
Glutathione Participation in the Prevention of Cardiovascular Diseases

 ,
, 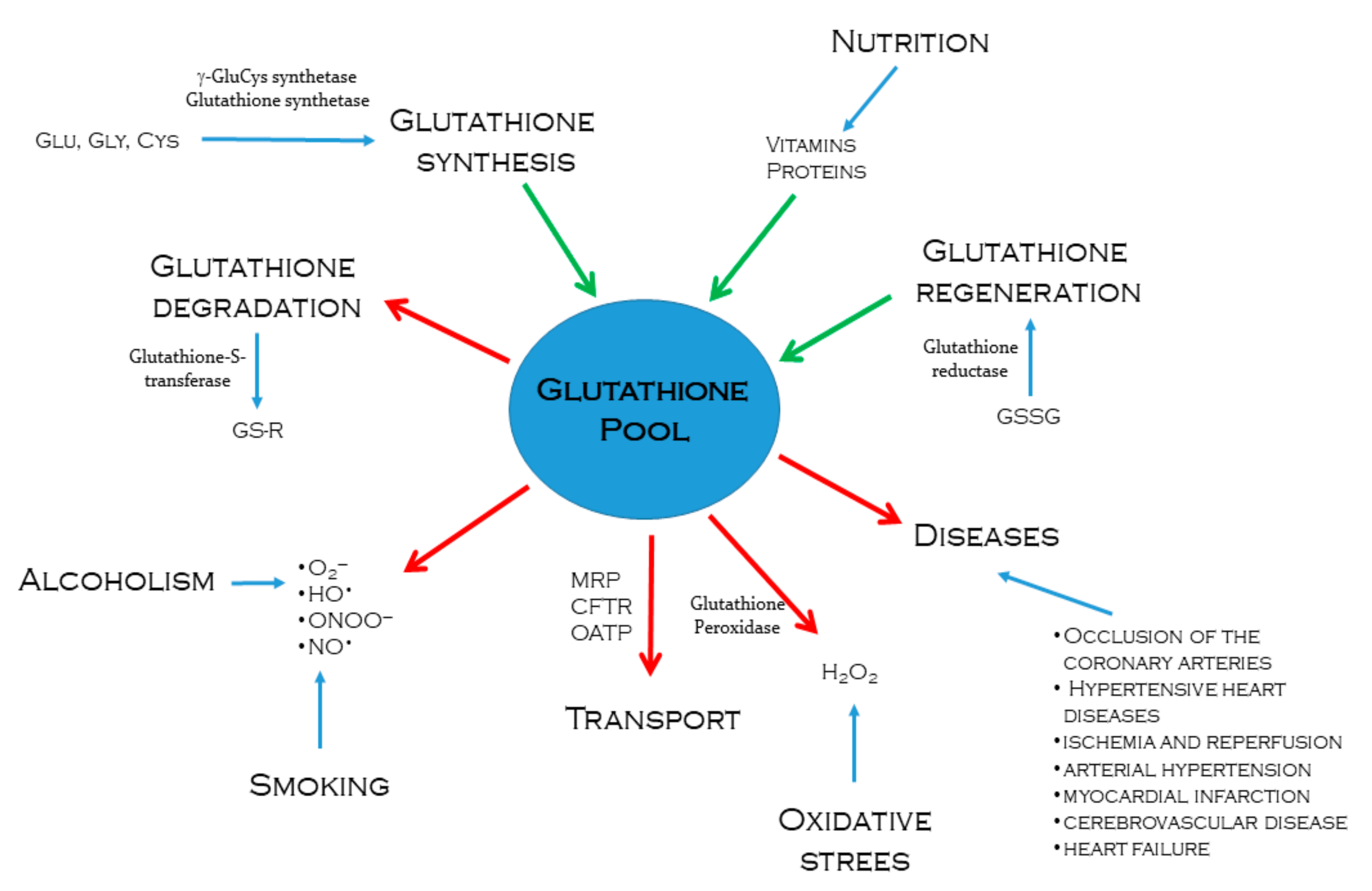{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ROS Excess and Antioxidant Mechanisms
2.1. Glutathione, a Key Intracellular Antioxidant
2.2. Determination of Glutathione in Biological Samples
2.3. Cellular Glutathione Synthesis
2.4. Regulation of Glutathione Levels Is Modulated by a Diversity of Factors
- Methionine as a precursor of cysteine. In cells, methionine is transformed into cysteine through the transsulfuration pathway, by the action of the enzyme cystathionine β-synthase and cystathionine γ-lyase or cystathionase [38,54,55,56]. Disruption of the transsulfuration pathway contributes to the pathology of several conditions, such as vascular dysfunction [56].
- N-acetylcysteine (NAC) functions as a precursor of the amino acid L-cysteine, fostering the intracellular production of GSH [45,57]. It has been shown that many types of cells can trap NAC, hydrolyze it and change it into L-cysteine, which is then incorporated into the cycle of gamma glutamyl, stimulating glutathione synthesis [58].
