
S-Denitrosylation: A Crosstalk between Glutathione and Redoxin Systems

 , , , ,
, , , ,
Abstract
:1. Introduction
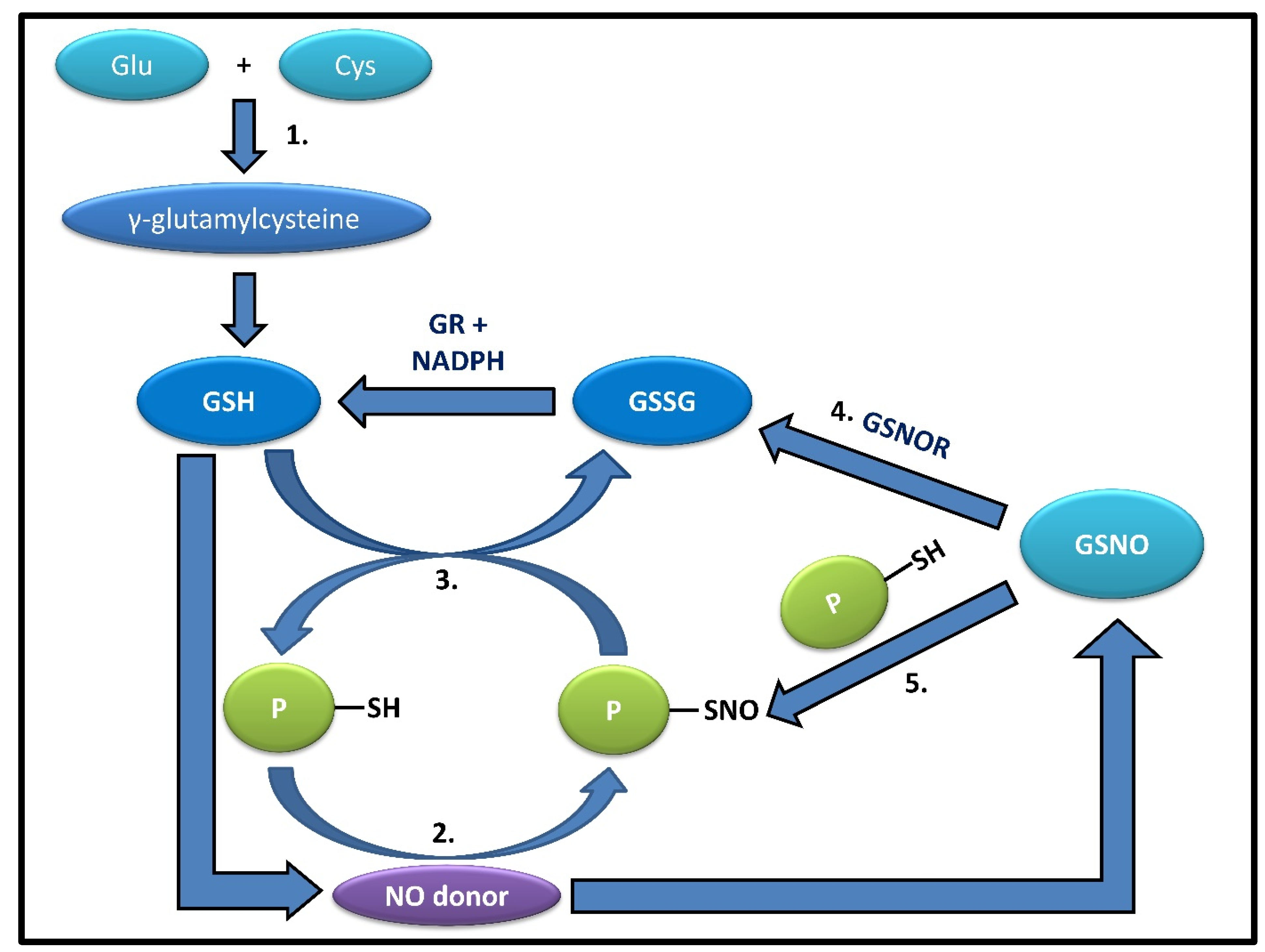
2. GSH Synthesis and Denitrosylation
2.1. GSH Synthesis
2.2. S-Nitrosylation of Glutathione
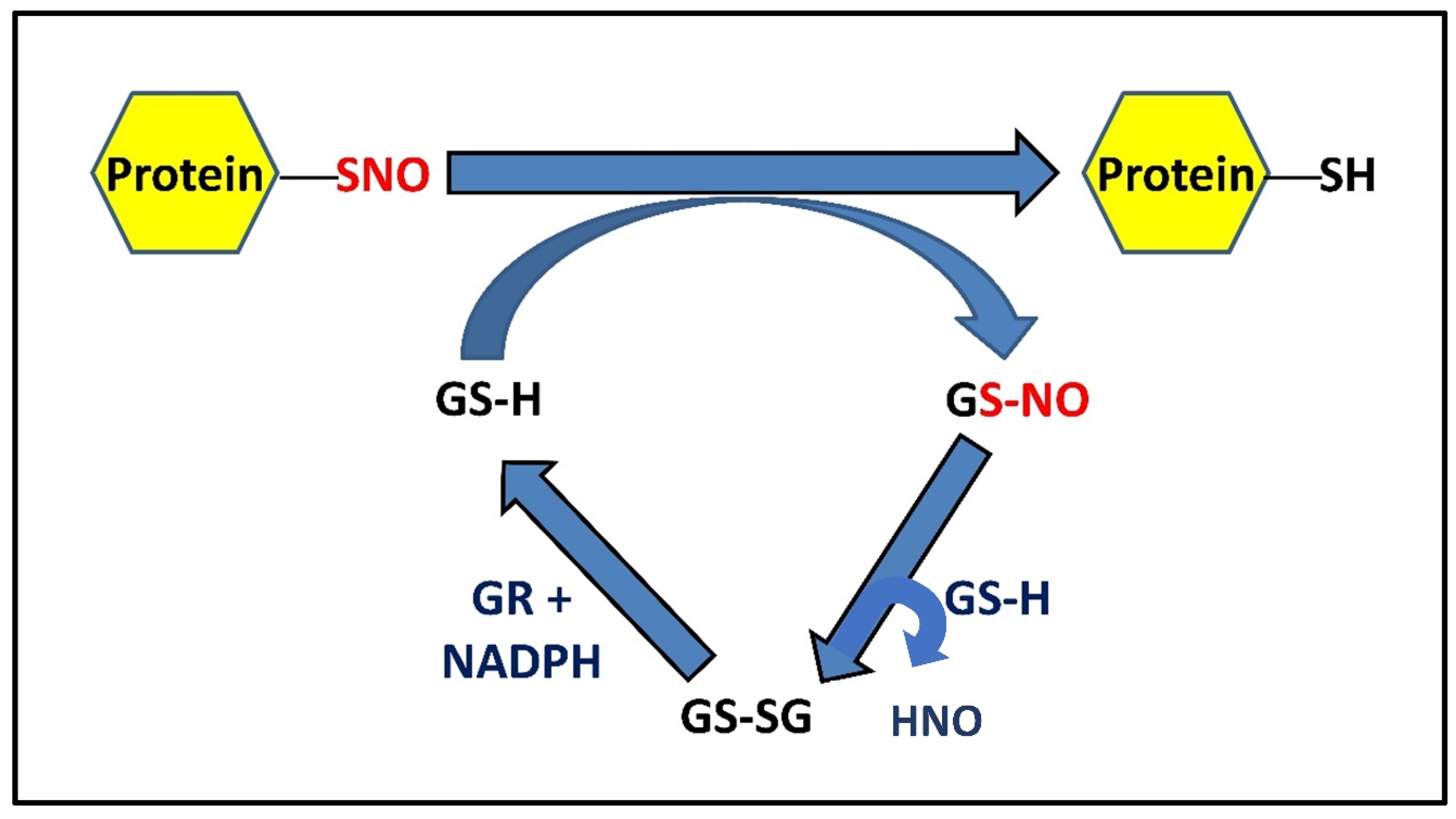
2.3. Role of GSH in S-Denitrosylation
2.4. PSNO Denitrosylation and Exceptions Amongst PSNOs
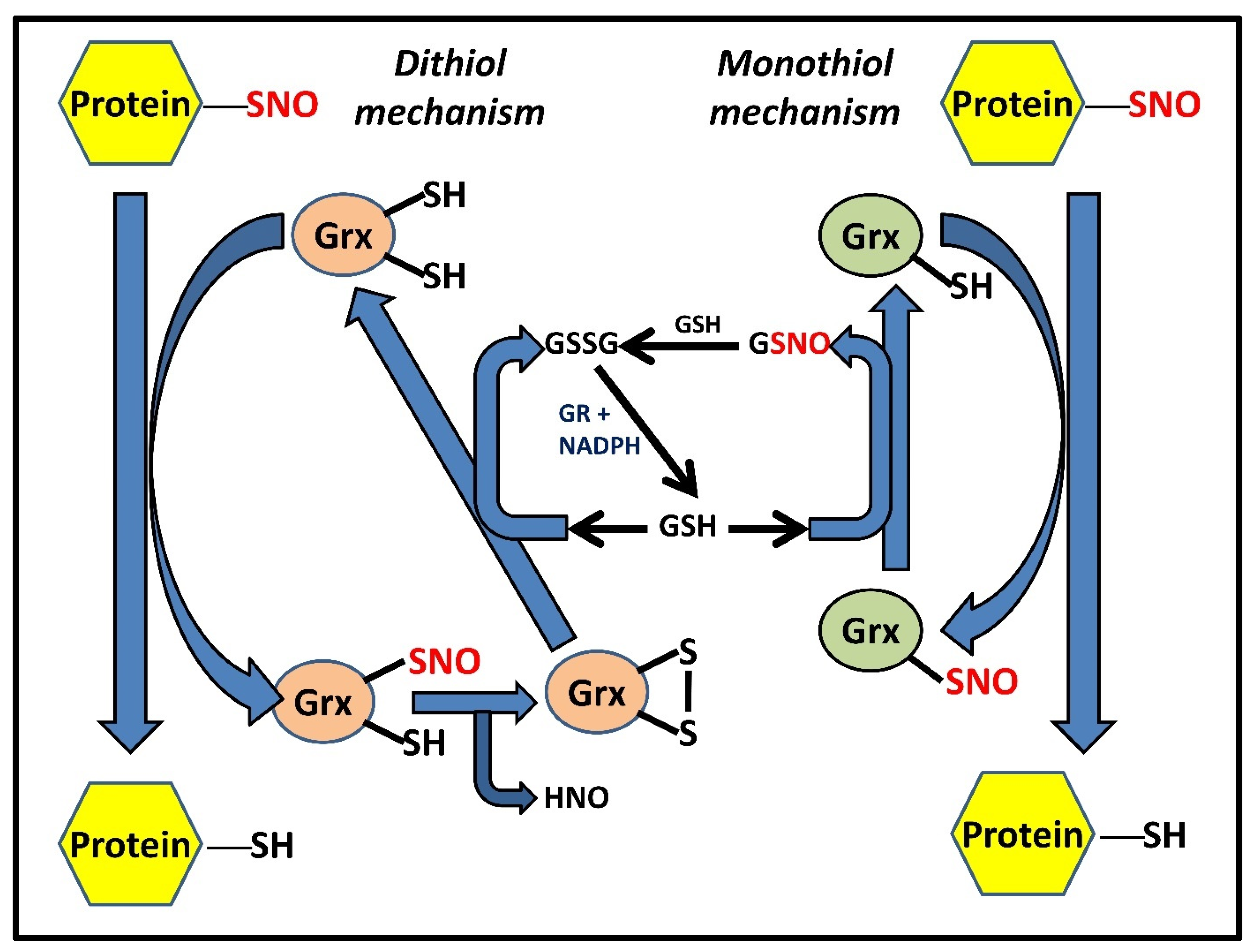
3. Grx-Mediated denitrosylation and Deglutathionylation
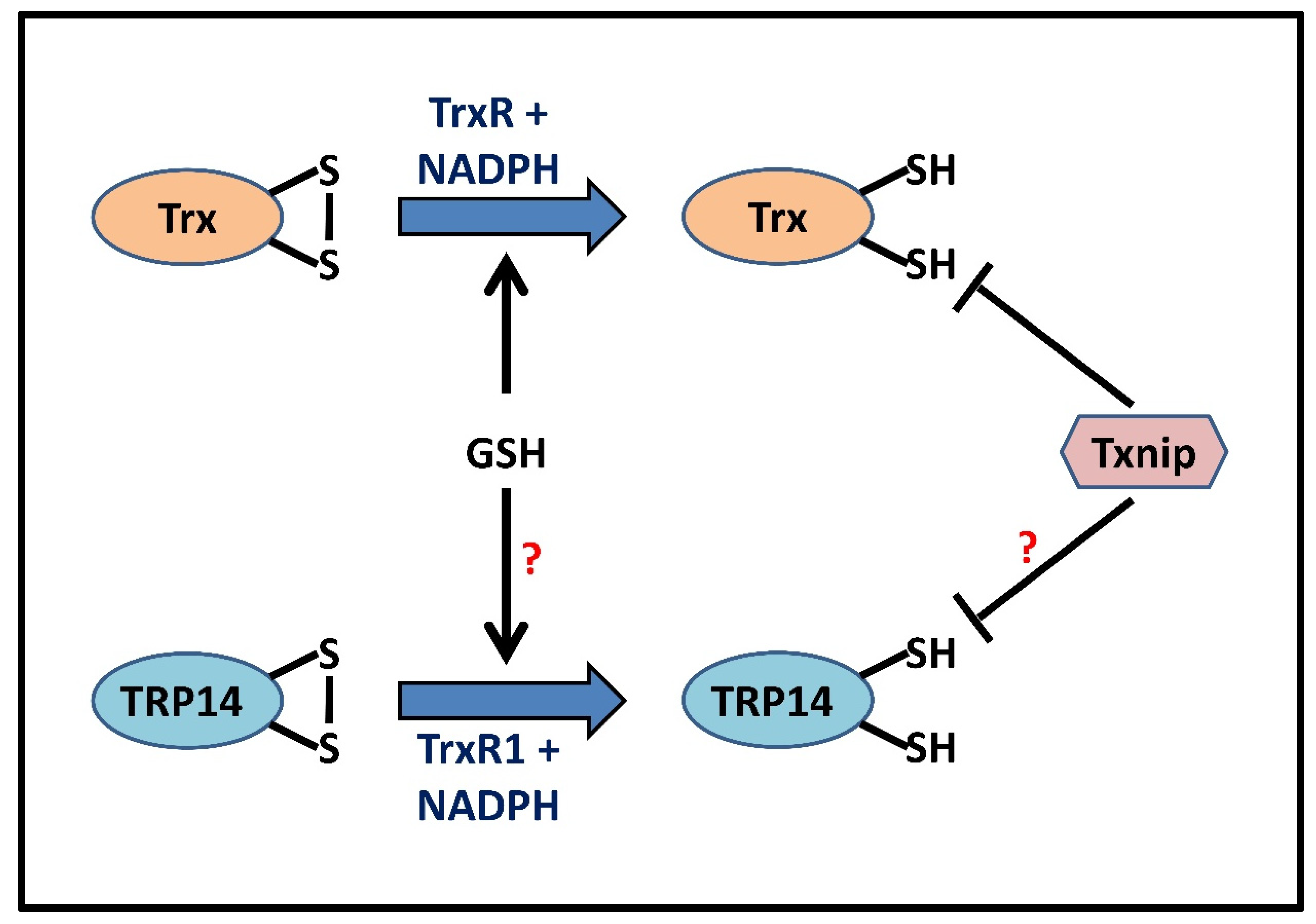
4. Trx Isoforms and Trx-Mediated Denitrosylation
5. Redundancy of Redoxin Systems
6. Substrate Specificity of GSH and Redoxin Systems
7. Drugs and Treatments: Current Potential and Challenges
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NO | nitric oxide |
| cGMP | cyclic GMP/Guanosine 3′,5′-cyclic monophosphate |
| NOSs | Nitric Oxide Synthases |
| nNOS | neuronal nitric oxide synthase |
| iNOS | inducible nitric oxide synthase |
| eNOS | endothelial nitric oxide synthase |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NOX | NADPH Oxidase |
| PSNOs | S-nitrosoproteins |
| GSNO | S-nitrosoglutathione |
| PTEN | phosphatase and tensin homolog |
| FTD | frontal temporal dementia |
| ALS | amyotrophic lateral sclerosis |
| GSH | glutathion/ϒ-glutamylcysteinylglycine |
| Trx | thioredoxin |
| Grx | glutaredoxin |
| GSSG | oxidized glutathione |
| GSNOR | GSNO reductase |
| KGDHC | α-ketoglutarate dehydrogenase complex |
| Prx6 | peroxiredoxin 6 |
| AMPK | 5′AMP-activated protein kinase |
| SERCA | sarcoplasmic reticulum Ca2+-ATPase |
| PDI | protein disulfide isomerase |
| GPx | glutathione peroxidase |
| GSH-EE | GSH Ethyl Ester |
| NPSH | Non-Protein Thiol |
| HNO | nitroxyl |
| GR | glutathione reductase |
| ADH-III | alcohol dehydrogenase III |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| TrxR | thioredoxin reductase |
| ATG | aurothioglucose |
| DHLA | dihydrolipoic acid |
| PDI | protein disulfide isomerase |
| TRP14 | thioredoxin-related protein 14 |
| LMW | low molecular weight |
| HMW | high molecular weight |
| XO | xanthine oxidase |
| SOD | superoxide dismutase |
| Msr | methionine sulfoxide reductase |
| I/R | ischemia/reperfusion |
| MEF | mouse embryonic fibroblast |
| LEC | lens epithelial cells |
| ARPE-19 | human retinal pigment epithelial cells |
| KO | knockout |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| MMP | mitochondrial membrane potential |
| iPSC | induced pluripotent stem cells |
| Hif-1α | hypoxia-inducible factor-1α |
| JNK | c-Jun NH2-terminal kinase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| HSP | heat shock protein |
| BCNU | 1:3-bis[2-chloroethyl]-1-nitrosourea |
| 2-AAPA | 2-acetylamino-3-[4-(2-acetylamino-2-carboxyethylsulfanylthiocarbonylamino)phenyl-thiocarbamoyl sulfanyl propionic acid |
| Akt | protein kinase B |
| VEGF | vascular endothelial growth factor |
| GST | glutathione-S-transferase |
| PSSG | glutathionylated protein |
| TXNIP | thioredoxin interacting protein, or thioredoxin binding protein-2, TBP-2, or Vit D3 upregulated protein |
| ASK1 | apoptosis signal-regulating kinase 1 |
| NHC complex | N-heterocyclic carbene complexes |
| RNR | ribonucleotide reductase |
| MQ | methylene quinuclidine |
References
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, R.; Holmgren, A. The role of thioredoxin in the regulation of cellular processes by S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Giulivi, C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic. Biol. Med. 2010, 49, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Ren, X. Thioredoxin and Glutaredoxin Systems under Oxidative and Nitrosative Stress. Ph.D. Thesis, Karolinska Institutet, Department of Medical Biochemistry and Biophysics, Stockholm, Sweden, 2017. [Google Scholar]
- Csillag, A.; Boldogh, I.; Pazmandi, K.; Magyarics, Z.; Gogolak, P.; Sur, S.; Rajnavolgyi, E.; Bacsi, A. Pollen-induced oxidative stress influences both innate and adaptive immune responses via altering dendritic cell functions. J. Immunol. 2010, 184, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.; Cardaci, S.; Montagna, C.; Giacomo, G.D.; Zio, D.D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397. [Google Scholar] [CrossRef]
- Walsh, G.M.; Leane, D.; Moran, N.; Keyes, T.E.; Forster, R.J.; Kenny, D.; O’Neill, S. S-nitrosylation of platelet alphaIIbbeta3 as revealed by Raman spectroscopy. Biochemistry 2007, 46, 6429–6436. [Google Scholar] [CrossRef]
- Que, L.G.; Liu, L.; Yan, Y.; Whitehead, G.S.; Gavett, S.H.; Schwartz, D.A.; Stamler, J.S. Protection from experimental asthma by an endogenous bronchodilator. Science 2005, 308, 1618–1621. [Google Scholar] [CrossRef]
- Zima, A.V.; Mazurek, S.R. Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev. Physiol. Biochem. Pharmacol. 2016, 171, 39–62. [Google Scholar]
- Sengupta, R.; Ryter, S.W.; Zuckerbraun, B.S.; Tzeng, E.; Billiar, T.R.; Stoyanovsky, D.A. Thioredoxin catalyzes the denitrosation of low-molecular-mass and protein S-nitrosothiols. Biochemistry 2007, 46, 8472–8483. [Google Scholar] [CrossRef]
- Ren, X.; Sengupta, R.; Lu, J.; Lundberg, J.O.; Holmgren, A. Characterization of mammalian glutaredoxin isoforms as S-denitrosylases. FEBS Lett. 2019, 593, 1799–1806. [Google Scholar] [CrossRef]
- Pader, I.; Sengupta, R.; Cebula, M.; Xu, J.; Lundberg, J.O.; Holmgren, A.; Johansson, K.; Arnér, E.S.J. Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc. Natl. Acad. Sci. USA 2014, 111, 6964–6969. [Google Scholar] [CrossRef]
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869. [Google Scholar] [CrossRef]
- Raj Rai, S.; Bhattacharyya, C.; Sarkar, A.; Chakraborty, S.; Sircar, E.; Dutta, S.; Sengupta, R. Glutathione: Role in oxidative/nitrosative stress, antioxidant defense, and treatments. ChemistrySelect 2021, 6, 4566–4590. [Google Scholar] [CrossRef]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 3150145. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Markovic, J.; Dasí, F.; Queval, G.; Schnaubelt, D.; Foyer, C.H.; Pallardó, F.V. Nuclear glutathione. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3304–3316. [Google Scholar] [CrossRef]
