
Melatonin as a Reducer of Neuro- and Vasculotoxic Oxidative Stress Induced by Homocysteine



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
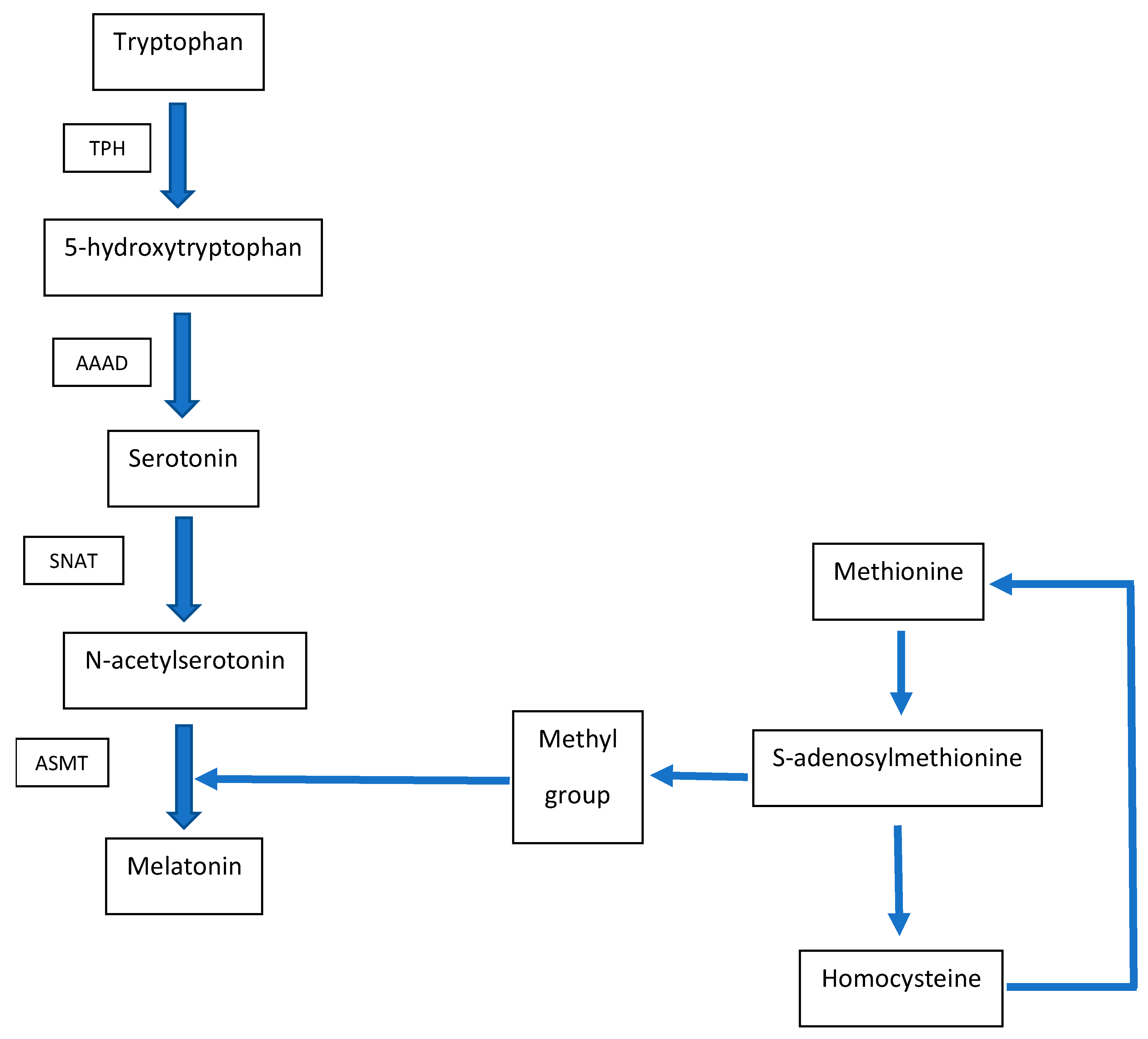
2. Metabolic Interconnection between Melatonin and Homocysteine
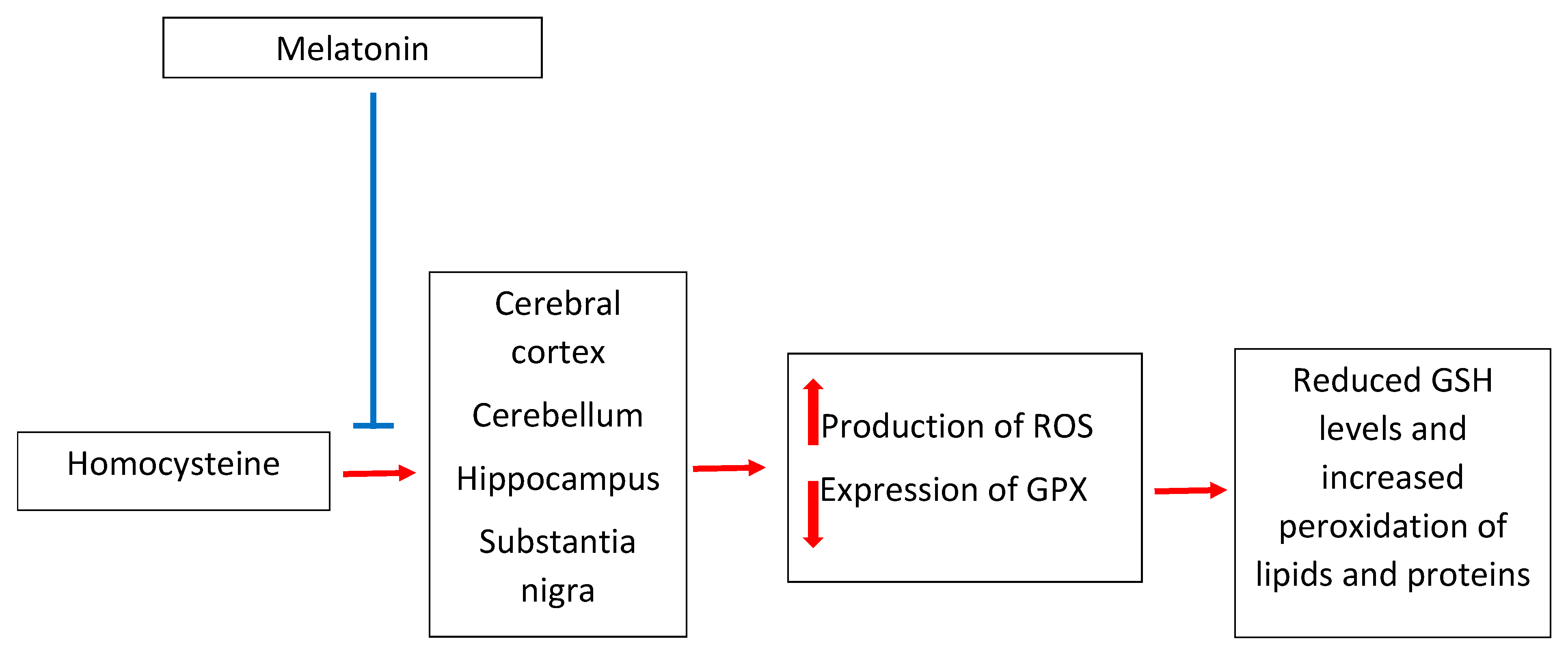
3. Melatonin and Homocysteine in Neurotoxicity
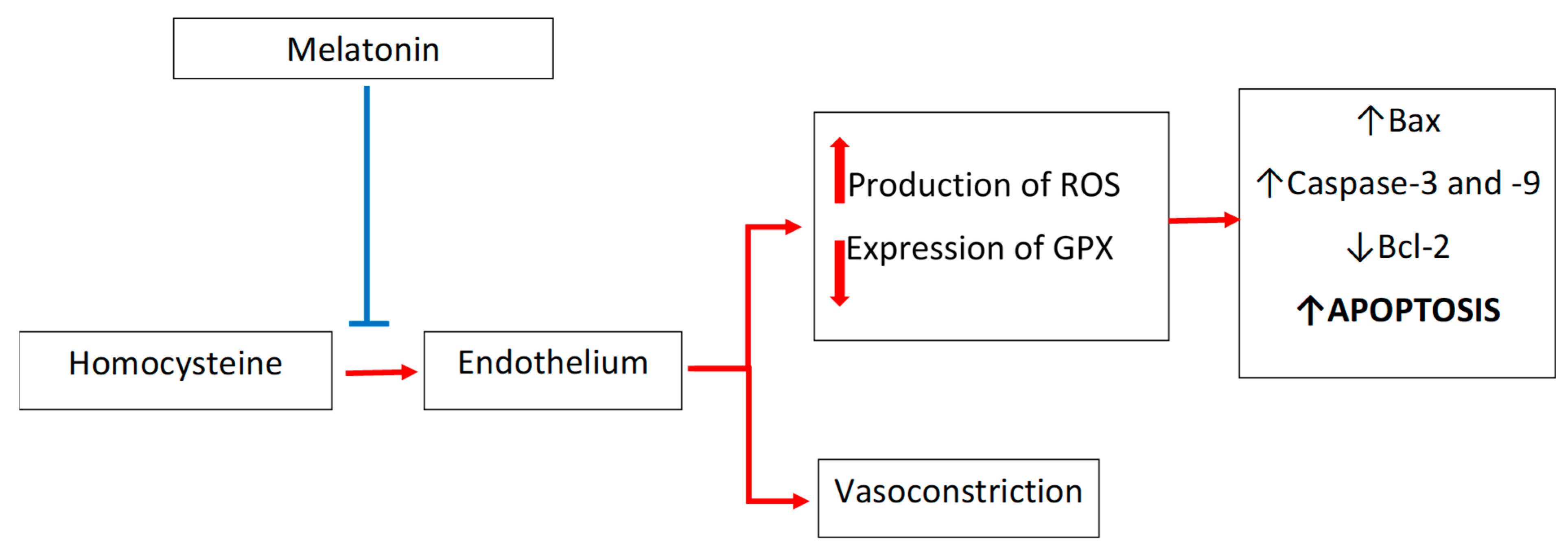
4. Melatonin and Homocysteine in Cardiovascular Diseases: Focus on Endothelium
5. Melatonin as a Regulator of Blood Homocysteine Levels
6. Conclusions
Funding
Conflicts of Interest
References
- Cajochen, C.; Kräuchi, K.; Wirz-Justice, A. Role of Melatonin in the Regulation of Human Circadian Rhythms and Sleep. J. Neuroendocrinol. 2003, 15, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zisapel, N. New Perspectives on the Role of Melatonin in Human Sleep, Circadian Rhythms and Their Regulation. Br. J. Pharmacol. 2018, 175, 3190–3199. [Google Scholar] [CrossRef]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a Master Regulator of Cell Death and Inflammation: Molecular Mechanisms and Clinical Implications for Newborn Care. Cell Death Dis. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Amaral, F.G.D.; Andrade-Silva, J.; Kuwabara, W.M.T.; Cipolla-Neto, J. New Insights into the Function of Melatonin and its Role in Metabolic Disturbances. Expert Rev. Endocrinol. Metab. 2019, 14, 293–300. [Google Scholar] [CrossRef]
- Baltatu, O.C.; Senar, S.; Campos, L.A.; Cipolla-Neto, J. Cardioprotective Melatonin: Translating from Proof-of-Concept Studies to Therapeutic Use. Int. J. Mol. Sci. 2019, 20, 4342. [Google Scholar] [CrossRef] [Green Version]
- Karolczak, K.; Watala, C. The Mystery behind the Pineal Gland: Melatonin Affects the Metabolism of Cholesterol. Oxid. Med. Cell. Longev. 2019, 2019, 4531865. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of Melatonin in the Reduction of Oxidative Stress. A review. J. Biomed. Sci. 2000, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D.; Martin, J.J. Homocysteine. Int. J. Biochem. Cell. Biol. 2000, 32, 385–389. [Google Scholar] [CrossRef]
- McCully, K.S. Homocysteine and the Pathogenesis of Atherosclerosis. Expert Rev. Clin. Pharmacol. 2015, 8, 211–219. [Google Scholar] [CrossRef]
- Obeid, R.; Herrmann, W. Mechanisms of Homocysteine Neurotoxicity in Neurodegenerative Diseases with Special Reference to Dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refsum, H.; Ueland, P.M.; Nygård, O.; Vollset, S.E. Homocysteine and Cardiovascular Disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef] [PubMed]
- Tchantchou, F. Homocysteine Metabolism and Various Consequences of Folate Deficiency. J. Alzheimers Dis. 2006, 9, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Desouza, C.; Keebler, M.; McNamara, D.B.; Fonseca, V. Drugs Affecting Homocysteine Metabolism: Impact on Cardiovascular Risk. Drugs 2002, 62, 605–616. [Google Scholar] [CrossRef]
- Refsum, H.; Smith, A.D.; Ueland, P.M.; Nexo, E.; Clarke, R.; McPartlin, J.; Johnston, C.; Engbaek, F.; Schneede, J.; McPartlin, C.; et al. Facts and Recommendations about Total Homocysteine Determinations: An Expert Opinion. Clin. Chem. 2004, 50, 3–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Ingrosso, D.; Lombardi, C.; Acanfora, F.; Satta, E.; Cesare, C.M.; Violetti, E.; Romano, M.M.; De Santo, N.G. Possible Mechanisms of Homocysteine Toxicity. Kidney Int. Suppl. 2003, 84, S137–S140. [Google Scholar] [CrossRef] [Green Version]
