


Conditions Conducive to the Glutathionylation of Complex I Subunit NDUFS1 Augment ROS Production following the Oxidation of Ubiquinone Linked Substrates, Glycerol-3-Phosphate and Proline



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Experimental
2.1. Chemicals
2.2. Isolation of Liver Mitochondria
2.3. Interrogation of Mitochondrial O2●−/H2O2 Production
2.4. Evaluation of O2●−/H2O2 Sources in Liver Mitochondria

2.5. GPD and PRODH Activity
2.6. Measurement of PSSG Adducts
2.7. BioGEE Switch Assays
2.8. Complex I Immunocapture

2.9. Seahorse XFe24 Assays
2.10. Regulation of Complex I Activity and ROS Production
2.11. Statistical Analyses and Diagrams
3. Results and Discussion
3.1. Disulfiram Augments O2●−/H2O2 Production by Liver Mitochondria Oxidizing UQ-Linked Substrates

3.2. GPD and PRODH Are not Glutathionylation Targets


3.3. Blocking Electron Flow through Glutathionylation of Complex I Subunit NDUFS1 Increases RET-Driven ROS Production during G3P and Pro Metabolism
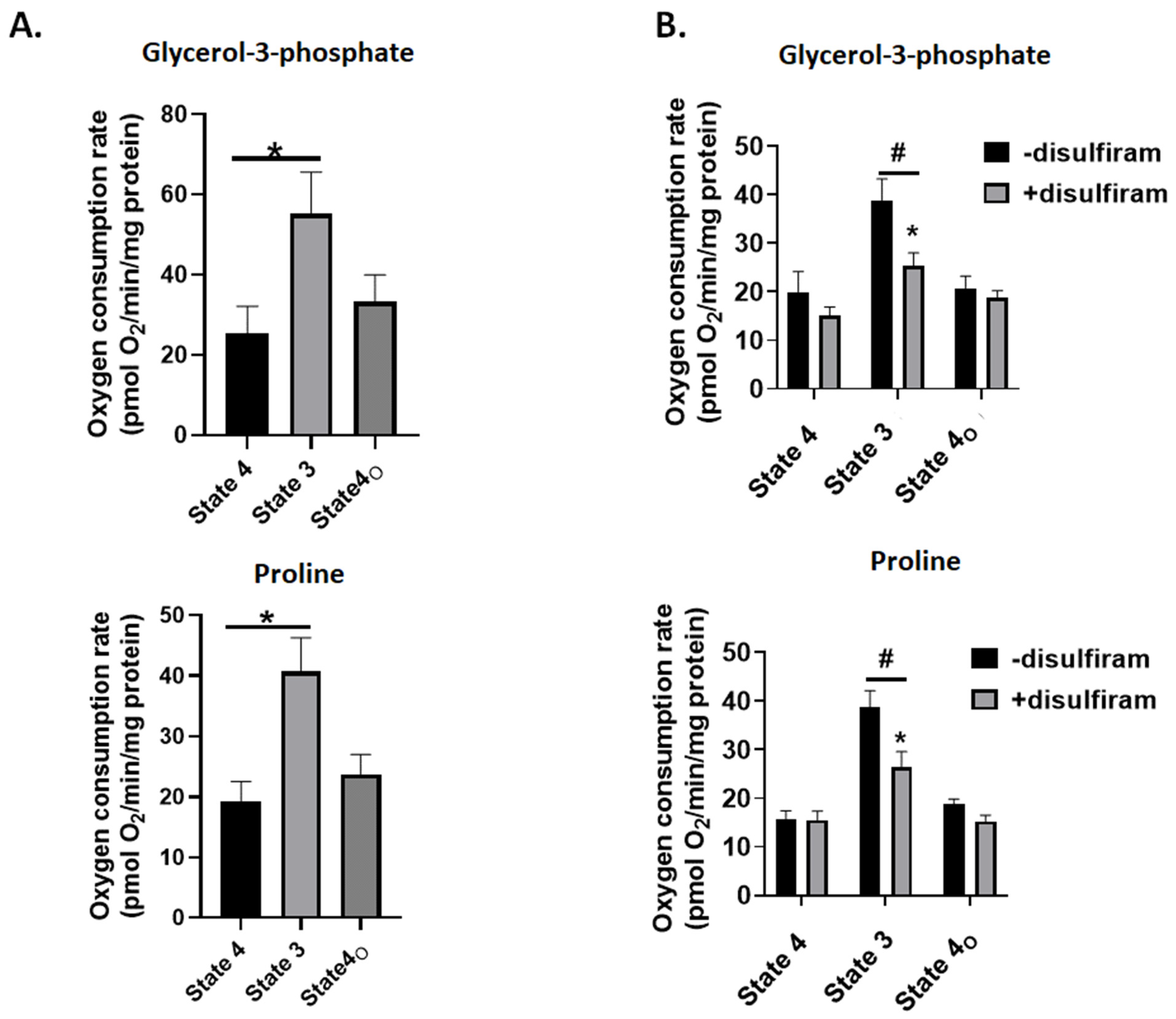
3.4. Disulfiram Treatment Inhibits G3P and Pro Fueled ATP Production