- Russell, T.M.; Azad, M.G.; Richardson, D.R. The Relationship of Glutathione-S-Transferase and Multi-Drug Resistance-Related Protein 1 in Nitric Oxide (NO) Transport and Storage. Molecules 2021, 26, 5784. [Google Scholar] [CrossRef]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell. Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J.; Choi, J. Gamma-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar]
- Corpas, F.J.; Alché, J.D.; Barroso, J.B. Current overview of S-nitrosoglutathione (Gsno) in higher plants. Front. Plant Sci. 2013, 4, 126. [Google Scholar] [CrossRef] [Green Version]
- Won, J.S.; Kim, J.; Annamalai, B.; Shunmugavel, A.; Singh, I.; Singh, A.K. Protective role of s-nitrosoglutathione (Gsno) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J. Alzheimer’s Dis. 2013, 34, 621–635. [Google Scholar] [CrossRef]
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (Gsnor) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 340–354. [Google Scholar] [CrossRef]
- Kalinina, E.; Novichkova, M. Glutathione in Protein Redox Modulation through S-Glutathionylation and S-Nitrosylation. Molecules 2021, 26, 435. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Robaczewska, J.; Kedziora-Kornatowska, K.; Kozakiewicz, M.; Zary-Sikorska, E.; Pawluk, H.; Pawliszak, W.; Kedziora, J. Role of glutathione metabolism and glutathione-related antioxidant defense systems in hypertension. J. Physiol. Pharmacol. 2016, 67, 331–337. [Google Scholar]
- Romero, J.M.; Bizzozero, O.A. Intracellular glutathione mediates the denitrosylation of protein nitrosothiols in the rat spinal cord. J. Neurosci. Res. 2009, 87, 701–709. [Google Scholar] [CrossRef]
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548. [Google Scholar] [CrossRef]
- Forrester, M.T.; Foster, M.W.; Stamler, J.S. Assessment and application of the biotin switch technique for examining protein s-nitrosylation under conditions of pharmacologically induced oxidative stress. J. Biol. Chem. 2007, 282, 13977–13983. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494. [Google Scholar] [CrossRef]
- Liu, L.; Yan, Y.; Zeng, M.; Zhang, J.; Hanes, M.A.; Ahearn, G.; McMahon, T.J.; Dickfeld, T.; Marshall, H.E.; Que, L.G.; et al. Essential roles of s-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 2004, 116, 617–628. [Google Scholar] [CrossRef]
- He, J.; Wang, T.; Wang, P.; Han, P.; Yin, Q.; Chen, C. A novel mechanism underlying the susceptibility of neuronal cells to nitric oxide: The occurrence and regulation of protein S -nitrosylation is the checkpoint: A novel mechanism for the susceptibility of neuronal cells to NO. J. Neurochem. 2007, 102, 1863–1874. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Ravagna, A.; Bella, R.; Butterfield, D.A.; Calvani, M.; Pennisi, G.; Giuffrida Stella, A.M. Disruption of thiol homeostasis and nitrosative stress in the cerebrospinal fluid of patients with active multiple sclerosis: Evidence for a protective role of acetylcarnitine. Neurochem. Res. 2003, 28, 1321–1328. [Google Scholar] [CrossRef]
- Chatterji, A.; Sengupta, R. Cellular S-denitrosylases: Potential role and interplay of Thioredoxin, TRP14, and Glutaredoxin systems in thiol-dependent protein denitrosylation. Int. J. Biochem. Cell Biol. 2021, 131, 105904. [Google Scholar] [CrossRef]
- Sircar, E.; Stoyanovsky, D.A.; Billiar, T.R.; Holmgren, A.; Sengupta, R. Analysis of glutathione mediated S-(De)nitrosylation in complex biological matrices by immuno-spin trapping and identification of two novel substrates. Nitric Oxide 2022, 118, 26–30. [Google Scholar] [CrossRef]
- Benhar, M.; Thompson, J.W.; Moseley, M.A.; Stamler, J.S. Identification of s-nitrosylated targets of thioredoxin using a quantitative proteomic approach. Biochemistry 2010, 49, 6963–6969. [Google Scholar] [CrossRef]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein s-nitrosylation: Determinants of specificity and enzymatic regulation of s-nitrosothiol-based signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef]
- Sengupta, R.; Holmgren, A. Thioredoxin and thioredoxin reductase in relation to reversible s-nitrosylation. Antioxid. Redox Signal. 2013, 18, 259–269. [Google Scholar] [CrossRef]
- Chatterji, A.; Banerjee, D.; Billiar, T.R.; Sengupta, R. Understanding the role of S-nitrosylation/nitrosative stress in inflammation and the role of cellular denitrosylases in inflammation modulation: Implications in health and diseases. Free Radic. Biol. Med. 2021, 172, 604–621. [Google Scholar] [CrossRef]
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.J.; Kim, H.; Choi, H.J.; Lee, S.; Kim, K. Protein glutathionylation in the pathogenesis of neurodegenerative diseases. Oxid. Med. Cell. Longev. 2017, 2017, 2818565. [Google Scholar] [CrossRef]
- Conibear, E.; Davis, N.G. Palmitoylation and depalmitoylation dynamics at a glance. J. Cell Sci. 2010, 123, 4007–4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Short, J.D.; Downs, K.; Tavakoli, S.; Asmis, R. Protein Thiol Redox Signaling in Monocytes and Macrophages. Antioxid. Redox Signal. 2016, 25, 816–835. [Google Scholar] [CrossRef]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995, 251, 8–28. [Google Scholar]
- Gilbert, H.F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 69–172. [Google Scholar]
- Stoyanovsky, D.A.; Scott, M.J.; Billiar, T.R. Glutathione and thioredoxin type 1 cooperatively denitrosate HepG2 cells-derived cytosolic S-nitrosoproteins. Org. Biomol. Chem. 2013, 11, 4433–4437. [Google Scholar] [CrossRef]
- Stamler, J.S.; Lamas, S.; Fang, F.C. Nitrosylation. the prototypic redox-based signaling mechanism. Cell 2001, 106, 675–683. [Google Scholar] [CrossRef]
- Dominko, K.; Đikić, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arh. Hig. Rada Toksikol. 2018, 69, 196018. [Google Scholar] [CrossRef]
- Sen, C.K. Redox signaling and the emerging therapeutic potential of thiol antioxidants. Biochem. Pharmacol. 1998, 55, 1747–1758. [Google Scholar] [CrossRef]
- Paige, J.S.; Xu, G.; Stancevic, B.; Jaffrey, S.R. Nitrosothiol reactivity profiling identifies s-nitrosylated proteins with unexpected stability. Chem. Biol. 2008, 15, 1307–1316. [Google Scholar] [CrossRef] [Green Version]
- Chatterji, A.; Sengupta, R. Stability of S-nitrosothiols and S-nitrosylated proteins: A struggle for cellular existence! J. Cell. Biochem. 2021, 122, 1579–1593. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Tyurin, V.A.; Anand, D.; Mandavia, D.N.; Gius, D.; Ivanova, J.; Pitt, B.; Billiar, T.R.; Kagan, V.E. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein s -nitrosothiols. J. Am. Chem. Soc. 2005, 127, 15815–15823. [Google Scholar] [CrossRef]