- Alpha-lipoic acid (α-AL) is considered the “universal antioxidant” because it is an amphipathic molecule that may act in both aqueous and hydrophobic environments. The organism, using NADH or NADPH, usually transforms it into dihydrolipoic acid (DHLA), reduced form of α-AL, which has an important antioxidant effect and can reduce GSSG directly into GSH increasing the intracellular levels of reduced glutathione [62,63]. DHLA may also be released into the extracellular space where it reduces cysteine, which can be taken and transported into the cell and used in the synthesis of glutathione [63,64,65].
- The diester of GSH is effectively transported into cells, hydrolyzed to GSH, thus increasing glutathione levels. It is four times more effective than GSH monoester [61].
2.5. Glutathione in Liver and Heart
2.6. Plasma Glutathione
2.7. Other Functions of Glutathione
2.8. Glutathione Transport Is Performed by Three Different Protein Families
- The sinusoidal transport system, found on the basolateral membrane of hepatocytes, releases GSH into the blood [106].
3. Role of Oxidative Stress in the Generation of Cardiovascular Diseases
3.1. Endothelial Dysfunction (ED)
3.2. Hypertension and Diabetes Mellitus (DM)
3.3. Atherosclerosis
3.4. Cardiac Hypertrophy
3.5. Ischemia-Reperfusion Injury
3.6. Heart Failure
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bachhawat, A.K.; Yadav, S.; Jainarayanan, A.K.; Dubey, P. Heart failure and the glutathione cycle: An integrated view. Biochem. J. 2020, 477, 3123–3130. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Asp. Med. 2009, 30, 13–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashikuni, Y.; Sainz, J.; Nakamura, K.; Takaoka, M.; Enomoto, S.; Iwata, H.; Tanaka, K.; Sahara, M.; Hirata, Y.; Nagai, R.; et al. The ATP-Binding Cassette Transporter ABCG2 Protects Against Pressure Overload–Induced Cardiac Hypertrophy and Heart Failure by Promoting Angiogenesis and Antioxidant Response. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 654–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Health 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. J. Cell. Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef]
- Griendling, K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.-R.; Harrison, D.G.; Bhatnagar, A. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.-S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef] [Green Version]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadet, J.; Wagner, J.R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014, 557, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Ahuir, A.; Manzanares-Estreder, S.; Proft, M. Pro- and Antioxidant Functions of the Peroxisome-Mitochondria Connection and Its Impact on Aging and Disease. Oxidative Med. Cell. Longev. 2017, 2017, 9860841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubczyk, K.; Kałduńska, J.; Dec, K.; Kawczuga, D.; Janda, K. Antioxidant properties of small-molecule non-enzymatic compounds. Pol. Merkur Lekarski 2020, 48, 128–132. [Google Scholar]
- Metere, A.; Giacomelli, L. Absorption, metabolism and protective role of fruits and vegetables polyphenols against gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5850–5858. [Google Scholar] [PubMed]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in theentire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal Dysfunction in Age-Related Diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. SnapShot: Reactive Oxygen Intermediates (ROI). Cell 2010, 140, 951. [Google Scholar] [CrossRef] [Green Version]
- Osorio Alves, J.; Matta Pereira, L.; Cabral Coutinho do Rego Monteiro, I.; Pontes Dos Santos, L.H.; Soares Marreiros Ferraz, A.; Carneiro Loureiro, A.C.; Calado Lima, C.; Leal-Cardoso, J.H.; Pires Carvalho, D.; Soares Fortunato, R.; et al. Strenuous Acute Exercise Induces Slow and Fast Twitch-Dependent NADPH Oxidase Expression in Rat Skeletal Muscle. Antioxidants 2020, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef]
- Krishnamurthy, P. Antioxidant Enzymes and Human Health. Antioxid. Enzym. 2012, 3, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxidative Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Awad, M.; Aldosari, S.; Abid, M.R. Genetic Alterations in Oxidant and Anti-Oxidant Enzymes in the Vascular System. Front. Cardiovasc. Med. 2018, 5, 107. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Del Valle, N.R.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Pahwa, R.; Goyal, A.; Bansal, P.; Jialal, I. Chronic Inflammation; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Vigneron, A.; Vousden, K.H. p53, ROS and senescence in the control of aging. Aging 2010, 2, 471–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamiec, M.; Skonieczna, M. UV radiation in HCT 116 cells influences intracellular H2O2 and glutathione levels, antioxidant expression, and protein glutathionylation. Acta Biochim. Pol. 2019, 66, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Choi, S.-W. Antioxidants in Food. Adv. Food Nutr. Res. 2014, 71, 1–53. [Google Scholar] [CrossRef]
