


Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
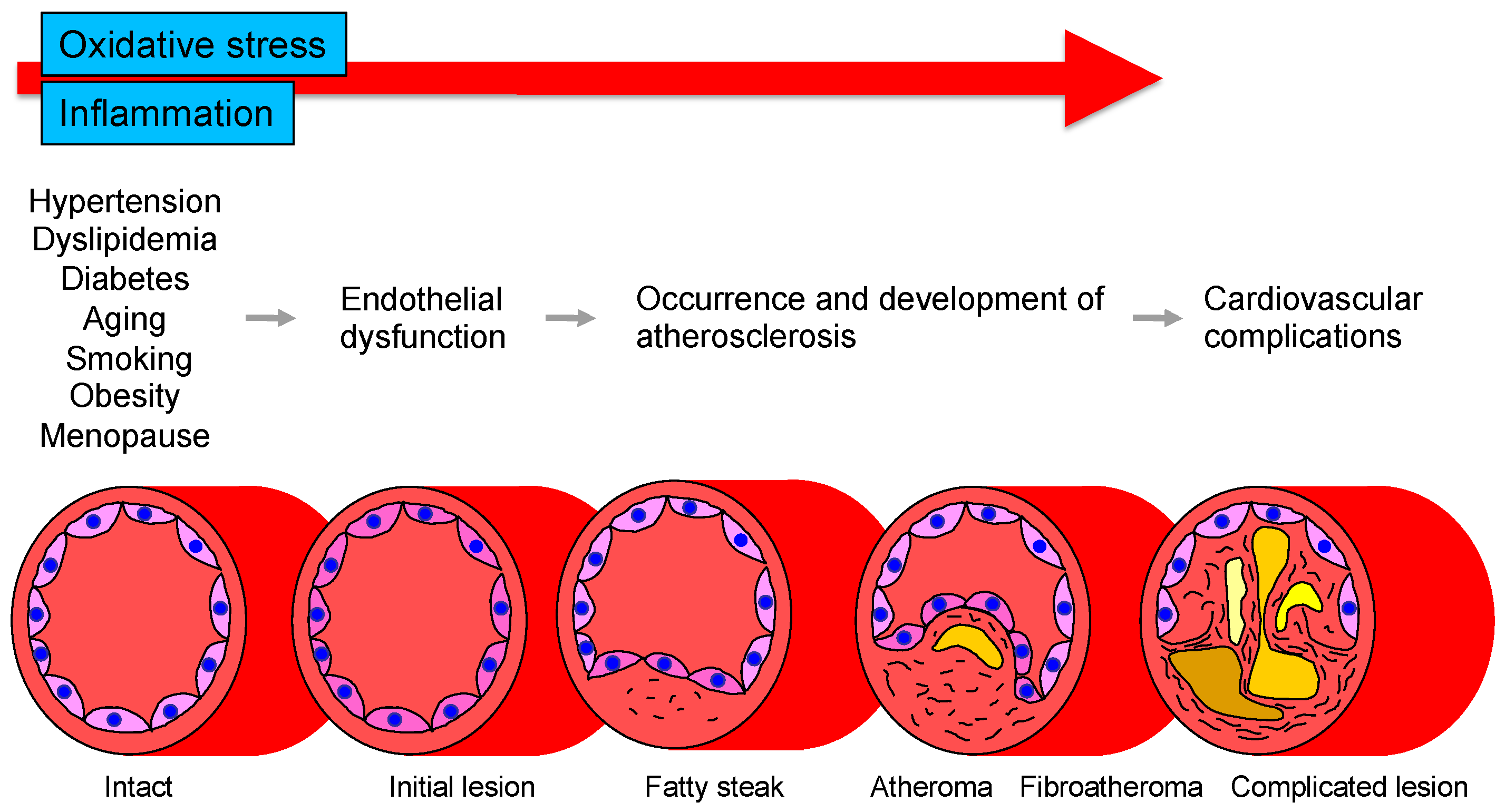
:1. Introduction
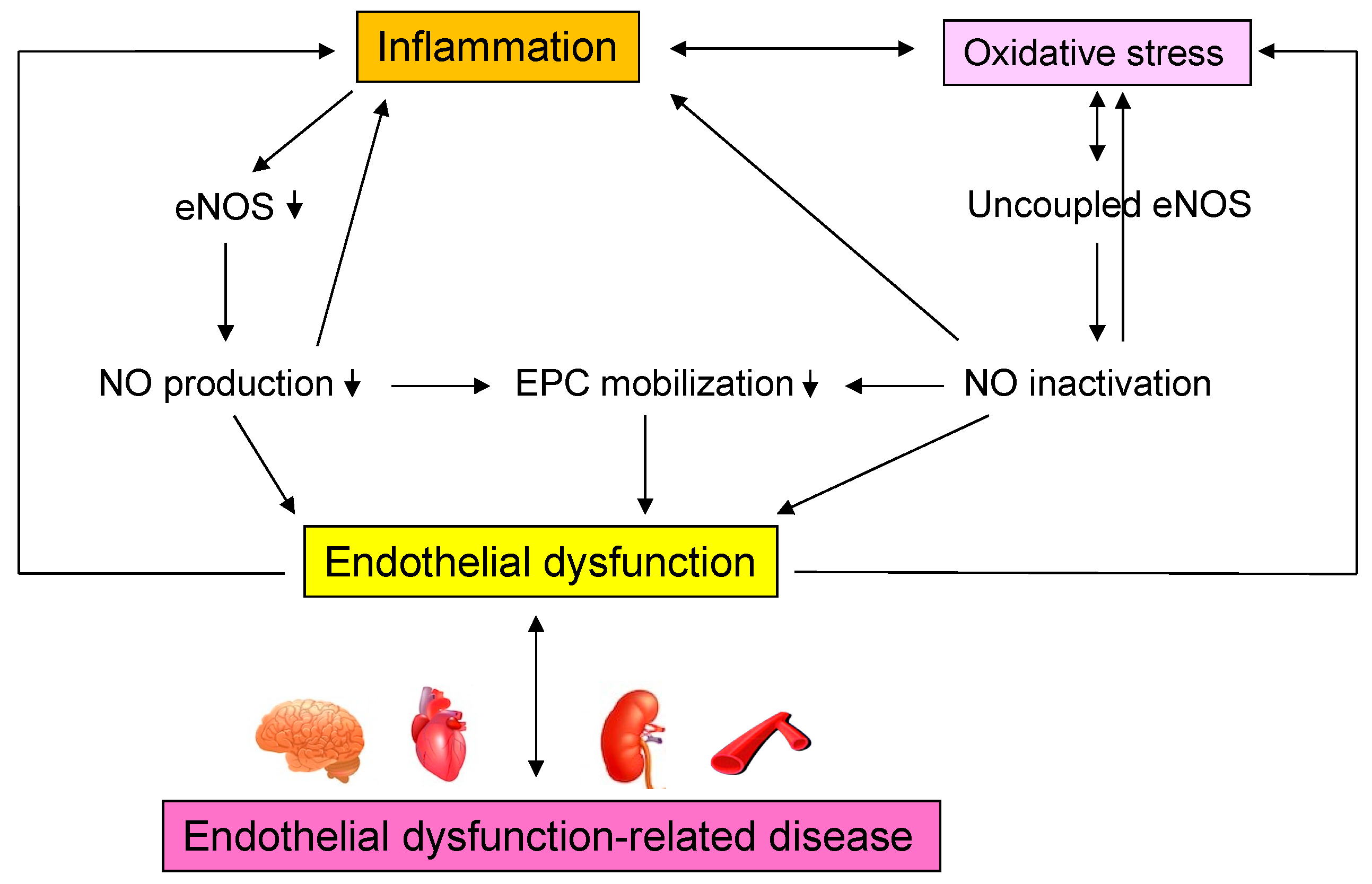
2. Vascular Endothelial Function
3. Oxidative Stress
3.1. Oxidative Stress and Vascular Injury
3.2. ROS Metabolism and Oxidative Stress
3.3. Enzymes Related to the Production of ROS
3.4. Oxidative Stress and Vascular Endothelial Dysfunction
3.5. Oxidative Stress and Vascular Smooth Muscle Hypertrophy and Remodeling
3.6. Oxidative Stress and Apoptosis
4. Inflammation
4.1. Helicobacter Pylori Infection
4.2. Periodontal Disease
4.3. Kawasaki Disease
4.4. Bürger’s Disease
4.5. Other Acute and Chronic Inflammatory Diseases
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Meigs, J.B.; Larson, M.G.; Fox, C.S.; Keaney, J.F., Jr.; Vasan, R.S.; Benjamin, E.J. Association of oxidative stress, insulin resistance, and diabetes risk phenotypes: The Framingham Offspring Study. Diabetes Care 2007, 30, 2529–2535. [Google Scholar] [CrossRef]
- Murthy, V.L.; Yu, B.; Wang, W.; Zhang, X.; Alkis, T.; Pico, A.R.; Yeri, A.; Bhupathiraju, S.N.; Bressler, J.; Ballantyne, C.M.; et al. Molecular Signature of Multisystem Cardiometabolic Stress and Its Association with Prognosis. JAMA Cardiol. 2020, 5, 1144–1153. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Schulz, E.; Münzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; McLoughlin, A.; Cummins, P.M. Shear-dependent attenuation of cellular ROS levels can suppress proinflammatory cytokine injury to human brain microvascular endothelial barrier properties. J. Cereb. Blood Flow Metab. 2015, 35, 1648–1656. [Google Scholar] [CrossRef]
- Li, J.M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1014–R1030. [Google Scholar] [CrossRef]
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016, 114, 110–120. [Google Scholar] [CrossRef]
- Lubrano, V.; Balzan, S. Roles of LOX-1 in microvascular dysfunction. Microvasc. Res. 2016, 105, 132–140. [Google Scholar] [CrossRef]
- Hwang, J.; Ing, M.H.; Salazar, A.; Lassègue, B.; Griendling, K.; Navab, M.; Sevanian, A.; Hsiai, T.K. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: Implication for native LDL oxidation. Circ. Res. 2003, 93, 1225–1232. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef]
- Lüscher, T.F. Imbalance of endothelium-derived relaxing and contracting factors. Am. J. Hypertens. 1990, 3, 317–330. [Google Scholar] [CrossRef]
- Vane, J.R.; Anggard, E.E.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36. [Google Scholar]
- Golshiri, K.; Ataei Ataabadi, E.; Portilla Fernandez, E.C.; Jan Danser, A.H.; Roks, A.J.M. The importance of the nitric oxide-cGMP pathway in age-related cardiovascular disease: Focus on phosphodiesterase-1 and soluble guanylate cyclase. Basic Clin. Pharmacol. Toxicol. 2020, 127, 67–80. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. Off. J. Jpn. Circ. Soc. 2009, 73, 411–418. [Google Scholar] [CrossRef]
- Higashi, Y.; Yoshizumi, M. Exercise and endothelial function: Role of endothelium-derived nitric oxide and oxidative stress in healthy subjects and hypertensive patients. Pharmacol. Ther. 2004, 102, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Mitchell, G.F.; Vasan, R.S.; Keaney, J.F., Jr.; Lehman, B.T.; Fan, S.; Osypiuk, E.; Vita, J.A. Clinical correlates and heritability of flow-mediated dilation in the community: The Framingham Heart Study. Circulation 2004, 109, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, E.; Cusi, K.; Ocampo, G.; Berria, R.; Buck, S.; Consoli, A.; Mandarino, L.J. Exercise-induced improvement in vasodilatory function accompanies increased insulin sensitivity in obesity and type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2006, 91, 4903–4910. [Google Scholar] [CrossRef]