- Johnson, W.M.; Yao, C.; Siedlak, S.L.; Wang, W.; Zhu, X.; Caldwell, G.A.; Wilson-Delfosse, A.L.; Mieyal, J.J.; Chen, S.G. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 1322–1335. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2013, 66, 75–87. [Google Scholar] [CrossRef]
- Arnér, E.S.J.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Eklund, H.; Gleason, F.K.; Holmgren, A. Structural and functional relations among thioredoxins of different species. Proteins 1991, 11, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, B.B.; Holmgren, A.; Jacquot, J.P.; Scheibe, R. Fifty years in the thioredoxin field and a bountiful harvest. Biochim. Biophys. Acta 2012, 1820, 1822–1829. [Google Scholar] [CrossRef] [PubMed]
- Hashemy, S.I.; Holmgren, A. Regulation of the Catalytic Activity and Structure of Human Thioredoxin 1 via Oxidation and S-Nitrosylation of Cysteine Residues. J. Biol. Chem. 2008, 283, 21890–21898. [Google Scholar] [CrossRef]
- Watson, W.H.; Pohl, J.; Montfort, W.R.; Stuchlik, O.; Reed, M.S.; Powis, G.; Jones, D.P. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 2003, 278, 33408–33415. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, H.; Zhang, X.; Lu, J.; Holmgren, A. Thioredoxin 1 Is Inactivated Due to Oxidation Induced by Peroxiredoxin under Oxidative Stress and Reactivated by the Glutaredoxin System. J. Biol. Chem. 2013, 288, 32241–32247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, N.; Oka, S.; Sadoshima, J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic. Biol. Med. 2017, 109, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 Inhibits Mitochondrial Reactive Oxygen Species Generation and Apoptosis Stress Kinase-1 Activity to Maintain Cardiac Function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef]
- Holzerova, E.; Danhauser, K.; Haack, T.B.; Kremer, L.S.; Melcher, M.; Ingold, I.; Kobayashi, S.; Terrile, C.; Wolf, P.; Schaper, J.; et al. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain 2016, 139, 346–354. [Google Scholar] [CrossRef]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules--from biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Lejnev, K.; Khomsky, L.; Bokvist, K.; Mistriel-Zerbib, S.; Naveh, T.; Farb, T.B.; Alsina-Fernandez, J.; Atlas, D. Thioredoxin-mimetic peptides (TXM) inhibit inflammatory pathways associated with high-glucose and oxidative stress. Free Radic. Biol. Med. 2016, 99, 557–571. [Google Scholar] [CrossRef]
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef]
- Lothrop, A.P.; Snider, G.W.; Ruggles, E.L.; Patel, A.S.; Lees, W.J.; Hondal, R.J. Selenium as an electron acceptor during the catalytic mechanism of thioredoxin reductase. Biochemistry 2014, 53, 654–663. [Google Scholar] [CrossRef]
- Arnér, E.S. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef]
- Lothrop, A.P.; Snider, G.W.; Ruggles, E.L.; Hondal, R.J. Why is mammalian thioredoxin reductase 1 so dependent upon the use of selenium? Biochemistry 2014, 53, 554–565. [Google Scholar] [CrossRef]
- Fritz-Wolf, K.; Urig, S.; Becker, K. The structure of human thioredoxin reductase 1 provides insights into C-terminal rearrangements during catalysis. J. Mol. Biol. 2007, 370, 116–127. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and Glutaredoxin Act as a Backup of Human Thioredoxin Reductase 1 to Reduce Thioredoxin 1 Preventing Cell Death by Aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219. [Google Scholar] [CrossRef]
- Jeong, W.; Yoon, H.W.; Lee, S.R.; Rhee, S.G. Identification and characterization of TRP14, a thioredoxin-related protein of 14 kDa. New insights into the specificity of thioredoxin function. J. Biol. Chem. 2004, 279, 3142–3150. [Google Scholar] [CrossRef]
- Woo, J.R.; Kim, S.J.; Jeong, W.; Cho, Y.H.; Lee, S.C.; Chung, Y.J.; Rhee, S.G.; Ryu, S.E. Structural Basis of Cellular Redox Regulation by Human TRP14. J. Biol. Chem. 2004, 279, 48120–48125. [Google Scholar] [CrossRef]
- Jeong, W.; Chang, T.S.; Boja, E.S.; Fales, H.M.; Rhee, S.G. Roles of TRP14, a thioredoxin-related protein in tumor necrosis factor-alpha signaling pathways. J. Biol. Chem. 2004, 279, 3151–3159. [Google Scholar] [CrossRef]
- Hong, S.; Huh, J.E.; Lee, S.Y.; Shim, J.K.; Rhee, S.G.; Jeong, W. TRP14 inhibits osteoclast differentiation via its catalytic activity. Mol. Cell Biol. 2014, 34, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, B.; Arnér, E.S.J. Thioredoxin-related protein of 14 kDa as a modulator of redox signalling pathways. Br. J. Pharmacol. 2019, 176, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.X.; Xu, T.M.; Zhou, Z.L.; Lv, X.J.; Liu, J.; Zhang, W.J.; Cui, M.H. TRP14 promotes resistance to cisplatin by inducing autophagy in ovarian cancer. Oncol. Rep. 2019, 42, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Murakawa, M.; Takahashi, S.; Tsubuki, S.; Kawashima, S.; Sakamaki, K.; Yonehara, S. Purification, Molecular Cloning, and Characterization of TRP32, a Novel Thioredoxin-related Mammalian Protein of 32 kDa. J. Biol. Chem. 1998, 273, 19160–19166. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, A.; Pelto-Huikko, M.; Gustafsson, J.A.; Miranda-Vizuete, A. Characterization of human thioredoxin-like-1: Potential involvement in the cellular response against glucose deprivation. FEBS Lett. 2006, 580, 960–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goroncy, A.K.; Koshiba, S.; Tochio, N.; Tomizawa, T.; Inoue, M.; Tanaka, A.; Sugano, S.; Kigawa, T.; Yokoyama, S. Solution structure of the C-terminal DUF1000 domain of the human thioredoxin-like 1 protein. Proteins 2010, 78, 2176–2180. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.M.; Madsen, L.; Prag, S.; Johnsen, A.H.; Semple, C.A.; Hendil, K.B.; Hartmann-Petersen, R. Thioredoxin Txnl1/TRP32 is a redox-active cofactor of the 26 S proteasome. J. Biol. Chem. 2009, 284, 15246–15254. [Google Scholar] [CrossRef]
- Ishii, T.; Funato, Y.; Miki, H. Thioredoxin-related protein 32 (TRP32) specifically reduces oxidized phosphatase of regenerating liver (PRL). J. Biol. Chem. 2013, 288, 7263–7270. [Google Scholar] [CrossRef]
- Qayyum, N.; Haseeb, M.; Kim, M.S.; Choi, S. Role of Thioredoxin-Interacting Protein in Diseases and Its Therapeutic Outlook. Int. J. Mol. Sci. 2021, 22, 2754. [Google Scholar] [CrossRef]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1, 25-dihydroxyvitamin D-3. Biochim. Biophys. Acta 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef]