- Tsao, R. Chemistry and Biochemistry of Dietary Polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef]
- Duncan, K.R.; Suzuki, Y.J. Vitamin E Nicotinate. Antioxidants 2017, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, S.; Dharmaraj, S. Selenium and selenoproteins: It’s role in regulation of inflammation. Inflammopharmacology 2020, 28, 667–695. [Google Scholar] [CrossRef] [PubMed]
- Ratnam, D.V.; Ankola, D.; Bhardwaj, V.; Sahana, D.; Kumar, M.R. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Release 2006, 113, 189–207. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.J.; Pappa, A.; Panayiotidis, M.I. The central role of glutathione in the pathophysiology of human diseases. Arch. Physiol. Biochem. 2007, 113, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, A. Mitochondrial changes associated with glutathione deficiency. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1995, 1271, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Giustarini, D.; Milzani, A.D.G.; Dalle-Donne, I.; Rossi, R. Red blood cells as a physiological source of glutathione for extracellular fluids. Blood Cells Mol. Dis. 2008, 40, 174–179. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Xia, T.; Bian, G.; Dong, L.; Tang, Z.; Wang, F. A highly sensitive assay for spectrofluorimetric determination of reduced glutathione using organic nano-probes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 61, 2533–2538. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- López-Mirabal, H.R.; Winther, J.R. Redox characteristics of the eukaryotic cytosol. Biochim. Biophys. Acta (BBA)-Bioenerg. 2008, 1783, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atmaca, G. Antioxidant Effects of Sulfur-Containing Amino Acids. Yonsei Med. J. 2004, 45, 776–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harfield, J.C.; Batchelor-McAuley, C.; Compton, R.G. Electrochemical determination of glutathione: A review. Analyst 2012, 137, 2285–2296. [Google Scholar] [CrossRef]
- Dickinson, D.A.; Forman, H.J. Cellular glutathione and thiols metabolism. Biochem. Pharmacol. 2002, 64, 1019–1026. [Google Scholar] [CrossRef]
- Mari, M.; Colell, A.; Morales, A.; Von Montfort, C.; Garcia-Ruiz, C.; Fernández-Checa, J.C. Redox Control of Liver Function in Health and Disease. Antioxid. Redox Signal. 2010, 12, 1295–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Paolicchi, A.; Dominici, S.; Pieri, L.; Maellaro, E.; Pompella, A. Glutathione catabolism as a signaling mechanism. Biochem. Pharmacol. 2002, 64, 1027–1035. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Vivancos, P.D.; Wolff, T.; Markovic, J.; Pallardó, F.V.; Foyer, C.H. A nuclear glutathione cycle within the cell cycle. Biochem. J. 2010, 431, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Lomaestro, B.M.; Malone, M. Glutathione in Health and Disease: Pharmacotherapeutic Issues. Ann. Pharmacother. 1995, 29, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Rudman, D.; Kutner, M.; Ansley, J.; Jansen, R.; Chipponi, J.; Bain, R.P. Hypotyrosinemia, Hypocystinemia, and Failure to Retain Nitrogen during Total Parenteral Nutrition of Cirrhotic Patients. Gastroenterology 1981, 81, 1025–1035. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Nakayama, J.; Sadakata, Y. Protective Effects of Exogenous Glutathione and Related Thiol Compounds against Drug-Induced Liver Injury. Biol. Pharm. Bull. 2011, 34, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sochman, J. N-acetylcysteine in acute cardiology: 10 years later. J. Am. Coll. Cardiol. 2002, 39, 1422–1428. [Google Scholar] [CrossRef] [Green Version]
- Vendemiale, G.; Altomare, E.; Trizio, T.; Le Grazie, C.; Di Padova, C.; Salerno, M.T.; Carrieri, V.; Albano, O. Effects of Oral S-Adenosyl-l-Methionine on Hepatic Glutathione in Patients with Liver Disease. Scand. J. Gastroenterol. 1989, 24, 407–415. [Google Scholar] [CrossRef]
- Anderson, M.; Luo, J.-L. Glutathione Therapy: From Prodrugs to Genes. Semin. Liver Dis. 1998, 18, 415–424. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [Green Version]
- Busse, E.; Zimmer, G.; Schopohl, B.; Kornhuber, B. Influence of alpha-lipoic acid on intracellular glutathione in vitro and in vivo. Arzneimittelforschung 1992, 42, 829–831. [Google Scholar] [PubMed]
- Moini, H.; Packer, L.; Saris, N.-E.L. Antioxidant and Prooxidant Activities of α-Lipoic Acid and Dihydrolipoic Acid. Toxicol. Appl. Pharmacol. 2002, 182, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, S.; Atalay, M.; Laaksonen, D.E.; Gul, M.; Roy, S.; Sen, C.K. α-Lipoic acid supplementation: Tissue glutathione homeostasis at rest and after exercise. J. Appl. Physiol. 1999, 86, 1191–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrick, L. Mercury toxicity and antioxidants: Part 1: Role of glutathione and alpha-lipoic acid in the treatment of mercury toxicity. Altern. Med. Rev. A J. Clin. Ther. 2002, 7, 456–471. [Google Scholar]