- Sanada, M.; Higashi, Y.; Nakagawa, K.; Sasaki, S.; Kodama, I.; Sakashita, T.; Tsuda, M.; Ohama, K. Hormone replacement effects on endothelial function measured in the forearm resistance artery in normocholesterolemic and hypercholesterolemic postmenopausal women. J. Clin. Endocrinol. Metab. 2002, 87, 4634–4641. [Google Scholar] [CrossRef]
- Kimura, M.; Goto, C.; Umemura, T.; Ueda, K.; Nishioka, K.; Jitsuiki, D.; Yoshizumi, M.; Chayama, K.; Higashi, Y. Repetition of ischemic preconditioning augments endothelium-dependent vasodilation in humans: Role of endothelium-derived nitric oxide and endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1403–1410. [Google Scholar] [CrossRef]
- Fujimura, N.; Jitsuiki, D.; Goto, C.; Soga, J.; Hidaka, T.; Umemura, T.; Nishioka, K.; Oshima, T.; Chayama, K.; Higashi, Y. Geranylgeranylacetone, Hsp90/AMPK/eNOS/NO pathway, and endothelial function in humans. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 153–160. [Google Scholar] [CrossRef]
- Hambrecht, R.; Wolf, A.; Gielen, S.; Linke, A.; Hofer, J.; Erbs, S.; Schoene, N.; Schuler, G. Effect of exercise on coronary endothelial function in patients with coronary artery disease. N. Engl. J. Med. 2000, 342, 454–460. [Google Scholar] [CrossRef]
- Goto, C.; Nishioka, K.; Umemura, T.; Jitsuiki, D.; Sakaguchi, A.; Kawamura, M.; Yoshizumi, M.; Chayama, K.; Higashi, Y. The effects of different intensities of acute exercise on systemic hemodynamics and forearm vascular function in humans. Am. J. Hypertens. 2007, 20, 825–830. [Google Scholar] [CrossRef]
- Sessa, W.C.; Pritchard, K.; Seyedi, N.; Wang, J.; Hintze, T.H. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ. Res. 1994, 74, 349–353. [Google Scholar] [CrossRef]
- Higashi, Y. Assessment of endothelial function: History, methodological aspects, and clinical perspectives. Int. Heart. J. 2015, 56, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Tomiyama, H.; Maruhashi, T.; Matsuzawa, Y.; Miyoshi, T.; Kabutoya, T.; Kario, K.; Sugiyama, S.; Munakata, M.; Uto, H.; et al. Physiological diagnosis criteria for vascular failure. Hypertension 2018, 72, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, C.; Wang, H.; Lu, M.; Li, Y.; Feng, H.; Lin, J.; Yuan, Z.; Wang, X. Ox-LDL promotes migration and adhesion of bone marrow-derived mesenchymal stem cells via regulation of MCP-1 expression. Mediators. Inflamm. 2013, 2013, 691023. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.H.; Chen, D.; Zhou, B.; Chen, A.D.; Wang, J.J.; Gao, X.Y.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. FNDC5 inhibits foam cell formation and monocyte adhesion in vascular smooth muscle cells via suppressing NFκB-mediated NLRP3 upregulation. Vascul. Pharmacol. 2019, 121, 106579. [Google Scholar] [CrossRef]
- Chioslm, G.M.; Steinberg, D. The oxidative modification hypothesis of atherogenesis: An overview. Free. Radic. Biol. Med. 2000, 28, 1815–1826. [Google Scholar] [CrossRef]
- Tai, S.C.; Robb, G.B.; Marsden, P.A. Endothelial nitric oxide synthase: A new paradigm for gene regulation in the injured blood vessel. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 405–412. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, S.; Shen, G.X. Glycation amplifies lipoprotein(a)-induced alterations in the generation of fibrinolytic regulators from human vascular endothelial cells. Atherosclerosis 2000, 150, 299–308. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef]