- Fournier, I.; Ploye, F.; Cottet-Emard, J.M.; Brun, J.; Claustrat, B. Folate Deficiency Alters Melatonin Secretion in Rats. J. Nutr. 2002, 132, 2781–2784. [Google Scholar] [CrossRef] [Green Version]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef]
- Rehman, T.; Shabbir, M.A.; Inam-Ur-Raheem, M.; Manzoor, M.F.; Ahmad, N.; Liu, Z.W.; Ahmad, M.H.; Siddeeg, A.; Abid, M.; Aadil, R.M. Cysteine and Homocysteine as Biomarker of Various Diseases. Food Sci. Nutr. 2020, 8, 4696–4707. [Google Scholar] [CrossRef]
- Galvin, J.E.; Howard, D.H.; Denny, S.S.; Dickinson, S.; Tatton, N. The Social and Economic Burden of Frontotemporal Degeneration. Neurology. 2017, 89, 2049–2056. [Google Scholar] [CrossRef] [Green Version]
- Mela, A.; Rdzanek, E.; Poniatowski, Ł.A.; Jaroszyński, J.; Furtak-Niczyporuk, M.; Gałązka-Sobotka, M.; Olejniczak, D.; Niewada, M.; Staniszewska, A. Economic Costs of Cardiovascular Diseases in Poland Estimates for 2015–2017 Years. Front. Pharmacol. 2020, 11, 1231. [Google Scholar] [CrossRef]
- Cattaneo, M. Hyperhomocysteinaemia and Atherothrombosis. Ann. Med. 2000, 32 (Suppl. 1), 46–52. [Google Scholar]
- Firoz, C.K.; Jabir, N.R.; Khan, M.S.; Mahmoud, M.; Shakil, S.; Damanhouri, G.A.; Zaidi, S.K.; Tabrez, S.; Kamal, M.A. An Overview on the Correlation of Neurological Disorders with Cardiovascular Disease. Saudi J. Biol. Sci. 2015, 22, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Cavalca, V.; Cighetti, G.; Bamonti, F.; Loaldi, A.; Bortone, L.; Novembrino, C.; De Franceschi, M.; Belardinelli, R.; Guazzi, M.D. Oxidative Stress and Homocysteine in Coronary Artery Disease. Clin. Chem. 2001, 47, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.; Mattson, M.P. Homocysteine Elicits a DNA Damage Response in Neurons that Promotes Apoptosis and Hypersensitivity to Excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhara, T.; Fukuo, K.; Yasuda, O.; Tsubakimoto, M.; Takemura, Y.; Kawamoto, H.; Yokoi, T.; Mogi, M.; Kaimoto, T.; Ogihara, T. Homocysteine Enhances Endothelial Apoptosis via Upregulation of Fas-mediated Pathways. Hypertension. 2004, 43, 1208–1213. [Google Scholar] [CrossRef]
- Karolczak, K.; Pieniazek, A.; Watala, C. Inhibition of Glutamate Receptors Reduces the Homocysteine-induced whole Blood Platelet Aggregation but does not Affect Superoxide Anion Generation or Platelet Membrane Fluidization. Platelets. 2017, 28, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Yang, W.; Hou, Z.; Yang, J. Homocysteine Enhances Cell Proliferation in Vascular Smooth Muscle Cells: Role of p38 MAPK and p47phox. Acta Biochim. Biophys. Sin. (Shanghai) 2010, 42, 908–915. [Google Scholar] [CrossRef] [Green Version]
- Pfanzagl, B.; Tribl, F.; Koller, E.; Möslinger, T. Homocysteine Strongly Enhances Metal-Catalyzed LDL Oxidation in the Presence of Cystine and Cysteine. Atherosclerosis 2003, 168, 39–48. [Google Scholar] [CrossRef]