- Jin, H.O.; Seo, S.K.; Kim, Y.S.; Woo, S.H.; Lee, K.H.; Yi, J.Y.; Lee, S.J.; Choe, T.B.; Lee, J.H.; An, S.; et al. TXNIP potentiates Redd1-induced mTOR suppression through stabilization of Redd1. Oncogene 2011, 30, 3792–3801. [Google Scholar] [CrossRef]
- Pan, M.; Zhang, F.; Qu, K.; Liu, C.; Zhang, J. TXNIP: A Double-Edged Sword in Disease and Therapeutic Outlook. Oxid. Med. Cell. Longev. 2022, 2022, 7805115. [Google Scholar] [CrossRef]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin structure and mechanism: Conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 1995, 3, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.T.; Fernandes, P.A.; Ramos, M.J. Theoretical study of the unusual protonation properties of the active site cysteines in thioredoxin. J. Phys. Chem. B 2006, 110, 5758–5761. [Google Scholar] [CrossRef]
- Kallis, G.B.; Holmgren, A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J. Biol. Chem. 1980, 255, 10261–10265. [Google Scholar] [CrossRef]
- Chivers, P.T.; Raines, R.T. General acid/base catalysis in the active site of Escherichia coli thioredoxin. Biochemistry 1997, 36, 15810–15816. [Google Scholar] [CrossRef]
- Carvalho, A.T.; Swart, M.; van Stralen, J.N.; Fernandes, P.A.; Ramos, M.J.; Bickelhaupt, F.M. Mechanism of thioredoxin-catalyzeddisulfide reduction. Activation of the buried thiol and role of the variable active-site residues. J. Phys. Chem. B 2008, 112, 2511–2523. [Google Scholar] [CrossRef]
- Wu, C.; Parrott, A.M.; Fu, C.; Liu, T.; Marino, S.M.; Gladyshev, V.N.; Jain, M.R.; Baykal, A.T.; Li, Q.; Oka, S.; et al. Thioredoxin 1-mediated post-translational modifications: Reduction, transnitrosylation, denitrosylation, and related proteomics methodologies. Antioxid. Redox Signal. 2011, 15, 2565–2604. [Google Scholar] [CrossRef]
- Benhar, M.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 2008, 320, 1050–1054. [Google Scholar] [CrossRef]
- Forrester, M.T.; Seth, D.; Hausladen, A.; Eyler, C.E.; Foster, M.W.; Matsumoto, A.; Benhar, M.; Marshall, H.E.; Stamler, J.S. Thioredoxin-interacting protein (Txnip) is a feedback regulator of S-nitrosylation. J. Biol. Chem. 2009, 284, 316160–316166. [Google Scholar] [CrossRef]
- Sengupta, R.; Billiar, T.R.; Kagan, V.E.; Stoyanovsky, D.A. Nitric oxide and thioredoxin type 1 modulate the activity of caspase 8 in HepG2 cells. Biochem. Biophys. Res. Commun. 2010, 391, 1127–1130. [Google Scholar] [CrossRef]
- Tian, H.; Wang, J.; Zhang, B.; Di, J.; Chen, F.; Li, H.; Li, L.; Pei, D.; Zheng, J. MDA-7/IL-24 induces Bcl-2 denitrosylation and ubiquitin-degradation involved in cancer cell apoptosis. PLoS ONE 2012, 75, e37200. [Google Scholar] [CrossRef]
- Wu, C.; Parrott, A.M.; Liu, T.; Jain, M.R.; Yang, Y.; Sadoshima, J.; Li, H. Distinction of thioredoxin transnitrosylation and denitrosylation target proteins by the ICAT quantitative approach. J. Proteom. 2011, 74, 2498–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnér, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef]
- Tan, S.X.; Greetham, D.; Raeth, S.; Grant, C.M.; Dawes, I.W.; Perrone, G.G. The thioredoxin-thioredoxin reductase system can function in vivo as an alternative system to reduce oxidized glutathione in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 6118–6126. [Google Scholar] [CrossRef]
- Rollins, M.F.; van der Heide, D.M.; Weisend, C.M.; Kundert, J.A.; Comstock, K.M.; Suvorova, E.S.; Capecchi, M.R.; Merrill, G.F.; Schmidt, E.E. Hepatocytes lacking thioredoxin reductase 1 have normal replicative potential during development and regeneration. J. Cell Sci. 2010, 123, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Trotter, E.W.; Grant, C.M. Non-reciprocal regulation of the redox state of the glutathione-glutaredoxin and thioredoxin systems. EMBO Rep. 2003, 4, 184–188. [Google Scholar] [CrossRef]
- Han, C.; Kim, M.J.; Ding, D.; Park, H.J.; White, K.; Walker, L.; Gu, T.; Tanokura, M.; Yamasoba, T.; Linser, P.; et al. GSR is not essential for the maintenance of antioxidant defenses in mouse cochlea: Possible role of the thioredoxin system as a functional backup for GSR. PLoS ONE 2017, 12, e0180817. [Google Scholar] [CrossRef]
- Mandal, P.K.; Schneider, M.; Kölle, P.; Kuhlencordt, P.; Förster, H.; Beck, H.; Bornkamm, G.W.; Conrad, M. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010, 70, 9505–9514. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Xiong, Y.; Ho, D.S.; Gao, J.; Chua, B.H.; Pai, H.; Mieyal, J.J. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 2007, 43, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Kronschläger, M.; Galichanin, K.; Ekström, J.; Lou, M.F.; Söderberg, P.G. Protective effect of the thioltransferase gene on in vivo UVR-300 nm-induced cataract. Investig. Ophthalmol. Vis. Sci. 2012, 53, 248–252. [Google Scholar] [CrossRef]
- Wu, H.; Lin, L.; Giblin, F.; Ho, Y.S.; Lou, M.F. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 2011, 51, 2108–2117. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; David, L.; Ho, Y.S.; Lou, M.F. Glutaredoxin 2 (Grx2) gene deletion induces early onset of age-dependent cataracts in mice. J. Biol. Chem. 2014, 289, 36125–36139. [Google Scholar] [CrossRef] [Green Version]
- Löfgren, S.; Fernando, M.R.; Xing, K.Y.; Wang, Y.; Kuszynski, C.A.; Ho, Y.S.; Lou, M.F. Effect of thioltransferase (glutaredoxin) deletion on cellular sensitivity to oxidative stress and cell proliferation in lens epithelial cells of thioltransferase knockout mouse. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4497–4505. [Google Scholar] [CrossRef]
- Liu, X.; Jann, J.; Xavier, C.; Wu, H. Glutaredoxin 1 (Grx1) Protects Human Retinal Pigment Epithelial Cells From Oxidative Damage by Preventing AKT Glutathionylation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2821–2832. [Google Scholar] [CrossRef]
- Yang, F.; Yi, M.; Liu, Y.; Wang, Q.; Hu, Y.; Deng, H. Glutaredoxin-1 Silencing Induces Cell Senescence via p53/p21/p16 Signaling Axis. J. Proteome Res. 2018, 17, 1091–1100. [Google Scholar] [CrossRef]
- Cater, M.A.; Materia, S.; Xiao, Z.; Wolyniec, K.; Ackland, S.M.; Yap, Y.W.; Cheung, N.S.; La Fontaine, S. Glutaredoxin1 protects neuronal cells from copper-induced toxicity. Biometals 2014, 27, 661–672. [Google Scholar] [CrossRef]