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548. [Google Scholar] [CrossRef]
- Allen, J.; Bradley, R.D. Effects of Oral Glutathione Supplementation on Systemic Oxidative Stress Biomarkers in Human Volunteers. J. Altern. Complement. Med. 2011, 17, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Buonocore, D.; Grosini, M.; Giardina, S.; Michelotti, A.; Carrabetta, M.; Seneci, A.; Verri, M.; Dossena, M.; Marzatico, F. Bioavailability Study of an Innovative Orobuccal Formulation of Glutathione. Oxidative Med. Cell. Longev. 2016, 2016, 3286365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S. Relationships Between Nutrition, Alcohol Use, and Liver Disease. Alcohol Res. Health J. Natl. Inst. Alcohol Abus. Alcohol. 2003, 27, 220–231. [Google Scholar]
- Guerra, J.I.E. Oxidation, between life and disease. An. Med. Interna 2001, 18, 1–4. [Google Scholar]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Kugiyama, K.; Motoyama, T.; Hirashima, O.; Ohgushi, M.; Soejima, H.; Misumi, K.; Kawano, H.; Miyao, Y.; Yoshimura, M.; Ogawa, H.; et al. Vitamin C attenuates abnormal vasomotor reactivity in spasm coronary arteries in patients with coronary spastic angina. J. Am. Coll. Cardiol. 1998, 32, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Kugiyama, K.; Sugiyama, S.; Soejima, H.; Kawano, H.; Sakamoto, T.; Takazoe, K.; Ogawa, H.; Doi, H.; Yasue, H. Increase in plasma levels of oxidized low-density lipoproteins in patients with coronary spastic angina. Atherosclerosis 2001, 154, 463–467. [Google Scholar] [CrossRef]
- Leopold, J.A.; Loscalzo, J. Oxidative Enzymopathies and Vascular Disease. Arter. Thromb. Vasc. Biol. 2005, 25, 1332–1340. [Google Scholar] [CrossRef]
- Blanco, R.A.; Ziegler, T.R.; Carlson, B.A.; Cheng, P.-Y.; Park, Y.; Cotsonis, G.A.; Accardi, C.J.; Jones, D.P. Diurnal variation in glutathione and cysteine redox states in human plasma. Am. J. Clin. Nutr. 2007, 86, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, S.; Rizvi, S.I. Day and Night GSH and MDA Levels in Healthy Adults and Effects of Different Doses of Melatonin on These Parameters. Int. J. Cell Biol. 2011, 2011, 404591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosower, N.S.; Kosower, E.M. The Glutathione Status of Cells. Adv. Clin. Chem. 1978, 54, 109–160. [Google Scholar] [CrossRef]
- Urata, Y.; Yamamoto, H.; Goto, S.; Tsushima, H.; Akazawa, S.; Yamashita, S.; Nagataki, S.; Kondo, T. Long Exposure to High Glucose Concentration Impairs the Responsive Expression of γ-Glutamylcysteine Synthetase by Interleukin-1β and Tumor Necrosis Factor-α in Mouse Endothelial Cells. J. Biol. Chem. 1996, 271, 15146–15152. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W.; Schulze-Osthoff, K.; Mihm, S.; Galter, D.; Schenk, H.; Eck, H.; Roth, S.; Gmünder, H. Functions of glutathione and glutathione disulfide in immunology and immunopathology. FASEB J. 1994, 8, 1131–1138. [Google Scholar] [CrossRef]
- Meister, A. Selective modification of glutathione metabolism. Science 1983, 220, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Aw, T.Y. Cellular Redox: A Modulator of Intestinal Epithelial Cell Proliferation. News Physiol. Sci. 2003, 18, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; García-Ruiz, C.; Colell, A.; Morales, A.; Mari, M.; Miranda, M.; Ardite, E. Oxidative stress: Role of mitochondria and protection by glutathione. BioFactors 1998, 8, 7–11. [Google Scholar] [CrossRef]
- Li, T.-K. The Glutathione and Thiol Content of Mammalian Spermatozoa and Seminal Plasma. Biol. Reprod. 1975, 12, 641–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Chen, Y.; Seth, S.; Furukawa, S.; Compans, R.; Jones, D.P. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 2003, 34, 928–936. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Burek, C.L.; Rose, N.R. Autoimmune thyroiditis and ROS. Autoimmun. Rev. 2008, 7, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-Z.; Yang, S.; Wu, G. Free radicals, antioxidants, and nutrition. Nutrition 2002, 18, 872–879. [Google Scholar] [CrossRef]
- Griffiths, H.R. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun. Rev. 2008, 7, 544–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Moon, J. Circulating Total Glutathione in Normal Tension Glaucoma Patients: Comparison with Normal Control Subjects. Korean J. Ophthalmol. 2012, 26, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Sahaf, B.; Heydari, K.; Herzenberg, L.A. The extracellular microenvironment plays a key role in regulating the redox status of cell surface proteins in HIV-infected subjects. Arch. Biochem. Biophys. 2005, 434, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Thomas, M.; Ghorpade, A.; Gendelman, H.E.; Banerjee, R. A Functional Transsulfuration Pathway in the Brain Links to Glutathione Homeostasis. J. Biol. Chem. 2006, 281, 35785–35793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.W.; Wang, Y. Deficient glutathione peroxidase activity in preeclampsia is associated with increased placental production of thromboxane and lipid peroxides. Am. J. Obstet. Gynecol. 1993, 169, 1456–1461. [Google Scholar] [CrossRef]