- Riddell, L.J.; Chisholm, A.; Williams, S.; Mann, J.I. Dietary Strategies for Lowering Homocysteine Concentrations. Am. J. Clin. Nutr. 2000, 71, 1448–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoef, P.; de Groot, L.C. Dietary Determinants of Plasma Homocysteine Concentrations. Semin. Vasc. Med. 2005, 5, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Thambyrajah, J.; Landray, M.J.; McGlynn, F.J.; Jones, H.J.; Wheeler, D.C.; Townend, J.N. Does Folic Acid Decrease Plasma Homocysteine and Improve Endothelial Function in Patients with Predialysis Renal Failure? Circulation 2000, 102, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.E.; Hornstra, J.M.; Kok, R.M.; Blom, H.J.; Smulders, Y.M. Folic Acid Supplementation does not Reduce Intracellular Homocysteine, and may Disturb Intracellular One-Carbon Metabolism. Clin. Chem. Lab. Med. 2013, 51, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- de Bree, A.; van Dusseldorp, M.; Brouwer, I.A.; van het Hof, K.H.; Steegers-Theunissen, R.P. Folate Intake in Europe: Recommended, Actual and Desired Intake. Eur. J. Clin. Nutr. 1997, 51, 643–660. [Google Scholar] [CrossRef] [Green Version]
- Ubbink, J.B. Should All Elderly People Receive Folate Supplements? Drugs Aging 1998, 13, 415–420. [Google Scholar] [CrossRef]
- Breilmann, J.; Pons-Kühnemann, J.; Brunner, C.; Richter, M.; Neuhäuser-Berthold, M. Effect of Antioxidant Vitamins on the Plasma Homocysteine Level in a Free-Living Elderly Population. Ann. Nutr. Metab. 2010, 57, 177–182. [Google Scholar] [CrossRef]
- Malinowska, J.; Olas, B. Effect of Resveratrol on Hemostatic Properties of Human Fibrinogen and Plasma during Model of Hyperhomocysteinemia. Thromb. Res. 2010, 126, e379–e382. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, J.; Kolodziejczyk, J.; Olas, B. The disturbance of Hemostasis Induced by Hyperhomocysteinemia; the Role of Antioxidants. Acta Biochim. Pol. 2012, 59, 185–194. [Google Scholar] [CrossRef]
- Malinowska, J.; Babicz, K.; Olas, B.; Stochmal, A.; Oleszek, W. Aronia Melanocarpa Extract Suppresses the Biotoxicity of Homocysteine and its Metabolite on the Hemostatic Activity of Fibrinogen and Plasma. Nutrition 2012, 28, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Qi, W. Biochemical Reactivity of Melatonin with Reactive Oxygen and Nitrogen Species: A Review of the Evidence. Cell. Biochem. Biophys. 2001, 34, 237–256. [Google Scholar] [CrossRef]
- Kotler, M.; Rodríguez, C.; Sáinz, R.M.; Antolín, I.; Menéndez-Peláez, A. Melatonin Increases Gene Expression for Antioxidant Enzymes in Rat Brain Cortex. J. Pineal Res. 1998, 24, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef]
- De Bree, A.; Verschuren, W.M.; Kromhout, D.; Kluijtmans, L.A.; Blom, H.J. Homocysteine Determinants and the Evidence to What Extent Homocysteine Determines the Risk of Coronary Heart Disease. Pharmacol. Rev. 2002, 54, 599–618. [Google Scholar] [CrossRef] [PubMed]
- García-Casal, M.N.; Osorio, C.; Landaeta, M.; Leets, I.; Matus, P.; Fazzino, F.; Marcos, E. High Prevalence of Folic Acid and Vitamin B12 Deficiencies in Infants, Children, Adolescents and Pregnant Women in Venezuela. Eur. J. Clin. Nutr. 2005, 59, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Gursu, M.F.; Cikim, G.; Canpolat, S.; Yasar, A.; Canatan, H.; Kelestimur, H. Effects of Pinealectomy on the Levels and the Circadian Rhythm of Plasma Homocysteine in Rats. J. Pineal Res. 2002, 33, 151–155. [Google Scholar] [CrossRef]