- Saeed, U.; Durgadoss, L.; Valli, R.K.; Joshi, D.C.; Joshi, P.G.; Ravindranath, V. Knockdown of cytosolic glutaredoxin 1 leads to loss of mitochondrial membrane potential: Implication in neurodegenerative diseases. PLoS ONE 2008, 3, e2459. [Google Scholar] [CrossRef]
- Dannenmann, B.; Lehle, S.; Hildebrand, D.G.; Kübler, A.; Grondona, P.; Schmid, V.; Holzer, K.; Fröschl, M.; Essmann, F.; Rothfuss, O.; et al. High glutathione and glutathione peroxidase-2 levels mediate cell-type-specific DNA damage protection in human induced pluripotent stem cells. Stem Cell Rep. 2015, 4, 886–898. [Google Scholar] [CrossRef]
- Enoksson, M.; Fernandes, A.P.; Prast, S.; Lillig, C.H.; Holmgren, A.; Orrenius, S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem. Biophys. Res. Commun. 2005, 327, 774–779. [Google Scholar] [CrossRef]
- Hellfritsch, J.; Kirsch, J.; Schneider, M.; Fluege, T.; Wortmann, M.; Frijhoff, J.; Dagnell, M.; Fey, T.; Esposito, I.; Kölle, P.; et al. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid. Redox Signal. 2015, 22, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Kiermayer, C.; Northrup, E.; Schrewe, A.; Walch, A.; de Angelis, M.H.; Schoensiegel, F.; Zischka, H.; Prehn, C.; Adamski, J.; Bekeredjian, R.; et al. Heart-Specific Knockout of the Mitochondrial Thioredoxin Reductase (Txnrd2) Induces Metabolic and Contractile Dysfunction in the Aging Myocardium. J. Am. Heart Assoc. 2015, 4, 2153. [Google Scholar] [CrossRef]
- Poerschke, R.L.; Moos, P.J. Thioredoxin reductase 1 knockdown enhances selenazolidine cytotoxicity in human lung cancer cells via mitochondrial dysfunction. Biochem. Pharmacol. 2011, 81, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Horstkotte, J.; Perisic, T.; Schneider, M.; Lange, P.; Schroeder, M.; Kiermayer, C.; Hinkel, R.; Ziegler, T.; Mandal, P.K.; David, R.; et al. Mitochondrial thioredoxin reductase is essential for early postischemic myocardial protection. Circulation 2011, 124, 2892–2902. [Google Scholar] [CrossRef]
- Yoo, M.H.; Xu, X.M.; Carlson, B.A.; Patterson, A.D.; Gladyshev, V.N.; Hatfield, D.L. Targeting thioredoxin reductase 1 reduction in cancer cells inhibits self-sufficient growth and DNA replication. PLoS ONE 2007, 2, e1112. [Google Scholar] [CrossRef]
- Watson, W.H.; Heilman, J.M.; Hughes, L.L.; Spielberger, J.C. Thioredoxin reductase-1 knock down does not result in thioredoxin-1 oxidation. Biochem. Biophys. Res. Commun. 2008, 368, 832–836. [Google Scholar] [CrossRef]
- Jakupoglu, C.; Przemeck, G.K.; Schneider, M.; Moreno, S.G.; Mayr, N.; Hatzopoulos, A.K.; de Angelis, M.H.; Wurst, W.; Bornkamm, G.W.; Brielmeier, M.; et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol. Cell. Biol. 2005, 25, 1980–1988. [Google Scholar] [CrossRef]
- Das, K.C. Thioredoxin-deficient mice, a novel phenotype sensitive to ambient air and hypersensitive to hyperoxia-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, 429–442. [Google Scholar] [CrossRef]
- Nalvarte, I.; Damdimopoulos, A.E.; Nystöm, C.; Nordman, T.; Miranda-Vizuete, A.; Olsson, J.M.; Eriksson, L.; Björnstedt, M.; Arnér, E.S.; Spyrou, G. Overexpression of enzymatically active human cytosolic and mitochondrial thioredoxin reductase in HEK-293 cells. Effect on cell growth and differentiation. J. Biol. Chem. 2004, 279, 54510–54517. [Google Scholar] [CrossRef]
- Stancill, J.S.; Hansen, P.A.; Mathison, A.J.; Schmidt, E.E.; Corbett, J.A. Deletion of Thioredoxin Reductase Disrupts Redox Homeostasis and Impairs β-Cell Function. Function 2022, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Harris-Gauthier, N.; Traa, A.; AlOkda, A.; Moldakozhayev, A.; Anglas, U.; Soo, S.K.; Van Raamsdonk, J.M. Mitochondrial thioredoxin system is required for enhanced stress resistance and extended longevity in long-lived mitochondrial mutants. Redox Biol. 2022, 53, 102335. [Google Scholar] [CrossRef]
- Krajewska, M.; Wang, H.G.; Krajewski, S.; Zapata, J.M.; Shabaik, A.; Gascoyne, R.; Reed, J.C. Immunohistochemical analysis of in vivo patterns of expression of CPP32 (Caspase-3), a cell death protease. Cancer Res. 1997, 57, 1605–1613. [Google Scholar] [PubMed]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubb, K.J.; Drummond, G.R.; Figtree, G.A. New opportunities for targeting redox dysregulation in cardiovascular disease. Cardiovasc. Res. 2020, 116, 532–544. [Google Scholar] [CrossRef]
- Kalinina, E.V.; Chernov, N.N.; Saprin, A.N. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry 2008, 73, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Bebber, C.M.; Thomas, E.S.; Stroh, J.; Chen, Z.; Androulidaki, A.; Schmitt, A.; Höhne, M.N.; Stüker, L.; de Pádua, A.C.; Khonsari, A.; et al. Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes. Nat. Commun. 2021, 12, 2048. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, X.; Wang, L.; Zhang, R.; Pu, X.; Wu, S.; Li, L.; Tong, P.; Wang, J.; Meng, Q.H.; et al. Inhibition of Thioredoxin/Thioredoxin Reductase Induces Synthetic Lethality in Lung Cancers with Compromised Glutathione Homeostasis. Cancer Res. 2019, 79, 125–132. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H.; Kang, G. Nitrosative Stress and Human Disease: Therapeutic Potential of Denitrosylation. Int. J. Mol. Sci. 2021, 22, 9794. [Google Scholar] [CrossRef]
- Ju, Y.J.; Lee, H.W.; Choi, J.W.; Choi, M.S. The Role of Protein S-Nitrosylation in Protein Misfolding-Associated Diseases. Life 2021, 11, 705. [Google Scholar] [CrossRef]
- Sliskovic, I.; Raturi, A.; Mutus, B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J. Biol. Chem. 2005, 280, 8733–8741. [Google Scholar] [CrossRef]
- Trujillo, M.; Alvarez, M.N.; Peluffo, G.; Freeman, B.A.; Radi, R. Xanthine oxidase-mediated decomposition of S-nitrosothiols. J. Biol. Chem. 1998, 273, 7828–7834. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Putative denitrosylase activity of Cu, Zn-superoxide dismutase. Free. Radic. Biol. Med. 2007, 43, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Guo, Z.; Li, J.; Wang, P.G. Seleno compounds and glutathione peroxidase catalyzed decomposition of S-nitrosothiols. Biochem. Biophys. Res. Commun. 1996, 228, 88–93. [Google Scholar] [CrossRef]
- Wang, J.; Pan, S.; Berk, B.C. Glutaredoxin mediates Akt and eNOS activation by flow in a glutathione reductase-dependent manner. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1283–1288. [Google Scholar] [CrossRef]