- Ballatori, N.; Hammond, C.L.; Cunningham, J.B.; Krance, S.M.; Marchan, R. Molecular mechanisms of reduced glutathione transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol. Appl. Pharmacol. 2005, 204, 238–255. [Google Scholar] [CrossRef]
- Rebbeor, J.F.; Connolly, G.C.; Ballatori, N. Inhibition of Mrp2- and Ycf1p-mediated transport by reducing agents: Evidence for GSH transport on rat Mrp. Biochim. Biophys. Acta (BBA)-Biomembr. 2002, 1559, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Palomero, J.; Galán, A.; Muñoz, M.; Tuñón, M.J.; González-Gallego, J.; Jiménez, R. Effects of aging and cyclosporin treatment on the hepatobiliary efflux of glutathione. Life Sci. 2003, 73, 3387–3397. [Google Scholar] [CrossRef] [PubMed]
- Matuz-Mares, D.; Hernández-Vázquez, A.; Riveros-Rosas, H.; Guinzberg, R.; Quesada-López, T.; Cárabez-Trejo, A.; Mora, O.; Piña, E. β- Adrenoceptors activate hepatic glutathione efflux through an unreported pathway. Arch. Biochem. Biophys. 2018, 644, 47–56. [Google Scholar] [CrossRef]
- Han, D.; Hanawa, N.; Saberi, B.; Kaplowitz, N. Mechanisms of Liver Injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Liver Physiol. 2006, 291, G1–G7. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, W.A.; Richie, J. Status of glutathione and other thiols and disulfides in human plasma. Biochem. Pharmacol. 2000, 60, 19–29. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The Quantitative Significance of the Transsulfuration Enzymes for H2S Production in Murine Tissues. Antioxid. Redox Signal. 2011, 15, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Ookhtens, M.; Lyon, I.; Fernández-Checa, J.C.; Kaplowitz, N. Inhibition of glutathione efflux in the perfused rat liver and isolated hepatocytes by organic anions and bilirubin. Kinetics, sidedness, and molecular forms. J. Clin. Investig. 1988, 82, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C.; Ge, J.L.; Huang, H.Y.; Kuhlenkamp, J.; Kaplowitz, N. Thiol-disulfide effects on hepatic glutathione transport. Studies in cultured rat hepatocytes and perfused livers. J. Clin. Investig. 1993, 92, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Ballatori, N.; Rebbeor, J. Roles of MRP2 and oatp1 in Hepatocellular Export of Reduced Glutathione. Semin. Liver Dis. 1998, 18, 377–387. [Google Scholar] [CrossRef]
- Hammond, C.L.; Lee, T.K.; Ballatori, N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J. Hepatol. 2001, 34, 946–954. [Google Scholar] [CrossRef]
- Rius, M.; Nies, A.T.; Hummel-Eisenbeiss, J.; Jedlitschky, G.; Keppler, D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology 2003, 38, 374–384. [Google Scholar] [CrossRef]
- Homolya, L.; Varadi, A.; Sarkadi, B. Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate. Biofactors 2003, 17, 103–114. [Google Scholar] [CrossRef]
- Nasr, R.; Lorendeau, D.; Khonkarn, R.; Dury, L.; Peres, B.; Boumendjel, A.; Cortay, J.C.; Falson, P.; Chaptal, V.; Baubichon-Cortay, H. Molecular analysis of the massive GSH transport mechanism mediated by the human Multidrug Resistant Protein 1/ABCC1. Sci. Rep. 2020, 10, 7616. [Google Scholar] [CrossRef] [PubMed]
- Leier, I.; Jedlitschky, G.; Buchholz, U.; Center, M.; Cole, S.P.; Deeley, R.G.; Keppler, D. ATP-dependent glutathione disulphide transport mediated by the MRP gene-encoded conjugate export pump. Biochem. J. 1996, 314 Pt 2, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Deeley, R.G.; Cole, S.P. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS Lett. 2006, 580, 1103–1111. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Hanigan, M.H. Enzymes Involved in Processing Glutathione Conjugates. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 323–366. [Google Scholar]
- Cantin, A.M.; White, T.B.; Cross, C.E.; Forman, H.J.; Sokol, R.J.; Borowitz, D. Antioxidants in cystic fibrosis. Conclusions from the CF antioxidant workshop, Bethesda, Maryland, November 11-12, 2003. Free Radic. Biol. Med. 2007, 42, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R. Assembly of functional CFTR chloride channels. Annu. Rev. Physiol. 2005, 67, 701–718. [Google Scholar] [CrossRef]