- Calvier, L.; Herz, J.; Hansmann, G. Interplay of Low-Density Lipoprotein Receptors, LRPs, and Lipoproteins in Pulmonary Hypertension. JACC Basic Transl. Sci. 2022, 7, 164–180. [Google Scholar] [CrossRef]
- Yamashita, N.; Hoshida, S.; Otsu, K.; Asahi, M.; Kuzuya, T.; Hori, M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J. Exp. Med. 1999, 189, 1699–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraci, F.M.; Didion, S.P. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. NADPH oxidase: An update. Blood 1999, 93, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Heyworth, P.G.; Curnutte, J.T.; Nauseef, W.M.; Volpp, B.D.; Pearson, D.W.; Clark, R.A. Neutrofil NADPH oxidase assembly: Translocation of p47phox and p67phox requires the interaction between p47phox and cytochrome b558. J. Clin. Investig. 1991, 87, 352–356. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Zafari, A.M.; Fukai, T.; Nishizaka, N.; Griendling, K.K. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 23317–23321. [Google Scholar] [CrossRef]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Touyz, R.M. Redox signaling in hypertension. Cardiovasc. Res. 2006, 71, 247–258. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 29–38. [Google Scholar] [CrossRef]
- Zalba, G.; Beaumont, F.J.; San Jose, G.; Fortuno, A.; Fortuno, M.A.; Etayo, J.C.; Diez, J. Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 2000, 35, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: A unifying hypothesis. Hypertension 2001, 37, 1047–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touyz, R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Matsuura, H.; Oshima, T.; Chayama, K. Endothelial function and oxidative stress in renovascular hypertension. N. Engl. J. Med. 2002, 346, 1954–1962. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, Ł.; Wojnar-Lason, K.; Kaczara, P.; Tworzydło, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-methyltransferase in endothelium protects against oxidant stress-induced endothelial injury. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef] [PubMed]
- Mateuszuk, Ł.; Campagna, R.; Kutryb-Zając, B.; Kuś, K.; Słominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Anter, E.; Keaney, J.F., Jr. Oxidative stress, antioxidants, and endothelial function. Curr. Med. Chem. 2004, 11, 1093–1104. [Google Scholar] [CrossRef]
- Hooper, L.; Kroon, P.A.; Rimm, E.B.; Cohn, J.S.; Harvey, I.; Le Cornu, K.A.; Ryder, J.J.; Hall, W.L.; Cassidy, A. Flavonoids, flavonoid-rich foods, and cardiovascular risk: A meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2008, 88, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cao, J.; Mao, Q.; Lu, X.; Zhou, X.; Fan, L. Influence of omega-3 polyunsaturated fatty acid-supplementation on platelet aggregation in humans: A meta-analysis of randomized controlled trials. Atherosclerosis 2013, 226, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Skyrme-Jones, R.A.; O’Brien, R.C.; Berry, K.L.; Meredith, I.T. Vitamin E supplementation improves endothelial function in type I diabetes mellitus: A randomized, placebo-controlled study. J. Am. Coll. Cardiol. 2000, 36, 94–102. [Google Scholar] [CrossRef]