- Jacobs, R.L.; House, J.D.; Brosnan, M.E.; Brosnan, J.T. Effects of Streptozotocin-Induced Diabetes and of Insulin Treatment on Homocysteine Metabolism in the Rat. Diabetes 1998, 47, 1967–1970. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Prudova, A.; Stabler, S.; Dayal, S.; Lentz, S.R.; Banerjee, R. Testosterone Regulation of Renal Cystathionine Beta-Synthase: Implications for Sex-Dependent Differences in Plasma Homocysteine Levels. Am. J. Physiol. Renal Physiol. 2007, 293, F594–F600. [Google Scholar] [CrossRef]
- Karolczak, K.; Kubalczyk, P.; Glowacki, R.; Pietruszynski, R.; Watala, C. Aldosterone Modulates Blood Homocysteine and CHOLesterol in Coronary Artery Disease Patients—A Possible Impact on Atherothrombosis? Physiol. Res. 2018, 67, 197–207. [Google Scholar] [CrossRef]
- Karolczak, K.; Konieczna, L.; Kostka, T.; Witas, P.J.; Soltysik, B.; Baczek, T.; Watala, C. Testosterone and Dihydrotestosterone Reduce Platelet Activation and Reactivity in Older Men and Women. Aging (Albany NY) 2018, 10, 902–929. [Google Scholar] [CrossRef] [PubMed]
- Bremner, W.F.; Holmes, E.W.; Kanabrocki, E.L.; Hermida, R.C.; Ayala, D.; Garbincius, J.; Third, J.L.; Ryan, M.D.; Johnson, M.; Foley, S.; et al. Circadian Rhythm of Serum Total Homocysteine in Men. Am. J. Cardiol. 2000, 86, 1153–1156. [Google Scholar] [CrossRef]
- Lavie, L.; Lavie, P. Daily Rhythms in Plasma Levels of Homocysteine. J. Circadian Rhythms 2004, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, P.J.; Galdieri, L.C.; Souza, F.G.; Andersen, M.L.; Benedito-Silva, A.A.; Tufik, S.; D’Almeida, V. Physiological Variation in Plasma Total Homocysteine Concentrations in Rats. Life Sci. 2005, 76, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michaelis, E.K. Selective Neuronal Vulnerability to Oxidative Stress in the Brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 Reasons Why the Brain is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Ataie, A.; Ataee, R.; Mansoury, Z.; Aghajanpour, M. Homocysteine Intracerebroventricular Injection Induces Apoptosis in the Substantia Nigra Cells and Parkinson’s Disease Like Behavior in Rats. Int. J. Mol. Cell. Med. 2013, 2, 80–85. [Google Scholar] [PubMed]
- Paul, R.; Dutta, A.; Phukan, B.C.; Mazumder, M.K.; Justin-Thenmozhi, A.; Manivasagam, T.; Bhattacharya, P.; Borah, A. Accumulation of Cholesterol and Homocysteine in the Nigrostriatal Pathway of Brain Contributes to the Dopaminergic Neurodegeneration in Mice. Neuroscience 2018, 388, 347–356. [Google Scholar] [CrossRef] [PubMed]
- den Heijer, T.; Vermeer, S.E.; Clarke, R.; Oudkerk, M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Homocysteine and Brain Atrophy on MRI of Non-Demented Elderly. Brain 2003, 126, 170–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.E.; Tian, Q.; Wei, W.; Peng, J.H.; Liu, G.P.; Zhou, X.W.; Wang, Q.; Wang, D.W.; Wang, J.Z. Homocysteine Induces Tau Phosphorylation by Inactivating Protein Phosphatase 2A in Rat Hippocampus. Neurobiol. Aging 2008, 29, 1654–1665. [Google Scholar] [CrossRef]