- Hudson, V.M. Rethinking cystic fibrosis pathology: The critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic. Biol. Med. 2001, 30, 1440–1461. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. The superfamily of organic anion transporting polypeptides. Biochim. Biophys. Acta 2003, 1609, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, D.; Nozawa, T.; Imai, K.; Nezu, J.; Tsuji, A.; Tamai, I. Involvement of human organic anion transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport across intestinal apical membrane. J. Pharmacol. Exp. Ther. 2003, 306, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briz, O.; Romero, M.R.; Martinez-Becerra, P.; Macias, R.I.; Perez, M.J.; Jimenez, F.; San Martin, F.G.; Marin, J.J. OATP8/1B3-mediated cotransport of bile acids and glutathione: An export pathway for organic anions from hepatocytes? J. Biol. Chem. 2006, 281, 30326–30335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugele, B.; Gebhardt, R. Heterogeneity of rat liver parenchyma in taurocholate uptake. Hepatol. Res. 2003, 27, 238–247. [Google Scholar] [CrossRef]
- Li, L.; Meier, P.J.; Ballatori, N. Oatp2 mediates bidirectional organic solute transport: A role for intracellular glutathione. Mol. Pharmacol. 2000, 58, 335–340. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Colell, A.; Morales, A.; Kaplowitz, N.; Fernández-Checa, J.C. Role of oxidative stress generated from the mitochondrial electron transport chain and mitochondrial glutathione status in loss of mitochondrial function and activation of transcription factor nuclear factor-kappa B: Studies with isolated mitochondria and rat hepatocytes. Mol. Pharmacol. 1995, 48, 825–834. [Google Scholar] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, J.; Lai, J.C.; Meister, A. High-affinity transport of glutathione is part of a multicomponent system essential for mitochondrial function. Proc. Natl. Acad. Sci. USA 1990, 87, 7185–7189. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Lash, L.H. Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J. Pharmacol. Exp. Ther. 1998, 285, 608–618. [Google Scholar]
- Chen, Z.; Putt, D.A.; Lash, L.H. Enrichment and functional reconstitution of glutathione transport activity from rabbit kidney mitochondria: Further evidence for the role of the dicarboxylate and 2-oxoglutarate carriers in mitochondrial glutathione transport. Arch. Biochem. Biophys. 2000, 373, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.; Murphy, M.P. The mitochondrial dicarboxylate and 2-oxoglutarate carriers do not transport glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, I.M.; Faux, S.P. Oxidative stress and cardiovascular disease: Novel tools give (free) radical insight. J. Mol. Cell. Cardiol. 2009, 47, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Kiyohara, Y.; Kato, I.; Kitazono, T.; Tanizaki, Y.; Kubo, M.; Ueno, H.; Ibayashi, S.; Fujishima, M.; Iida, M. Relationship between plasma glutathione levels and cardiovascular disease in a defined population: The Hisayama study. Stroke 2004, 35, 2072–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “Redox Homeostasis” and Its Relation to Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 5028181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preville, X.; Salvemini, F.; Giraud, S.; Chaufour, S.; Paul, C.; Stepien, G.; Ursini, M.V.; Arrigo, A.-P. Mammalian Small Stress Proteins Protect against Oxidative Stress through Their Ability to Increase Glucose-6-phosphate Dehydrogenase Activity and by Maintaining Optimal Cellular Detoxifying Machinery. Exp. Cell Res. 1999, 247, 61–78. [Google Scholar] [CrossRef]
- Brewer, A.C.; Murray, T.V.; Arno, M.; Zhang, M.; Anilkumar, N.P.; Mann, G.E.; Shah, A. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 2011, 51, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Brewer, A.; Schröder, K.; Santos, C.X.C.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, V.; Padmaja, G.; Kuppusamy, P.; Kumar, V.K. Oxidative stress in cardiovascular disease. Indian J. Biochem. Biophys. 2009, 46, 421–440. [Google Scholar]
- Higashi, Y.; Maruhashi, T.; Noma, K.; Kihara, Y. Oxidative stress and endothelial dysfunction: Clinical evidence and therapeutic implications. Trends Cardiovasc. Med. 2014, 24, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Vallejo, S.; Sanchez, F.J.; Sandoval, E.; Blanco, E.; Cannata, P.; Peiro, C.; Sanchez-Ferrer, C.F.; Lamas, S. Role of glutathione biosynthesis in endothelial dysfunction and fibrosis. Redox Biol. 2018, 14, 88–99. [Google Scholar] [CrossRef]
- Conklin, D.J.; Haberzettl, P.; Prough, R.A.; Bhatnagar, A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1586–H1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera, M.; Torramade-Moix, S.; Martin-Rodriguez, S.; Cases, A.; Cruzado, J.M.; Rivera, J.; Escolar, G.; Palomo, M.; Diaz-Ricart, M. Antioxidant and Anti-Inflammatory Strategies Based on the Potentiation of Glutathione Peroxidase Activity Prevent Endothelial Dysfunction in Chronic Kidney Disease. Cell Physiol. Biochem. 2018, 51, 1287–1300. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef] [PubMed]