- Seefeldt, T.; Zhao, Y.; Chen, W.; Raza, A.S.; Carlson, L.; Herman, J.; Stoebner, A.; Hanson, S.; Foll, R.; Guan, X. Characterization of a novel dithiocarbamate glutathione reductase inhibitor and its use as a tool to modulate intracellular glutathione. J. Biol. Chem. 2009, 284, 2729–2737. [Google Scholar] [CrossRef]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Wang, X.; Matthees, D.; Hu, Y.; Guan, X. Effects of glutathione reductase inhibition on cellular thiol redox state and related systems. Arch. Biochem. Biophys. 2009, 485, 56–62. [Google Scholar] [CrossRef]
- Chrestensen, C.A.; Starke, D.W.; Mieyal, J.J. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 2000, 275, 26556–26565. [Google Scholar] [CrossRef]
- Choong, G.; Liu, Y.; Xiao, W.; Templeton, D.M. Cadmium-induced glutathionylation of actin occurs through a ROS-independent mechanism: Implications for cytoskeletal integrity. Toxicol. Appl. Pharmacol. 2013, 272, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Sabens, E.A.; Distler, A.M.; Mieyal, J.J. Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: Implications for therapy of Parkinson’s disease. Biochemistry 2010, 49, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, U.; Bala, A.; Jao, S.C.; Starke, D.W.; Jordan, T.W.; Mieyal, J.J. Selective inactivation of glutaredoxin by sporidesmin and other epidithiopiperazinediones. Biochemistry 2006, 45, 8978–8987. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, S.; Mailloux, R.J.; Chan, H.M. Methylmercury alters glutathione homeostasis by inhibiting glutaredoxin 1 and enhancing glutathione biosynthesis in cultured human astrocytoma cells. Toxicol. Lett. 2016, 256, 1–10. [Google Scholar] [CrossRef]
- Chen, J.C.; Zhang, Y.; Jie, X.M.; She, J.; Dongye, G.Z.; Zhong, Y.; Deng, Y.Y.; Wang, J.; Guo, B.Y.; Chen, L.M. Ruthenium(II) salicylate complexes inducing ROS-mediated apoptosis by targeting thioredoxin reductase. J. Inorg. Biochem. 2019, 193, 112–123. [Google Scholar] [CrossRef]
- Oehninger, L.; Küster, L.N.; Schmidt, C.; Muñoz-Castro, A.; Prokop, A.; Ott, I. A chemical-biological evaluation of rhodium(I) N-heterocyclic carbene complexes as prospective anticancer drugs. Chemistry 2013, 19, 17871–17880. [Google Scholar] [CrossRef]
- Prast-Nielsen, S.; Cebula, M.; Pader, I.; Arnér, E.S. Noble metal targeting of thioredoxin reductase--covalent complexes with thioredoxin and thioredoxin-related protein of 14 kDa triggered by cisplatin. Free Radic. Biol. Med. 2010, 49, 1765–1778. [Google Scholar] [CrossRef]
- Santini, C.; Pellei, M.; Papini, G.; Morresi, B.; Galassi, R.; Ricci, S.; Tisato, F.; Porchia, M.; Rigobello, M.P.; Gandin, V.; et al. In vitro antitumour activity of water soluble Cu(I), Ag(I) and Au(I) complexes supported by hydrophilic alkyl phosphine ligands. J. Inorg. Biochem. 2011, 105, 232–240. [Google Scholar] [CrossRef]
- Jastrząb, A.; Skrzydlewska, E. Thioredoxin-dependent system. Application of inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 362–371. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J.; Peng, S.; Liu, R.; Li, X.; Hou, Y.; Han, X.; Fang, J. Thioredoxin reductase inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 547–556. [Google Scholar] [CrossRef]
- Lu, J.; Papp, L.V.; Fang, J.; Rodriguez-Nieto, S.; Zhivotovsky, B.; Holmgren, A. Inhibition of Mammalian thioredoxin reductase by some flavonoids: Implications for myricetin and quercetin anticancer activity. Cancer Res. 2006, 66, 4410–4418. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, B.; Duan, D.; Wu, J.; Fang, J. Curcumin targeting the thioredoxin system elevates oxidative stress in HeLa cells. Toxicol. Appl. Pharmacol. 2012, 262, 341–348. [Google Scholar] [CrossRef]
- Parrilha, G.L.; Ferraz, K.S.; Lessa, J.A.; de Oliveira, K.N.; Rodrigues, B.L.; Ramos, J.P.; Souza-Fagundes, E.M.; Ott, I.; Beraldo, H. Metal complexes with 2-acetylpyridine-N(4)-orthochlorophenylthiosemicarbazone: Cytotoxicity and effect on the enzymatic activity of thioredoxin reductase and glutathione reductase. Eur. J. Med. Chem. 2014, 84, 537–544. [Google Scholar] [CrossRef]
- Arnér, E.S.; Nakamura, H.; Sasada, T.; Yodoi, J.; Holmgren, A.; Spyrou, G. Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum (II) and its major metabolite, the glutathione-platinum complex. Free Radic. Biol. Med. 2001, 31, 1170–1178. [Google Scholar] [CrossRef]
- Yu, Y.; Suryo, R.Y.; Hawkins, C.L.; Richardson, D.R. The potent and novel thiosemicarbazone chelators di-2-pyridylketone-4, 4-dimethyl-3-thiosemicarbazone and 2-benzoylpyridine-4, 4-dimethyl-3-thiosemicarbazone affect crucial thiol systems required for ribonucleotide reductase activity. Mol. Pharmacol. 2011, 79, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Johnson, F.D.; Ferrarone, J.; Liu, A.; Brandstädter, C.; Munuganti, R.; Farnsworth, D.A.; Lu, D.; Luu, J.; Sihota, T.; Jansen, S.; et al. Characterization of a small molecule inhibitor of disulfide reductases that induces oxidative stress and lethality in lung cancer cells. Cell Rep. 2022, 38, 110343. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Jovanović, M.; Podolski-Renić, A.; Krasavin, M.; Pešić, M. The Role of the Thioredoxin Detoxification System in Cancer Progression and Resistance. Front. Mol. Biosci. 2022, 9, 883297. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Yu, S.; Zhao, G. Recent advances in the development of thioredoxin reductase inhibitors as anticancer agents. Curr. Drug Targets 2012, 13, 1432–1444. [Google Scholar] [CrossRef]
- Gamberi, T.; Chiappetta, G.; Fiaschi, T.; Modesti, A.; Sorbi, F.; Magherini, F. Upgrade of an old drug: Auranofin in innovative cancer therapies to overcome drug resistance and to increase drug effectiveness. Med. Res. Rev. 2022, 42, 1111–1146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, K.S.; Park, S.J.; Min, K.H.; Lee, M.H.; Lee, K.A.; Bartov, O.; Atlas, D.; Lee, Y.C. A novel dithiol amide CB3 attenuates allergic airway disease through negative regulation of p38 mitogen-activated protein kinase. Am. J. Respir. Crit. Care Med. 2011, 183, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Bartov, O.; Sultana, R.; Butterfield, D.A.; Atlas, D. Low molecular weight thiol amides attenuate MAPK activity and protect primary neurons from Abeta(1-42) toxicity. Brain Res. 2006, 1069, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Kronenfeld, G.; Engelman, R.; Weisman-Shomer, P.; Atlas, D.; Benhar, M. Thioredoxin-mimetic peptides as catalysts of S-denitrosylation and anti-nitrosative stress agents. Free Radic. Biol. Med. 2015, 79, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Atlas, D. Emerging therapeutic opportunities of novel thiol-amides, NAC-amide (AD4/NACA) and thioredoxin mimetics (TXM-Peptides) for neurodegenerative-related disorders. Free Radic. Biol. Med. 2021, 176, 120–141. [Google Scholar] [CrossRef] [PubMed]