- Gallucci, M.; Zanardo, A.; Bendini, M.; Di Paola, F.; Boldrini, P.; Grossi, E. Serum Folate, Homocysteine, Brain Atrophy, and Auto-CM System: The Treviso Dementia (TREDEM) study. J. Alzheimers Dis. 2014, 38, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Yu, M.S.; Yang, X.F.; So, K.F.; Yuen, W.H.; Chang, R.C. Neuroprotective Effects of Polysaccharides from Wolfberry, the Fruits of Lycium barbarum, against Homocysteine-Induced Toxicity in Rat Cortical Neurons. J. Alzheimers Dis. 2010, 19, 813–827. [Google Scholar] [CrossRef] [Green Version]
- Gröger, A.; Kolb, R.; Schäfer, R.; Klose, U. Dopamine Reduction in the Substantia Nigra of Parkinson’s Disease Patients Confirmed by in Vivo Magnetic Resonance Spectroscopic Imaging. PLoS ONE 2014, 9, e84081. [Google Scholar] [CrossRef] [PubMed]
- Sowell, E.R.; Jernigan, T.L.; Mattson, S.N.; Riley, E.P.; Sobel, D.F.; Jones, K.L. Abnormal Development of the Cerebellar Vermis in Children Prenatally Exposed to Alcohol: Size Reduction in Lobules I-V. Alcohol. Clin. Exp. Res. 1996, 20, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, W.; Ladenheim, B.; Cutler, R.G.; Kruman, I.I.; Cadet, J.L.; Mattson, M.P. Dietary Folate Deficiency and Elevated Homocysteine Levels Endanger Dopaminergic Neurons in Models of Parkinson’s Disease. J. Neurochem. 2002, 80, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.M.; Sohn, B.K.; Choi, H.J.; Byun, M.S.; Seo, E.H.; Han, J.Y.; Kim, Y.K.; Yoon, E.J.; Lee, J.M.; Park, J.; et al. Association of Homocysteine with Hippocampal Volume Independent of Cerebral Amyloid and Vascular Burden. Neurobiol. Aging. 2014, 35, 1519–1525. [Google Scholar] [CrossRef]
- Sibarov, D.A.; Giniatullin, R.; Antonov, S.M. High Sensitivity of Cerebellar Neurons to Homocysteine is Determined by Expression of Glun2c and Glun2d Subunits of NMDA Receptors. Biochem. Biophys. Res. Commun. 2018, 506, 648–652. [Google Scholar] [CrossRef]
- Osuna, C.; Reiter, R.J.; García, J.J.; Karbownik, M.; Tan, D.X.; Calvo, J.R.; Manchester, L.C. Inhibitory Effect of Melatonin on Homocysteine-Induced Lipid Peroxidation in Rat Brain Homogenates. Pharmacol. Toxicol. 2002, 90, 32–37. [Google Scholar] [CrossRef]
- Ortega-Gutiérrez, S.; Fuentes-Broto, L.; García, J.J.; López-Vicente, M.; Martínez-Ballarín, E.; Miana-Mena, F.J.; Millán-Plano, S.; Reiter, R.J. Melatonin Reduces Protein and Lipid Oxidative Damage Induced by Homocysteine in Rat Brain Homogenates. J. Cell. Biochem. 2007, 102, 729–735. [Google Scholar] [CrossRef]
- Paul, R.; Phukan, B.C.; Justin Thenmozhi, A.; Manivasagam, T.; Bhattacharya, P.; Borah, A. Melatonin Protects against Behavioral Deficits, Dopamine Loss and Oxidative Stress in Homocysteine Model of Parkinson’s Disease. Life Sci. 2018, 192, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Antolín, I.; Mayo, J.C.; Sainz, R.M.; del Brío Mde, L.; Herrera, F.; Martín, V.; Rodríguez, C. Protective Effect of Melatonin in A Chronic Experimental Model of Parkinson’s Disease. Brain Res. 2002, 943, 163–173. [Google Scholar] [CrossRef]
- Yildirim, F.B.; Ozsoy, O.; Tanriover, G.; Kaya, Y.; Ogut, E.; Gemici, B.; Dilmac, S.; Ozkan, A.; Agar, A.; Aslan, M. Mechanism of the Beneficial Effect of Melatonin in Experimental Parkinson’s Disease. Neurochem. Int. 2014, 79, 1–11. [Google Scholar] [CrossRef]