- Engler, M.M.; Engler, M.B.; Malloy, M.J.; Chiu, E.Y.; Schloetter, M.C.; Paul, S.M.; Stuehlinger, M.; Lin, K.Y.; Cooke, J.P.; Morrow, J.D.; et al. Antioxidant vitamins C and E improve endothelial function in children with hyperlipidemia: Endothelial Assessment of Risk from Lipids in Youth (EARLY) Trial. Circulation 2003, 108, 1059–1063. [Google Scholar] [CrossRef]
- Mune, M.; Uto-Kondo, H.; Iteya, I.; Fujii, Y.; Ikeda, S.; Ikewaki, K. Vitamin E supplementation improves high-densitiy lipoprotein and endothelial functions in end-stage kidney disease patients undergoing hemodialysis. Clin. Nephrol. 2018, 90, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Dalan, R.; Goh, L.L.; Lim, C.J.; Seneviratna, A.; Liew, H.; Seow, C.J.; Xia, L.; Chew, D.E.K.; Leow, M.K.S.; Boehm, B.O. Impact of Vitamin E supplementation on vascular function in haptoglobin genotype stratified diabetes patients (EVAS Trial): A randomised controlled trial. Nutr. Diabetes 2020, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.S.; Yip, T.W.C.; Chook, P.; Koon, K.V.; Leong, H.C.; Feng, X.H.; Lee, A.P.W.; Kwok, T.C.Y. Vitamins B-12 and C Supplementation Improves Arterial Reactivity and Structure in Passive Smokers: Implication in Prevention of Smoking-Related Atherosclerosis. J. Nutr. Health Aging 2021, 25, 248–254. [Google Scholar] [CrossRef]
- Sahin, G.; Yalcin, A.U.; Akcar, N. Effect of N-acetylcysteine on endothelial dysfunction in dialysis patients. Blood Purif. 2007, 25, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, K.; Kjerrulf, M.; Jungersten, L.; Johansson, K.; Långström, G.; Kalies, I.; Lenkei, R.; Walldius, G.; Lind, L. The new oral immunomodulating drug DiNAC induces brachial artery vasodilatation at rest and during hyperemia in hypercholesterolemic subjects, likely by a nitric oxide-dependent mechanism. Atherosclerosis 2008, 196, 275–282. [Google Scholar] [CrossRef]
- Khandouzi, N.; Zahedmehr, A.; Mohammadzadeh, A.; Sanati, H.R.; Nasrollahzadeh, J. Effect of flaxseed consumption on flow-mediated dilation and inflammatory biomarkers in patients with coronary artery disease: A randomized controlled trial. Eur. J. Clin. Nutr. 2019, 73, 258–265. [Google Scholar] [CrossRef]
- Volek, J.S.; Judelson, D.A.; Silvestre, R.; Yamamoto, L.M.; Spiering, B.A.; Hatfield, D.L.; Vingren, J.L.; Quann, E.E.; Anderson, J.M.; Maresh, C.M.; et al. Effects of carnitine supplementation on flow-mediated dilation and vascular inflammatory responses to a high-fat meal in healthy young adults. Am. J. Cardiol. 2008, 102, 1413–1417. [Google Scholar] [CrossRef]
- Shafabakhsh, R.; Milajerdi, A.; Reiner, Ž.; Kolahdooz, F.; Amirani, E.; Mirzaei, H.; Barekat, M.; Asemi, Z. The effects of catechin on endothelial function: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food. Sci. Nutr. 2020, 60, 2369–2378. [Google Scholar] [CrossRef]
- Littarru, G.P.; Tiano, L. Bioenergetic and antioxidant properties of coenzyme Q10: Recent developments. Mol. Biotechnol. 2007, 37, 31–37. [Google Scholar] [CrossRef]
- Nishioka, K.; Hidaka, T.; Nakamura, S.; Umemura, T.; Jitsuiki, D.; Soga, J.; Goto, C.; Chayama, K.; Yoshizumi, M.; Higashi, Y. Pycnogenol, French maritime pine bark extract, augments endothelium-dependent vasodilation in humans. Hypertens. Res. 2007, 30, 775–780. [Google Scholar] [CrossRef]