- Lagman, M.; Ly, J.; Saing, T.; Kaur Singh, M.; Vera Tudela, E.; Morris, D.; Chi, P.T.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS ONE 2015, 10, e0118436. [Google Scholar] [CrossRef] [Green Version]
- Rybka, J.; Kupczyk, D.; Kedziora-Kornatowska, K.; Motyl, J.; Czuczejko, J.; Szewczyk-Golec, K.; Kozakiewicz, M.; Pawluk, H.; Carvalho, L.A.; Kedziora, J. Glutathione-related antioxidant defense system in elderly patients treated for hypertension. Cardiovasc. Toxicol. 2011, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Damy, T.; Kirsch, M.; Khouzami, L.; Caramelle, P.; Le Corvoisier, P.; Roudot-Thoraval, F.; Dubois-Rande, J.L.; Hittinger, L.; Pavoine, C.; Pecker, F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE 2009, 4, e4871. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Volkova, N.; Coleman, R.; Aviram, M. Anti-oxidant and anti-atherogenic properties of liposomal glutathione: Studies in vitro, and in the atherosclerotic apolipoprotein E-deficient mice. Atherosclerosis 2007, 195, e61–e68. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Yin, M.-C.; Hsu, C.-C.; Lin, M.-P. Effect of five cysteine-containing compounds on three lipogenic enzymes in Balb/cA mice consuming a high saturated fat diet. Lipids 2004, 39, 843–848. [Google Scholar] [CrossRef]
- De Mattia, G.; Bravi, M.C.; Laurenti, O.; Cassone-Faldetta, M.; Proietti, A.; De Luca, O.; Armiento, A.; Ferri, C. Reduction of oxidative stress by oral N-acetyl-L-cysteine treatment decreases plasma soluble vascular cell adhesion molecule-1 concentrations in non-obese, non-dyslipidaemic, normotensive, patients with non-insulin-dependent diabetes. Diabetology 1998, 41, 1392–1396. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Andrews, N.P.; Padder, F.A.; Husain, M.; Quyyumi, A.A. Glutathione reverses endothelial dysfunction and improves nitric oxide bioavailability. J. Am. Coll. Cardiol. 1999, 34, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: A review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef]
- Sag, C.M.; Santos, C.X.; Shah, A.M. Redox regulation of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 73, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, D.R.; Gomes, M.J.; Rosa, C.M.; Pagan, L.U.; Damatto, F.C.; Damatto, R.L.; DePra, I.; Campos, D.H.; Fernandez, A.A.; Martinez, P.F.; et al. N-Acetylcysteine Influence on Oxidative Stress and Cardiac Remodeling in Rats During Transition from Compensated Left Ventricular Hypertrophy to Heart Failure. Cell. Physiol. Biochem. 2017, 44, 2310–2321. [Google Scholar] [CrossRef] [PubMed]
- Ramezani-Aliakbari, F.; Badavi, M.; Dianat, M.; Mard, S.A.; Ahangarpour, A. The Effects of Trimetazidine on QT-interval Prolongation and Cardiac Hypertrophy in Diabetic Rats. Arq. Bras. Cardiol. 2018, 112, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chuang, C.-C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. BioMed Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwakarma, V.K.; Upadhyay, P.K.; Gupta, J.K.; Yadav, H.N. Pathophysiologic role of ischemia reperfusion injury: A review. J. Indian Coll. Cardiol. 2017, 7, 97–104. [Google Scholar] [CrossRef]
- Terentyev, D.; Györke, I.; Belevych, A.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; De Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox Modification of Ryanodine Receptors Contributes to Sarcoplasmic Reticulum Ca 2+ Leak in Chronic Heart Failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Fushimi, K.; Kouchi, H.; Mihara, K.; Miyazaki, M.; Ohe, T.; Namba, M. Inhibitory Effects of Antioxidants on Neonatal Rat Cardiac Myocyte Hypertrophy Induced by Tumor Necrosis Factor-α and Angiotensin II. Circulation 1998, 98, 794–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuster, G.M.; Lancel, S.; Zhang, J.; Communal, C.; Trucillo, M.P.; Lim, C.C.; Pfister, O.; Weinberg, E.; Cohen, R.A.; Liao, R. Redox-mediated reciprocal regulation of SERCA and Na+–Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic. Biol. Med. 2010, 48, 1182–1187. [Google Scholar] [CrossRef] [Green Version]
- Kovács, V.; Gasz, B.; Balatonyi, B.; Járomi, L.; Kisfali, P.; Borsiczky, B.; Jancsó, G.; Marczin, N.; Szabados, S.; Melegh, B.; et al. Polymorphisms in glutathione S-transferase are risk factors for perioperative acute myocardial infarction after cardiac surgery: A preliminary study. Mol. Cell. Biochem. 2014, 389, 79–84. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 809–824. [Google Scholar] [CrossRef]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B. Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef]