- Medeiros, C.A.; Carvalhedo de Bruin, P.F.; Lopes, L.A.; Magalhães, M.C.; de Lourdes Seabra, M.; de Bruin, V.M. Effect of Exogenous Melatonin on Sleep and Motor Dysfunction in Parkinson’s Disease. A Randomized, Double Blind, Placebo-Controlled Study. J. Neurol. 2007, 254, 459–464. [Google Scholar] [CrossRef]
- Fertl, E.; Auff, E.; Doppelbauer, A.; Waldhauser, F. Circadian Secretion Pattern of Melatonin in Parkinson’s Disease. J. Neural. Transm. Park. Dis. Dement. Sect. 1991, 3, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bordet, R.; Devos, D.; Brique, S.; Touitou, Y.; Guieu, J.D.; Libersa, C.; Destée, A. Study of Circadian Melatonin Secretion Pattern at Different Stages of Parkinson’s Disease. Clin. Neuropharmacol. 2003, 26, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Dowling, G.A.; Mastick, J.; Colling, E.; Carter, J.H.; Singer, C.M.; Aminoff, M.J. Melatonin for Sleep Disturbances in Parkinson’s Disease. Sleep Med. 2005, 6, 459–466. [Google Scholar] [CrossRef]
- Willis, G.L.; Armstrong, S.M. A Therapeutic Role for Melatonin Antagonism in Experimental Models of Parkinson’s Disease. Physiol. Behav. 1999, 66, 785–795. [Google Scholar] [CrossRef]
- Tapias, V.; Cannon, J.R.; Greenamyre, J.T. Melatonin Treatment Potentiates Neurodegeneration in A Rat Rotenone Parkinson’s Disease Model. J. Neurosci. Res. 2010, 88, 420–427. [Google Scholar] [CrossRef]
- Bouzouf, M.; Martinez-Cruz, F.; Molinero, P.; Guerrero, J.M.; Osuna, C. Melatonin Prevents Hyperhomocysteinemia and Neural Lipid Peroxidation Induced by Methionine Intake. Curr. Neurovasc. Res. 2005, 2, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Ozer, M.; Yasar, A.; Koz, S.T.; Tuzcu, M. Melatonin Prevents Oxidative Stress and Inhibits Reactive Gliosis Induced by Hyperhomocysteinemia in Rats. Biochemistry (Mosc) 2006, 71 (Suppl. 1), S91–S95. [Google Scholar] [CrossRef]
- Stickel, F.; Choi, S.W.; Kim, Y.I.; Bagley, P.J.; Seitz, H.K.; Russell, R.M.; Selhub, J.; Mason, J.B. Effect of Chronic Alcohol Consumption on Total Plasma Homocysteine Level in Rats. Alcohol. Clin. Exp. Res. 2000, 24, 259–264. [Google Scholar] [CrossRef]
- Bleich, S.; Bleich, K.; Kropp, S.; Bittermann, H.J.; Degner, D.; Sperling, W.; Rüther, E.; Kornhuber, J. Moderate Alcohol Consumption in Social Drinkers Raises Plasma Homocysteine Levels: A Contradiction to the ‘French Paradox’? Alcohol Alcohol. 2001, 36, 189–192. [Google Scholar] [CrossRef]
- Sakuta, H.; Suzuki, T. Alcohol Consumption and Plasma Homocysteine. Alcohol 2005, 37, 73–77. [Google Scholar] [CrossRef]
- Bagheri, F.; Goudarzi, I.; Lashkarbolouki, T.; Elahdadi Salmani, M. Melatonin Prevents Oxidative Damage Induced by Maternal Ethanol Administration and Reduces Homocysteine in the Cerebellum of Rat Pups. Behav. Brain Res. 2015, 287, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Ozer, M.; Yasar, A.; Tuzcu, M.; Koz, S.T. Melatonin Improves Learning and Memory Performances Impaired by Hyperhomocysteinemia in Rats. Brain Res. 2005, 1046, 187–194. [Google Scholar] [CrossRef]
- Bhatia, P.; Singh, N. Homocysteine excess: Delineating the Possible Mechanism of Neurotoxicity and Depression. Fundam. Clin. Pharmacol. 2015, 29, 522–528. [Google Scholar] [CrossRef]
- Brown, A.S.; Susser, E.S. Homocysteine and Schizophrenia: From Prenatal to Adult Life. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 1175–1180. [Google Scholar] [CrossRef]
