Response of the antioxidant defense system to tert-butyl hydroperoxide and hydrogen peroxide in a human hepatoma cell line (HepG2). J. Biochem. Mol. Toxicol. 2005, 19, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Marangolo, M.; McGee, M.M.; Tipton, K.F.; Williams, D.C.; Zisterer, D.M. Oxidative stress induces apoptosis in C6 glioma cells: Involvement of mitogen-activated protein kinases and nuclear factor kappa B. Neurotox. Res. 2001, 3, 397–409. [Google Scholar] [CrossRef]
- Levesque, S.; Taetzsch, T.; Lull, M.E.; Kodavanti, U.; Stadler, K.; Wagner, A.; Johnson, J.O.; Duke, L.; Kodavanti, P.; Surace, M.J.; et al. Diesel exhaust activates and primes microglia: Air pollution, neuroinflammation, and regulation of dopaminergic neurotoxicity. Environ. Health Perspect. 2011, 119, 1149. [Google Scholar] [CrossRef] [Green Version]
- Kreyling, W.G.; Semmler-Behnke, M.; Seitz, J.; Scymczak, W.; Wenk, A.; Mayer, P.; Takenaka, S.; Oberdörsteret, G. Size dependence of the translocation of inhaled iridium and carbon nanoparticle aggregates from the lung of rats to the blood and secondary target organs. Inhal. Toxicol. 2009, 21, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Stalmach, A.; Williamson, G.; Crozier, A. Impact of dose on the bioavailability of coffee chlorogenic acids in humans. Food Funct. 2014, 5, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Benda, P.; Lightbody, J.; Sato, G.; Levine, L.; Sweet, W. Differentiated rat glial cell strain in tissue culture. Science 1968, 161, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J.; Fujimoto, K.; Marasa, J.C.; Agrawal, H.C. Relation of C-6 glial cells in culture to myelin. Biochem. J. 1975, 152, 701–703. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.G.; Xu, J.; Li, Z.G.; Ren, G.G.; Yang, Z. In vitro toxicity of multi-walled carbon nanotubes in C6 rat Glioma cells. NeuroToxicology 2012, 33, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, I.; Weiner, L.; Vogel, Z. Delta9-tetrahydrocannabinol increases C6 glioma cell death produced by oxidative stress. Neuroscience 2005, 134, 567–574. [Google Scholar] [CrossRef]
- Robb, S.J.; Connor, J.R. An in vitro model for analysis of oxidative death in primary mouse astrocytes. Brain Res. 1998, 788, 125–132. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.Y.; Jiang, Q.; Li, K.R.; Zhao, Y.X.; Cao, C.; Yao, J. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic. Biol. Med. 2014, 69, 219–228. [Google Scholar] [CrossRef]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Chen, L.; Wang, H.; Huang, M.; Sun, X.; Kann, J.; Du, J.; Li, Y. Protective effect of supplementation with Ginseng, Lilii Bulbus and Poria against PM2.5 in air pollution-induced cardiopulmonary damage among adults. Phytother. Res. 2020, 1–11. [Google Scholar] [CrossRef]
- Rice-Evans, C.A. Flavonoid antioxidants. Curr. Med. Chem. 2001, 8, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Danesi, F.; Del Rio, D.; Mena, P. The importance of studying cell metabolism when testing the bioactivity of phenolic compounds. Trends Food Sci. Technol. 2017, 69, 230–242. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef]
- Tsai, M.H.; Hsu, L.F.; Lee, C.W.; Chiang, Y.C.; Lee, M.H.; How, J.M.; Wu, C.M.; Huang, C.L.; Lee, I.T. Resveratrol inhibits urban particulate matter-induced COX-2/PGE2 release in human fibroblast-like synoviocytes via the inhibition of activation of NADPH oxidase/ROS/NF-κB. Int. J. Biochem. Cell Biol. 2017, 88, 113–123. [Google Scholar] [CrossRef]
- Wu, W.; Peden, D.B.; McConnell, R.; Fruin, S.; Sanchez, D.D. Glutathione-S-transferase M1 regulation of diesel exhaust particle-induced pro-inflammatory mediator expression in normal human bronchial epithelial cells. Part. Fibre Toxicol. 2012, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.I.; Tsai, C.H.; Sun, Y.L.; Hsieh, W.Y.; Lin, Y.C.; Chen, C.Y.; Lin, C.S. Instillation of particulate matter 2.5 induced acute lung injury and attenuated the injury recovery in ACE2 knockout mice. Int. J. Biol Sci. 2018, 14, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Li, P.G.; Xu, J.W.; Ikeda, K.; Kobayakawa, A.; Kayano, Y.; Mitani, T.; Ikami, T.; Yamori, Y. Caffeic Acid Inhibits Vascular Smooth Muscle Cell Proliferation Induced by Angiotensin II in Stroke-Prone Spontaneously Hypertensive Rats. Hypertens. Res. 2005, 28, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharikadze, N.; Jojua, N.; Sepashvili, M.; Zhuravliova, E.; Mikeladze, D.G. Mitochondrial Target of Nobiletin’s Action. NPC Nat. Prod. Commun. 2016, 11, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.S.; Hwang, Y.J.; Dong, Z. ERK1 directly interacts with JNK1 leading to regulation of JNK1/c-Jun activity and cell transformation. J. Cell Biochem. 2017, 118, 2357–2370. [Google Scholar] [CrossRef]