- Kajikawa, M.; Maruhashi, T.; Hidaka, T.; Nakano, Y.; Kurisu, S.; Matsumoto, T.; Iwamoto, Y.; Kishimoto, S.; Matsui, S.; Aibara, Y.; et al. Coffee with a high content of chlorogenic acids and low content of hydroxyhydroquinone improves postprandial endothelial dysfunction in patients with borderline and stage 1 hypertension. Eur. J. Nutr. 2019, 58, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelemen, M.; Vaidya, D.; Waters, D.D.; Howard, B.V.; Cobb, F.; Younes, N.; Tripputti, M.; Ouyang, P. Hormone therapy and antioxidant vitamins do not improve endothelial vasodilator function in postmenopausal women with established coronary artery disease: A substudy of the Women’s Angiographic Vitamin and Estrogen (WAVE) trial. Atherosclerosis 2005, 179, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Simons, L.A.; von Konigsmark, M.; Simons, J.; Stocker, R.; Celermajer, D.S. Vitamin E ingestion does not improve arterial endothelial dysfunction in older adults. Atherosclerosis 1999, 143, 193–199. [Google Scholar] [CrossRef]
- Creager, M.A.; Roddy, M.A.; Boles, K.; Stamler, J.S. N-acetylcysteine does not influence the activity of endothelium-derived relaxing factor in vivo. Hypertension 1997, 29, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.; Lubos, E.; Messow, C.M.; Sinning, C.R.; Zeller, T.; Wild, P.S.; Peetz, D.; Handy, D.E.; Munzel, T.; Loscalzo, J.; et al. Selenium supplementation improves antioxidant capacity in vitro and in vivo in patients with coronary artery disease The SElenium Therapy in Coronary Artery disease Patients (SETCAP) Study. Am. Heart J. 2008, 156, 1201.e1–1201.e11. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.L.; Evans, M.A.; Chan, W.; Nawaz, H.; Comerford, B.P.; Hoxley, M.L.; Njike, V.Y.; Sarrel, P.M. Oats, antioxidants and endothelial function in overweight, dyslipidemic adults. J. Am. Coll. Nutr. 2004, 23, 397–403. [Google Scholar] [CrossRef]
- Van-Assche, T.; Huygelen, V.; Crabtree, M.J.; Antoniades, C. Gene therapy targeting inflammation in atherosclerosis. Curr. Pharm. Des. 2011, 17, 4210–4223. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Regulation of redox glutathione levels and gene transcription in lung inflammation: Therapeutic approaches. Free. Radic. Biol. Med. 2000, 28, 1405–1420. [Google Scholar] [CrossRef]
- Ramprasath, T.; Selvam, G.S. Potential impact of genetic variants in Nrf2 regulated antioxidant genes and risk prediction of diabetes and associated cardiac complications. Curr. Med. Chem. 2013, 20, 4680–4693. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxid. Med. Cell Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- Rao, G.N.; Berk, B.C. Active oxyhen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res. 1992, 70, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Griendling, K.K.; Sorescu, D.; Lassegue, B.; Ushio-Fukai, M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathopysiology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Touys, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant effects of vitamin C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SFR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Zeiher, A.M. Reactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factors. Regul. Pept. 2000, 90, 19–25. [Google Scholar] [CrossRef]
- Anuranjani; Bala, M. Concerted action of Nrf2-ARE pathway, MRN complex, HMGB1 and inflammatory cytokines—Implication in modification of radiation damage. Redox Biol. 2014, 2, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Kuo, F.C.; Chao, C.T.; Lin, S.H. The Dynamics and Plasticity of Epigenetics in Diabetic Kidney Disease: Therapeutic Applications Vis-à-Vis. Int. J. Mol. Sci. 2022, 23, 843. [Google Scholar] [CrossRef]