- Ghanizadeh, A.; Singh, A.B.; Berk, M.; Torabi-Nami, M. Homocysteine as a Potential Biomarker in Bipolar Disorders: A Critical Review and Suggestions for Improved Studies. Expert Opin. Ther. Targets. 2015, 19, 927–939. [Google Scholar] [CrossRef]
- Tao, H.; Chen, X.; Zhou, H.; Fu, J.; Yu, Q.; Liu, Y. Changes of Serum Melatonin, Interleukin-6, Homocysteine, and Complement C3 and C4 Levels in Patients with Depression. Front. Psychol. 2020, 11, 1271. [Google Scholar] [CrossRef]
- Chen, M.; Mei, Q.; Xu, J.; Lu, C.; Fang, H.; Liu, X. Detection of Melatonin and Homocysteine Simultaneously in Ulcerative Colitis. Clin. Chim. Acta. 2012, 413, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Khazan, M.; Nasiri, S.; Riahi, S.M.; Robati, R.M.; Hedayati, M. Measurement of Melatonin, Indole-Dioxygenase, IL-6, IL-18, Ferritin, CRP, and Total Homocysteine Levels during Herpes Zoster. J. Med. Virol. 2020, 92, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Khoubnasabjafari, M.; Ansarin, K.; Jouyban, A. Reliability of Malondialdehyde as a Biomarker of Oxidative Stress in Psychological Disorders. Bioimpacts 2015, 5, 123–127. [Google Scholar]
- Temple, M.E.; Luzier, A.B.; Kazierad, D.J. Homocysteine as a Risk Factor for Atherosclerosis. Ann. Pharmacother. 2000, 34, 57–65. [Google Scholar] [CrossRef]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of Hyperhomocysteinemia in Endothelial Dysfunction and Atherothrombotic Disease. Cell Death Differ. 2004, 11 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [Green Version]
- Papatheodorou, L.; Weiss, N. Vascular Oxidant Stress and Inflammation in Hyperhomocysteinemia. Antioxid. Redox Signal. 2007, 9, 1941–1958. [Google Scholar] [CrossRef]
- Aykutoglu, G.; Tartik, M.; Darendelioglu, E.; Ayna, A.; Baydas, G. Melatonin and Vitamin E Alleviate Homocysteine-Induced Oxidative Injury and Apoptosis in Endothelial Cells. Mol. Biol. Rep. 2020, 47, 5285–5293. [Google Scholar] [CrossRef]
- Aminzadeh, A.; Mehrzadi, S. Melatonin Attenuates Homocysteine-Induced Injury in Human Umbilical Vein Endothelial Cells. Fundam. Clin. Pharmacol. 2018, 32, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Okatani, Y.; Wakatsuki, A.; Reiter, R.J. Melatonin Counteracts Potentiation by Homocysteine of Kcl-Induced Vasoconstriction in Human Umbilical Artery: Relation to Calcium Influx. Biochem. Biophys. Res. Commun. 2001, 280, 940–944. [Google Scholar] [CrossRef]
- Okatani, Y.; Wakatsuki, A.; Reiter, R.J. Protective Effect of Melatonin against Homocysteine-Induced Vasoconstriction of Human Umbilical Artery. Biochem. Biophys. Res. Commun. 2000, 277, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Okatani, Y.; Wakatsuki, A.; Reiter, R.J. Melatonin Suppresses Homocysteine Enhancement of Serotonin-Induced Vasoconstriction in the Human Umbilical Artery. J. Pineal Res. 2001, 31, 242–247. [Google Scholar] [CrossRef]
- Murawska-Cialowicz, E.; Januszewska, L.; Zuwala-Jagiello, J.; Milczarska, J.; Zawadzki, M.; Paprocka-Borowicz, M.; Wierzbicka-Damska, I. Melatonin Decreases Homocysteine Level in Blood of Rats. J. Physiol. Pharmacol. 2008, 59, 717–729. [Google Scholar]
- Baydas, G.; Yilmaz, O.; Celik, S.; Yasar, A.; Gursu, M.F. Effects of Certain Micronutrients and Melatonin on Plasma Lipid, Lipid Peroxidation, and Homocysteine Levels in Rats. Arch. Med. Res. 2002, 33, 515–519. [Google Scholar] [CrossRef]