- Kim, J.K.; Jang, H.D. Nrf2-Mediated HO-1 induction coupled with the ERK signaling pathway contributes to indirect antioxidant capacity of caffeic acid phenethyl ester in HepG2 cells. J. Mol. Sci. 2014, 15, 12149–12165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.J.; Ryoo, I.G.; Lee, Y.J.; Kwak, M.K. Role of the Nrf2-heme oxygenase-1 pathway in silver nanoparticle-mediated cytotoxicity. Toxicol. Appl. Pharmacol. 2012, 258, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Staurengo-Ferrari, L.; Badaro-Garcia, S.; Hohmann, M.S.; Manchope, M.F.; Zaninelli, T.H.; Casagrande, R.; Verri, W.A., Jr. Contribution of Nrf2 modulation to the mechanism of action of analgesic and anti-inflammatory drugs in pre-clinical and clinical stages. Front. Pharmacol. 2019, 9, 1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J. Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol. Sci. 2004, 83, 313–328. [Google Scholar] [CrossRef]
- Nguyen, T. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2002, 278, 4536–4541. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, B.; Li, S.; Zeng, L.; Chen, Y.; Fang, J. Mangiferin increases Nrf2 protein stability by inhibiting its ubiquitination and degradation in human HL60 myeloid leukemia cells. Int. J. Mol. Med. 2014, 33, 1348–1354. [Google Scholar] [CrossRef] [Green Version]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.; Talbot, D.A.; Brand, M.D. Superoxide Activates Mitochondrial Uncoupling Protein 2 from the Matrix Side Studies Using Targeted Antioxidants. J. Biol. Chem. 2002, 277, 47129–47135. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Qiu, X.; Zimmermann, R.; Rudich, Y. Particulate Matter Toxicity Is Nrf2 and Mitochondria Dependent: The Roles of Metals and Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2020, 33, 1110–1120. [Google Scholar] [CrossRef]
- Pallauf, K.; Duckstein, N.; Hasler, M.; Klotz, L.O.; Rimbach, G. Flavonoids as Putative Inducers of the Transcription Factors Nrf2, FoxO, and PPARγ. Oxid. Med. Cell. Longev. 2017, 4397340. [Google Scholar] [CrossRef] [Green Version]
- Zane, B.A.; Horvath, B.; Barnstable, C.J.; Elsworth, J.; Yang, L.; Beal, M.F.; Roth, R.H.; Matthews, R.T.; Horvath, T.L. Uncoupling Protein-2 Is Critical for Nigral Dopamine Cell Survival in a Mouse Model of Parkinson’s Disease. J. Neurosci. 2005, 25, 184–191. [Google Scholar] [CrossRef]
- Li, L.X.; Skorpen, F.; Egeberg, K.; Hals Jørgensen, I.; Grill, V. Uncoupling Protein-2 Participates in Cellular Defense against Oxidative Stress in Clonal β-Cells. Biochem. Biophys. Res. Commun. 2001, 282, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Alves-Guerra, M.C.; Rousset, S.; Pecqueur, C.; Mallat, Z.; Blanc, J.; Tedgui, A.; Bouillaud, F.; Cassard-Doulcier, A.; Ricquier, D.; Miroux, B. Bone Marrow Transplantation Reveals the in Vivo Expression of the Mitochondrial Uncoupling Protein 2 in Immune and Nonimmune Cells during Inflammation. J. Biol. Chem. 2003, 278, 42307–42312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, S.; Mozo, J.; Dujardin, G.; Emre, Y.; Masscheleyn, S.; Ricquier, D.; Cassard-Doulcier, A.M. UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Lett. 2007, 581, 479–482. [Google Scholar] [CrossRef]
- Martini, D.; Del Bò, C.; Tassotti, M.; Riso, P.; Del Rio, D.; Brighenti, F.; Porrini, M. Coffee Consumption and Oxidative Stress: A Review of Human Intervention Studies. Molecules 2016, 21, 979. [Google Scholar] [CrossRef] [Green Version]
- Rathod, M.A.; Patel, D.; Das, A.; Tipparaju, S.R.; Shinde, S.S.; Anderson, R.F. Inhibition of radical-induced DNA strand breaks by water-soluble constituents of coffee: Phenolics and caffeine metabolites. Free Radic. Res. 2013, 47, 480–487. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botto, L.; Bulbarelli, A.; Lonati, E.; Cazzaniga, E.; Tassotti, M.; Mena, P.; Del Rio, D.; Palestini, P. Study of the Antioxidant Effects of Coffee Phenolic Metabolites on C6 Glioma Cells Exposed to Diesel Exhaust Particles. Antioxidants 2021, 10, 1169. https://doi.org/10.3390/antiox10081169
Botto L, Bulbarelli A, Lonati E, Cazzaniga E, Tassotti M, Mena P, Del Rio D, Palestini P. Study of the Antioxidant Effects of Coffee Phenolic Metabolites on C6 Glioma Cells Exposed to Diesel Exhaust Particles. Antioxidants. 2021; 10(8):1169. https://doi.org/10.3390/antiox10081169
Chicago/Turabian StyleBotto, Laura, Alessandra Bulbarelli, Elena Lonati, Emanuela Cazzaniga, Michele Tassotti, Pedro Mena, Daniele Del Rio, and Paola Palestini. 2021. "Study of the Antioxidant Effects of Coffee Phenolic Metabolites on C6 Glioma Cells Exposed to Diesel Exhaust Particles" Antioxidants 10, no. 8: 1169. https://doi.org/10.3390/antiox10081169
APA StyleBotto, L., Bulbarelli, A., Lonati, E., Cazzaniga, E., Tassotti, M., Mena, P., Del Rio, D., & Palestini, P. (2021). Study of the Antioxidant Effects of Coffee Phenolic Metabolites on C6 Glioma Cells Exposed to Diesel Exhaust Particles. Antioxidants, 10(8), 1169. https://doi.org/10.3390/antiox10081169