- Bachnoff, N.; Trus, M.; Atlas, D. Alleviation of oxidative stress by potent and selective thioredoxin-mimetic peptides. Free Radic. Biol. Med. 2011, 50, 1355–1367. [Google Scholar] [CrossRef]
- Sengupta, R.; Coppo, L.; Sircar, E.; Mishra, P.; Holmgren, A. S-denitrosylation by the C-terminal swinging arm of R1 subunit: A novel mechanism to restore ribonucleotide reductase activity. Chemistry 2021, 6, 1845–1851. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Target | Genetic Manipulation Induced Functional Changes | References |
|---|---|---|---|
| Grx system | Grx1 knockout | -Lack of susceptibility of Grx1 deficient mice towards I/R heart and lungs injury in vivo -Limited role of Grx1 in hyperoxia-induced lung injury -Desensitizes Grx1 deficient MEFs towards H2O2 and diamide-induced apoptotic cell death; however, increases vulnerability to diquat and paraquat-induced cell damage | [113] |
| Grx1 knockout | -Increases lens susceptibility to UVR-B-induced oxidative stress -Highlights in vivo impact of Grx1 in protection from UVR-induced subcapsular and cortical cataract | [114] | |
| Grx2 knockout | -Exhibits reduced tolerance to oxidative stress -Suppression of mitochondrial protein complex I and IV activity upon S-glutathionylation and enhanced ATP loss -Accelerates early onset and progression of cataract -Impairs cell viability and membrane integrity under H2O2-induced stress in primary LECs | [115,116,117] | |
| Grx1 overexpression and knockdown | -Offers better resistance to H2O2-induced Akt glutathionylation and Caspase-3 mediated cell viability loss -Activation of Akt upon phosphorylation followed by decreased Bax and increased Bcl-2 expression levels -Sensitization of Grx-1 KO ARPE-19 cells to apoptosis | [118] | |
| Grx1 knockdown and overexpression | -Decrease in GSH/GSSG ratio and cellular GSH levels -Increased ROS accumulation and glutathionylation in DJ-1 and HSP60 -Inactivation of DNA replication and damage repair pathways -Induces cell cycle arrest via p53/p21/p16 signaling axis activation in 293T and U87 cells -Decrease in cellular ROS levels upon overexpression | [119] | |
| Grx1 knockout | -Induces cellular copper retention and reduction in copper-induced oxidative stress tolerance -Highlights role of Grx1 in ATP7A-mediated copper export, maintenance of neuronal copper homeostasis in AD, PD, and ALS -Offers protection from copper-mediated oxidative injury | [120] | |
| Downregulation of Grx1 and Grx1 overexpression | -Mitochondrial dysfunction and loss of MMP and mitochondrial membrane integrity upon oxidative insults in neuroblastoma cell lines -Oxidative modifications in redox-sensitive VDAC and increase in ROS generation -Desensitizes cells against L-BOAA cytotoxicity upon Grx1 upregulation | [121] | |
| Depletion of GSH and Gpx2 levels, Knockdown of Gpx2 or GSTA2 combined with GSH depletion | -Impairs iPSC-specific resistance to mt and nuclear DNA damage upon H2O2 exposure -Strong increase in DNA damage (GSH-depleted fibroblasts) and considerable increase in ROS levels (GSH-depleted fibroblasts and iPSC) | [122] | |
| Overexpressed mtGrx2 and truncated cytosolic Grx2 (tGrx2) | -Lower susceptibility to apoptosis in both forms -Lesser susceptibility of mtGrx2 than tGrx2 towards apoptosis -Inhibition of cytochrome c release, caspase3 activation, and cardiolipin loss | [123] | |
| Trx and TrxR | TrxR2 (Txnrd2) knockout | -Inactivation of Hif-1α signaling, Hif-1α degradation, decreased VEGF levels, increased JNK activation -Delays angiogenic switch, reduces tumor growth, and impairs angiogenesis -Displays higher ROS levels than wt MEFs despite strong, compensatory upregulation of Grx2 | [124] |
| Heart-specific TrxR2 (Txnrd2) knockout | -Deregulated autophagic activity in cardiomyocytes -Decreases O2 consumption, elevates ROS production, loss of morphological and functional integrity of KO mitochondria -Heightened catalase, HSP25, HSP60, and GSH levels in KO myocardium | [125] | |
| TrxR1 knockdown | -Increase in some selenocysteine prodrugs-mediated cytotoxicity in vivo -Increase in ROS generation, selenocompounds-induced mitochondrial membrane depolarization, DNA strand breaks, and AIF-induced cell death in human lung cancer cells | [126] | |
| Heart-specific TrxR1 and TrxR2 knockout, Trx2 overexpression | -Aggravated systolic dysfunction and myocardial cell death -Attenuates oxidative stress, mitochondrial impairment, loss of membrane integrity, and larger infarct size in clinical testings -Exhibits more rate-limiting relevance of TrxR2 as compared to Trx2 | [127] | |
| TrxR1 knockdown | -Reversal of numerous malignant properties, including tumorigenicity in malignant mouse cell line and mouse model -Defective S-phase cell progression in serum-deficient medium -Decrease in DNA polymerase α expression leading to inhibition of DNA replication | [128] | |
| TrxR1 knockdown | -Alters relative levels of reduced (decrease) and oxidized (40% increase) Trx1 in KO cells upon H2O2 exposure -Unaffected redox status of Trx1 under normal KO cells | [129] | |
| TrxR1 inactivation | -Leads to early embryonic lethality -Severe growth retardation (homozygous mutant)in vivo and in vitro, unaffected embryonic fibroblasts (ex vivo) -Reveals higher significance of TrxR2 in cardiogenesis and TrxR1 in other developing tissues | [130] | |
| Trx-deficient mice | -Increases endogenous Trx oxidation, p53 and Gadd45α expression, and synthesis of proinflammatory cytokines in normal O2 levels -Exhibits significant mortality in hyperoxic conditions -Decrease in aconitase and NADPH activity, impairs mitochondrial energy metabolism | [131] | |
| Cytosolic TrxR1 and mitochondrial TrxR2 overexpression | -Downregulates GPx expression and activity levels upon elevated TrxR1 levels -Induces novel expression of epithelial markers in HEK-293 cells for cellular differentiation | [132] | |