- Kantar, Ş.; Türközkan, N.; Bircan, F.S.; Paşaoğlu, Ö.T. Beneficial Effects of Melatonin on Serum Nitric Oxide, Homocysteine, and ADMA Levels in Fructose-Fed Rats. Pharm. Biol. 2015, 53, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Ghosh, C. Interactions Between Melatonin, Reactive Oxygen Species, and Nitric Oxide. Ann. N. Y. Acad. Sci. 1999, 893, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Jiménez, S.; Pina-Beltrán, B.; Gimeno-Martos, S.; Carvajal-Serna, M.; Casao, A.; Pérez-Pe, R. NADPH Oxidase 5 and Melatonin: Involvement in Ram Sperm Capacitation. Front. Cell. Dev. Biol. 2021, 9, 655794. [Google Scholar] [CrossRef] [PubMed]
- Simões, D.; Riva, P.; Peliciari-Garcia, R.A.; Cruzat, V.F.; Graciano, M.F.; Munhoz, A.C.; Taneda, M.; Cipolla-Neto, J.; Carpinelli, A.R. Melatonin Modifies Basal and Stimulated Insulin Secretion via NADPH Oxidase. J. Endocrinol. 2016, 231, 235–244. [Google Scholar] [CrossRef]
- Edirimanne, V.E.; Woo, C.W.; Siow, Y.L.; Pierce, G.N.; Xie, J.Y.; O., K. Homocysteine Stimulates NADPH Oxidase-Mediated Superoxide Production Leading to Endothelial Dysfunction in Rats. Can. J. Physiol. Pharmacol. 2007, 85, 1236–1247. [Google Scholar] [CrossRef]
- Alvarez-Maqueda, M.; El Bekay, R.; Monteseirín, J.; Alba, G.; Chacón, P.; Vega, A.; Santa María, C.; Tejedo, J.R.; Martín-Nieto, J.; Bedoya, F.J.; et al. Homocysteine Enhances Superoxide Anion Release and NADPH Oxidase Assembly by Human Neutrophils. Effects on MAPK Activation and Neutrophil Migration. Atherosclerosis 2004, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Zhang, A.Y.; Janscha, J.L.; Li, P.L.; Zou, A.P. Homocysteine Activates NADH/NADPH Oxidase through Ceramide-Stimulated Rac Gtpase Activity in Rat Mesangial Cells. Kidney Int. 2004, 66, 1977–1987. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.M.; Kruzliak, P.; Adamcikova, Z.; Zulli, A. Role of Nox Inhibitors Plumbagin, ML090 and gp91ds-tat peptide on Homocysteine Thiolactone Induced Blood Vessel Dysfunction. Clin. Exp. Pharmacol. Physiol. 2015, 42, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.M.; Rai, S.; Kruzliak, P.; Hayes, A.; Zulli, A. Putative Nox2 Inhibitors Worsen Homocysteine-Induced Impaired Acetylcholine-Mediated Relaxation. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 856–864. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karolczak, K.; Watala, C. Melatonin as a Reducer of Neuro- and Vasculotoxic Oxidative Stress Induced by Homocysteine. Antioxidants 2021, 10, 1178. https://doi.org/10.3390/antiox10081178
Karolczak K, Watala C. Melatonin as a Reducer of Neuro- and Vasculotoxic Oxidative Stress Induced by Homocysteine. Antioxidants. 2021; 10(8):1178. https://doi.org/10.3390/antiox10081178
Chicago/Turabian StyleKarolczak, Kamil, and Cezary Watala. 2021. "Melatonin as a Reducer of Neuro- and Vasculotoxic Oxidative Stress Induced by Homocysteine" Antioxidants 10, no. 8: 1178. https://doi.org/10.3390/antiox10081178
APA StyleKarolczak, K., & Watala, C. (2021). Melatonin as a Reducer of Neuro- and Vasculotoxic Oxidative Stress Induced by Homocysteine. Antioxidants, 10(8), 1178. https://doi.org/10.3390/antiox10081178