- Brasier, A.R.; Recinos, A.; Eledrisi, M.S. Vascular inflammation and the renin angiotensin system. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1257–1266. [Google Scholar] [CrossRef]
- Pueyo, M.E.; Gonzalez, W.; Nicoletti, A.; Savoie, F.; Arnal, J.F.; Michel, J.B. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via NFkB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 645–651. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Selwyn, A.P. Endothelial function, inflammation, and prognosis in cardiovascular disease. Am. J. Med. 2003, 115 (Suppl. 8A), 99S–106S. [Google Scholar] [CrossRef]
- Oshima, T.; Ozono, R.; Yano, Y.; Oishi, Y.; Teragawa, H.; Higashi, Y.; Yoshizumi, M.; Kambe, M. Association of helicobacter pylori infection with systemic inflammation and endothelial dysfunction in healthy male subjects. J. Am. Coll. Cardiol. 2005, 45, 1219–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, Y.; Jitsuiki, D.; Goto, C.; Umemura, T.; Nishioka, K.; Hidaka, T.; Takemoto, T.; Nakamura, S.; Nakagawa, K.; Oshima, T.; et al. Periodontal infection is associated with endothelial dysfunction in humans. Hypertension 2008, 51, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Goto, C.; Hidaka, T.; Soga, J.; Nakamura, S.; Fujii, Y.; Hata, T.; Idei, N.; Fujimura, N.; Chayama, K.; et al. Oral infection-inflammatory pathway, periodontitis, is a risk factor for endothelial dysfunction in patients with coronary artery disease. Atherosclerosis 2009, 206, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Kadono, T.; Sugiyama, H.; Hoshiai, M.; Osada, M.; Tan, T.; Naitoh, A.; Watanabe, M.; Koizumi, K.; Nakazawa, S. Endothelial function evaluated by flow-mediated dilatation in pediatric vascular disease. Pediatr. Cardiol. 2005, 26, 385–390. [Google Scholar] [CrossRef]
- Ishii, M.; Ueno, T.; Ikeda, H.; Iemura, M.; Sugimura, T.; Furui, J.; Sugahara, Y.; Muta, H.; Akagi, T.; Nomura, Y.; et al. Sequential follow-up results of catheter intervention for coronary artery lesions after Kawasaki disease: Quantitative coronary artery angiography and intravascular ultrasound imaging study. Circulation 2002, 105, 3004–3010. [Google Scholar] [CrossRef]
- Taguchi, A.; Sanada, M.; Suei, Y.; Ohtsuka, M.; Lee, R.; Tanimoto, K.; Tsuda, M.; Ohama, K.; Yoshizumi, M.; Higashi, Y. Tooth loss is associated with an increased risk of hypertension in postmenopausal women. Hypertension 2004, 43, 1297–1300. [Google Scholar] [CrossRef]
- Taguchi, A.; Miki, M.; Muto, A.; Kubokawa, K.; Migita, K.; Higashi, Y.; Yoshinari, N. Association between oral health and the risk of lacunar infarction in Japanese adult males and females. Gerontology 2013, 59, 499–506. [Google Scholar] [CrossRef]
- Dewaki, N.; Ishioka, Y.; Uchida, K.; Yoshida, A.; Tagiuchi, A.; Higashi, Y.; Yoshinari, N. Association between carotid artery calcification and periodontal disease progression in Japanese men and women—Cross-sectional study. J. Clin. Med. 2020, 9, 3365. [Google Scholar] [CrossRef]
- Mitani, Y.; Sawada, H.; Hayakawa, H.; Aoki, K.; Ohashi, H.; Matsumura, M.; Kuroe, K.; Shimpo, H.; Nakano, M.; Komada, Y. Elevated levels of high-sensitivity C-reactive protein and serum amyloid-A late after Kawasaki disease: Association between inflammation and late coronarysequelae in Kawasaki disease. Circulation 2005, 111, 38–43. [Google Scholar] [CrossRef]
- Idei, N.; Nishioka, K.; Soga, J.; Hidaka, T.; Hata, T.; Fujii, Y.; Fujimura, N.; Maruhashi, T.; Mikami, S.; Teragawa, H.; et al. Vascular function and circulating progenitor cell in thromboangiitis obliterans (Buerger disease) and atherosclerosis obliterans. Hypertension 2011, 57, 70–78. [Google Scholar] [CrossRef]