| ß-cell-specific TrxR1 knockout | -Increases sensitivity to oxidative damage -Lowers glucose-stimulated or membrane depolarization stimulated insulin secretion -Upregulates Nrf2-related antioxidant genes, altered expression of heme and GSH-related genes -Downregulates β-cell function and identity regulating factors | [133] | |
| Trx2, TrxR2, Trx1, and TrxR1 disruption and deletion | -Sensitizes cells to elevated levels of ROS in nuo-6 and isp-1 mutants -Significant increase in ROS levels upon loss of TrxR2 and Trx1 -Significant shortening of lifespan, decreased heat stress resistance, decreased resistance to osmotic stress and bacterial pathogens | [134] |
| Class of Inhibitors | Name of Compound/Complex/Drug Candidate | Outcome of Inhibition and Potential Consequences | References |
|---|---|---|---|
| Grx, Grx-system inhibitors | BCNU | -Dramatically reduced Grx activity -Inhibition of flow-induced Akt and eNOS phosphorylation (activation) in endothelial cells -Inhibition of atheroprotective effect of Akt-eNOS-NO signaling pathway | [148] |
| 2-AAPA | -Irreversible inactivation of intracellular GR activity in a time- and concentration-dependent manner -Induces thiol oxidative stress -Reduction in enzyme activity, i.e., minimal inhibition against GPx and GST -Exhibits potential anticancer and antimalarial activity | [149,150] | |
| Cadmium | -Inhibition of thioltransferase (GSH-dependent dethiolase) activity in a dose-dependent manner -Inhibition of cellular GR activity -Accumulation of PSSG substrates in mouse neuronal (HT4) cells -Mediates apoptosis in lysates of H9 and Jurkat cells -Inhibition of Grx1 and Grx2 | [151,152] | |
| Levodopa | -Inactivation of Grx activity in a time- and dose-dependent manner by dopaquinone adduct formation -Decreases TR and Trx content -Impairs the overall thiol homeostasis -Ensures apoptotic cell death in dopaminergic neurons in PD | [153] | |
| Sporidesmin | -Selective inactivation of Grx1 activity in a concentration-, time- and oxygen-dependent manner in the absence of GSH -Exhibits cytotoxic effects upon thiol modifications in selective target proteins | [154] | |
| Methylmercury | -Induces oxidative stress by disrupting GSH homeostasis in astrocytoma cells -Significant decrease in GSH/GSSG −50% decrease in Grx1 activity | [155] | |
| Trx, TrxR inhibitors | Metal complexes or compounds containing Sn(IV), Ru(II, III, IV), Rh(I), Ag(I), Cu(I), Pt(I), Au(I) | -Selectively inhibits TrxR activity (TrxR1 and TrxR2) -Triggers mitochondrial dysfunction, DNA damage, cell cycle arrest, and apoptosis in cancer cell lines -Induces paraptosis (Cu complexes) -Time- and dose-dependent inhibition of Trx, TrxR, NADPH (Cisplatin (CDDP) -Triggers intracellular covalent complex formation of TrxR1 with Trx1 or TRP14, cytotoxicity, and anticancer efficacy | [156,157,158,159,160] |
| TXNIP | -Negative regulator of Trx1 activity -Increases mitochondrial ROS accumulation -Activation of NLRP3 inflammasome -Binding of Txnip with Trx2 reduces Trx2-Ask1 interaction, further activating Ask1 for apoptosis | [136] | |
| Dichalcogenides and sulfur/selenium-containing compounds | -Interaction with TrxR enzymes, as inhibitors or substrates -Irreversibly alkylates Trx1 (PX-12) -Inhibits proliferation of cancer cells and reduces tumor size (Ethaselen) -Promotes oxidative stress-induced apoptosis of cancer cells (Chaetocin, gliotoxin, etc.) | [161] | |
| Polyphenolic compounds (curcumin, quercetin, myricetin, flavonoids, etc.) | -Induces alkylation of both cysteine and selenocysteine -Strong inhibition of mammalian TrxR activity -Significantly increases ROS production and induces oxidative stress-mediated cancer cell death | [162,163] | |
| Michael acceptors | -Inhibition of TrxR by covalent interaction with catalytic selenocysteine residue in enzyme’s active site -Exhibits pronounced anticancer efficacy | [160] | |
| Miscellaneous | Coordination Sb(III) and Au(III) complexes) | -Strong inhibition of TrxR at sub-micromolar concentration -Inhibits GR at higher concentrations -Pronounced cytotoxic effect against MCF-7 and HT-29 cells | [164] |
| GS-Pt chelate complex (purified glutathione adduct of cisplatin) | -Potentially inhibits Grx (human and bacterial) as well as Trx (mammalian) system | [165] | |
| Dp44mT and Bp44mT (Thiosemicarbazone iron chelators) | -Potent iron chelation efficacy -Inhibitory effect on RNR by altering thiol redoxin systems -Relative decrease in cellular GSH, TrxR activity, and Trx oxidation -Significant reduction of Grx activity -Exhibits potent and selective antitumor activity | [166] | |
| APR-246 (mutant p53-targeting compound)- MQ (Michael acceptor) | -Loss of free thiols due to covalent binding of MQ with thiols -Potentially inhibits Trx1 and Grx1, and RNR in vitro and in living cells -Exhibits GSH-dependent inhibitory efficiency -Triggers apoptosis-mediated tumor cell death due to mutant p53 reactivation | [167] | |
| LCS3 | -Selective inhibition of lung cancer cells -Induces ROS and activates Nrf2 pathway -Mediates synergistic inhibition of GSH/Trx pathways | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, S.; Sircar, E.; Bhattacharyya, C.; Choudhuri, A.; Mishra, A.; Dutta, S.; Bhatta, S.; Sachin, K.; Sengupta, R. S-Denitrosylation: A Crosstalk between Glutathione and Redoxin Systems. Antioxidants 2022, 11, 1921. https://doi.org/10.3390/antiox11101921
Chakraborty S, Sircar E, Bhattacharyya C, Choudhuri A, Mishra A, Dutta S, Bhatta S, Sachin K, Sengupta R. S-Denitrosylation: A Crosstalk between Glutathione and Redoxin Systems. Antioxidants. 2022; 11(10):1921. https://doi.org/10.3390/antiox11101921
Chicago/Turabian StyleChakraborty, Surupa, Esha Sircar, Camelia Bhattacharyya, Ankita Choudhuri, Akansha Mishra, Sreejita Dutta, Sneha Bhatta, Kumar Sachin, and Rajib Sengupta. 2022. "S-Denitrosylation: A Crosstalk between Glutathione and Redoxin Systems" Antioxidants 11, no. 10: 1921. https://doi.org/10.3390/antiox11101921
APA StyleChakraborty, S., Sircar, E., Bhattacharyya, C., Choudhuri, A., Mishra, A., Dutta, S., Bhatta, S., Sachin, K., & Sengupta, R. (2022). S-Denitrosylation: A Crosstalk between Glutathione and Redoxin Systems. Antioxidants, 11(10), 1921. https://doi.org/10.3390/antiox11101921