- Zhang, P.; Li, T.; Wu, X.; Nice, E.C.; Huang, C.; Zhang, Y. Oxidative stress and diabetes: Antioxidative strategies. Front. Med. 2020, 14, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Causer, A.; Shute, J.K.; Cummings, M.H.; Shepherd, A.; Gruet, M.; Costello, J.; Bailey, S.; Lindley, M.; Pearson, C.; Connett, G.; et al. Circulating biomarkers of antioxidant status and oxidative stress in people with cystic fibrosis: A systematic review and meta-analysis. Redox Biol. 2020, 32, 101436. [Google Scholar] [CrossRef] [PubMed]
- Scholte, B.J.; Horati, H.; Veltman, M.; Vreeken, R.J.; Garratt, L.W.; Tiddens, H.A.; Janssens, H.M.; Stick, S. Oxidative stress and abnormal bioactive lipids in early cystic fibrosis lung disease. J. Cyst. Fibros. 2019, 18, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Hemmati-Dinarvand, M.; Saedi, S.; Valilo, M.; Kalantary-Charvadeh, A.; Sani, M.A.; Kargar, R.; Safari, H.; Samadi, N. Oxidative stress and Parkinson’s disease: Conflict of oxidant-antioxidant systems. Neurosci. Lett. 2019, 709, 134296. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J.-W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trist, B.; Hare, D.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [Green Version]
- Martin-Subero, M.; Anderson, G.; Kanchanatawan, B.; Berk, M.; Maes, M. Comorbidity between depression and inflammatory bowel disease explained by immune-inflammatory, oxidative, and nitrosative stress; tryptophan catabolite; and gut–brain pathways. CNS Spectr. 2015, 21, 184–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free. Radic. Biol. Med. 2018, 125, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2016, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Khanfar, A.; Al Qaroot, B. Could glutathione depletion be the Trojan horse of COVID-19 mortality? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12500–12509. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants 2020, 9, 624. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.; Pazdro, R. Impact of Supplementary Amino Acids, Micronutrients, and Overall Diet on Glutathione Homeostasis. Nutrients 2019, 11, 1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettermann, E.L.; Hartman, T.J.; Easley, K.; Ferranti, E.P.; Jones, D.P.; Quyyumi, A.A.; Vaccarino, V.; Ziegler, T.R.; Alvarez, J.A. Higher Mediterranean Diet Quality Scores and Lower Body Mass Index Are Associated with a Less-Oxidized Plasma Glutathione and Cysteine Redox Status in Adults. J. Nutr. 2018, 148, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2221–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yubero-Serrano, E.M.; Fernandez-Gandara, C.; Garcia-Rios, A.; Rangel-Zuñiga, O.A.; Gutierrez-Mariscal, F.M.; Torres-Peña, J.D.; Marin, C.; Lopez-Moreno, J.; Castaño, J.P.; Delgado-Lista, J.; et al. Mediterranean diet and endothelial function in patients with coronary heart disease: An analysis of the CORDIOPREV randomized controlled trial. PLoS Med. 2020, 17, e1003282. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Nakano, H.; Fajardo, V.M.; Nakano, A. The role of glucose in physiological and pathological heart formation. Dev. Biol. 2021, 475, 222–233. [Google Scholar] [CrossRef]
- Gould, R.; Zhou, Y.; Yakaitis, C.L.; Love, K.; Reeves, J.; Kong, W.; Coe, E.; Xiao, Y.; Pazdro, R. Heritability of the aged glutathione phenotype is dependent on tissue of origin. Mamm. Genome 2018, 29, 619–631. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Romá-Mateo, C.; Pérez-Machado, G.; Peiró-Chova, L.; Pallardó, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef]
- Forman, H.J. Glutathione—From antioxidant to post-translational modifier. Arch. Biochem. Biophys. 2016, 595, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Hou, Y.; Zhou, W.; Keerthiga, R.; Fu, A. Mitochondrial transplantation therapy inhibit carbon tetrachloride-induced liver injury through scavenging free radicals and protecting hepatocytes. Bioeng. Transl. Med. 2020, 6, e10209. [Google Scholar] [CrossRef] [PubMed]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS ONE 2016, 11, e0160889. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matuz-Mares, D.; Riveros-Rosas, H.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Glutathione Participation in the Prevention of Cardiovascular Diseases. Antioxidants 2021, 10, 1220. https://doi.org/10.3390/antiox10081220
Matuz-Mares D, Riveros-Rosas H, Vilchis-Landeros MM, Vázquez-Meza H. Glutathione Participation in the Prevention of Cardiovascular Diseases. Antioxidants. 2021; 10(8):1220. https://doi.org/10.3390/antiox10081220
Chicago/Turabian StyleMatuz-Mares, Deyamira, Héctor Riveros-Rosas, María Magdalena Vilchis-Landeros, and Héctor Vázquez-Meza. 2021. "Glutathione Participation in the Prevention of Cardiovascular Diseases" Antioxidants 10, no. 8: 1220. https://doi.org/10.3390/antiox10081220
APA StyleMatuz-Mares, D., Riveros-Rosas, H., Vilchis-Landeros, M. M., & Vázquez-Meza, H. (2021). Glutathione Participation in the Prevention of Cardiovascular Diseases. Antioxidants, 10(8), 1220. https://doi.org/10.3390/antiox10081220