- Fujii, Y.; Fujimura, N.; Mikami, S.; Maruhashi, T.; Kihara, Y.; Chayama, K.; Noma, K.; Higashi, Y. Flow-mediated vasodilation is augmented in corkscrew collateral artery compared with that in a native artery in patients with thromboangiitis obliterans (Bureger disease). J. Vas. Surg. 2011, 54, 1689–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellon, X.; Bogdanova, V. Chronic Inflammatory Diseases and Endothelial Dysfunction. Aging Dis. 2016, 7, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Moroni, L.; Selmi, C.; Angelini, C.; Meroni, P.L. Evaluation of Endothelial Function by Flow-Mediated Dilation: A Comprehensive Review in Rheumatic Disease. Arch. Immunol. Ther. Exp. 2017, 65, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Çiftel, M.; Yilmaz, O. İnvestigation of endothelial dysfunction in children with acute rheumatic fever. Ann. Pediatr. Cardiol. 2020, 13, 199–204. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Xaplanteris, P.; Sambatakou, H.; Mariolis, E.; Bratsas, A.; Christoforatou, E.; Miliou, A.; Aznaouridis, K.; Stefanadis, C. Acute systemic inflammation induced by influenza A (H1N1) vaccination causes a deterioration in endothelial function in HIV-infected patients. HIV. Med. 2011, 12, 594–601. [Google Scholar] [CrossRef]
- Kirollos, S.; Skilton, M.; Patel, S.; Arnott, C.A. Systematic Review of Vascular Structure and Function in Pre-eclampsia: Non-invasive Assessment and Mechanistic Links. Front. Cardiovasc. Med. 2019, 6, 166. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, Y.; Wen, Y.; Xi, J.; Chen, T.; Lu, Y.; Wang, L.; Liu, Y.; Si, F. Ultrasound evaluation of endothelial dysfunction in immunoglobulin-resistant children with acute Kawasaki disease. Clin. Rheumatol. 2022. [Google Scholar] [CrossRef]
- Becker, L.; Prado, K.; Foppa, M.; Martinelli, N.; Aguiar, C.; Furian, T.; Clausell, N.; Rohde, L.E. Endothelial dysfunction assessed by brachial artery ultrasound in severe sepsis and septic shock. J. Crit. Care 2012, 316, e9–e14. [Google Scholar] [CrossRef]
- Nfon, C.K.; Toka, F.N.; Kenney, M.; Pacheco, J.M.; Golde, W.T. Loss of plasmacytoid dendritic cell function coincides with lymphopenia and viremia during foot-and-mouth disease virus infection. Viral. Immunol. 2010, 23, 29–41. [Google Scholar] [CrossRef]
- Mäki-Petäjä, K.M.; Booth, A.D.; Hall, F.C.; Wallace, S.M.; Brown, J.; McEniery, C.M.; Wilkinson, I.B. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J. Am. Coll. Cardiol. 2007, 50, 852–858. [Google Scholar] [CrossRef]
- Kotani, K.; Miyamoto, M.; Ando, H. The Effect of Treatments for Rheumatoid Arthritis on Endothelial Dysfunction Evaluated by Flow-Mediated Vasodilation in Patients with Rheumatoid Arthritis. Curr. Vasc. Pharmacol. 2017, 15, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Hussain, K.S.; Gulati, R.; Satheesh, S.; Negi, V.S. Early-onset subclinical cardiovascular damage assessed by non-invasive methods in children with Juvenile Idiopathic Arthritis: Analytical cross-sectional study. Rheumatol. Int. 2021, 41, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Pacholczak-Madej, R.; Kuszmiersz, P.; Bazan-Socha, S.; Kosałka-Wêgiel, J.; Iwaniec, T.; Zarêba, L.; Kielczewski, S.; Rams, A.; Walocha, J.A.; Musiał, J.; et al. Endothelial dysfunction in patients with systemic sclerosis. Postepy. Dermatol. Alergol. 2020, 37, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Valer, P.; Paul, B.; Eugenia, B.; Camelia, B. Annexin A5 as independent predictive biomarker for subclinical atherosclerosis and endothelial dysfunction in systemic lupus erythematosus patients. Clin. Lab. 2013, 59, 359–367. [Google Scholar] [CrossRef]
- Rodrigues, F.M.; Bacchiega, A.B.; Bacchiega, B.C.; Gomes Ochtrop, M.L.; Levy, R.A. Evaluation of endothelial function in patients with Behçet’s disease in remission: A cross-sectional study. Eur. J. Rheumatol. 2022, 9, 139–143. [Google Scholar] [CrossRef]
- Elcioglu, O.C.; Afsar, B.; Bakan, A.; Takir, M.; Ozkok, A.; Oral, A.; Kostek, O.; Basci, S.; Kanbay, A.; Toprak, A.E.; et al. Chronic rhinosinusitis, endothelial dysfunction, and atherosclerosis. Am. J. Rhinol. Allergy. 2016, 30, 58–61. [Google Scholar] [CrossRef]
- Saha, A.; Bagchi, A.; Chatterjee, S.; Dutta, S.; Misra, S.; Bhattacharjee, D.; Chatterjee, S.; Mondal, S.; Ghosh, P.; Chatterjee, M.; et al. Phenotypic characterization of circulating endothelial cells induced by inflammation and oxidative stress in ankylosing spondylitis. Free. Radic. Res. 2021, 55, 520–532. [Google Scholar] [CrossRef]
- Senzaki, K.; Okada, Y.; Ochi, H.; Ochi, M.; Takei, S.I.; Miura, S.; Igase, M.; Ohyagi, Y. Vascular endothelial dysfunction associated with severity in multiple sclerosis. Mult. Scler. Relat. Disord. 2021, 54, 103135. [Google Scholar] [CrossRef]
- Andreozzi, M.; Giugliano, F.P.; Strisciuglio, T.; Pirozzi, E.; Papparella, A.; Caprio, A.M.; Miele, E.; Strisciuglio, C.; Perrone Filardi, P. The Role of Inflammation in the Endothelial Dysfunction in a Cohort of Pediatric Patients with Inflammatory Bowel Disease. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 330–335. [Google Scholar] [CrossRef]
- Alibaz-Oner, F.; Yurdakul, S.; Aytekin, S.; Direskeneli, H. Impaired endothelial function in patients with Takayasu’s arteritis. Acta. Cardiol. 2014, 69, 45–49. [Google Scholar] [CrossRef]
- Karoli, R.; Fatima, J.; Shukla, V.; Dhillon, K.S.; Khanduri, S.; Maini, S.; Chandra, A. A study of cardio-metabolic risk profile in patients with psoriasis. J. Assoc. Physicians India 2013, 61, 798–803. [Google Scholar] [PubMed]
- Clarenbach, C.F.; Sievi, N.A.; Kohler, M. Annual progression of endothelial dysfunction in patients with COPD. Respir. Med. 2017, 132, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, A.; Wardyn, K.A.; Życińska, K. Peripheral Arterial Tonometry in Pulmonary Vasculitis. Adv. Exp. Med. Biol. 2018, 1040, 1–5. [Google Scholar] [PubMed]
- Vallbracht, K.B.; Schwimmbeck, P.L.; Seeberg, B.; Kuhl, U.; Schultheiss, H.P. Endothelial dysfunction of peripheral arteries in patients with immunohistologically confirmed myocardial inflammation correlates with endothelial expression of human leukocyte antigens and adhesion molecules in myocardial biopsies. J. Am. Coll. Cardiol. 2002, 40, 515–520. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higashi, Y. Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease. Antioxidants 2022, 11, 1958. https://doi.org/10.3390/antiox11101958
Higashi Y. Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease. Antioxidants. 2022; 11(10):1958. https://doi.org/10.3390/antiox11101958
Chicago/Turabian StyleHigashi, Yukihito. 2022. "Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease" Antioxidants 11, no. 10: 1958. https://doi.org/10.3390/antiox11101958
APA StyleHigashi, Y. (2022). Roles of Oxidative Stress and Inflammation in Vascular Endothelial Dysfunction-Related Disease. Antioxidants, 11(10), 1958. https://doi.org/10.3390/antiox11101958