4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shelton, M.D.; Chock, P.B.; Mieyal, J.J. Glutaredoxin: Role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005, 7, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 1941, 10–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Starke, D.W.; Leonberg, A.K.; Ospina, S.M.; Mieyal, J.J. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: Implications for intracellular roles. Biochemistry 2008, 47, 11144–11157. [Google Scholar] [CrossRef] [Green Version]
- Kramer, P.A.; Duan, J.; Gaffrey, M.J.; Shukla, A.K.; Wang, L.; Bammler, T.K.; Qian, W.J.; Marcinek, D.J. Fatiguing contractions increase protein S-glutathionylation occupancy in mouse skeletal muscle. Redox Biol. 2018, 17, 367–376. [Google Scholar] [CrossRef] [PubMed]
- McGarry, D.J.; Chakravarty, P.; Wolf, C.R.; Henderson, C.J. Altered protein S-glutathionylation identifies a potential mechanism of resistance to acetaminophen-induced hepatotoxicity. J. Pharmacol. Exp. Ther. 2015, 355, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustarini, D.; Galvagni, F.; Tesei, A.; Farolfi, A.; Zanoni, M.; Pignatta, S.; Milzani, A.; Marone, I.M.; Dalle-Donne, I.; Nassini, R.; et al. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures. Free Radic. Biol. Med. 2015, 89, 972–981. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Young, A.; Gill, R.M. Protein S-Glutathionylation and the Regulation of Cellular Functions. In Oxidative Stress: Oxidative Eustress and Distress; Sies, H., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; p. 878. [Google Scholar]
- Mailloux, R.J. Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 2020, 32, 101472. [Google Scholar] [CrossRef]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef]
- Gill, R.M.; O’Brien, M.; Young, A.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation lowers superoxide/hydrogen peroxide release from skeletal muscle mitochondria through modification of complex I and inhibition of pyruvate uptake. PLoS ONE 2018, 13, e0192801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: Potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008, 283, 24801–24815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Lin, L.; Giblin, F.; Ho, Y.S.; Lou, M.F. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 2011, 51, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Kuksal, N.; Gardiner, D.; Qi, D.; Mailloux, R.J. Partial loss of complex I due to NDUFS4 deficiency augments myocardial reperfusion damage by increasing mitochondrial superoxide/hydrogen peroxide production. Biochem. Biophys. Res. Commun. 2018, 498, 214–220. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Mezera, V.; Dighe, P.; Melov, S.; Gerencser, A.A.; Sweis, R.F.; Pliushchev, M.; Wang, Z.; Esbenshade, T.; McKibben, B.; et al. Superoxide produced by mitochondrial site IQ inactivates cardiac succinate dehydrogenase and induces hepatic steatosis in Sod2 knockout mice. Free Radic. Biol. Med. 2021, 164, 223–232. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2014, 2, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.E.; Kennedy, C.R.; Rippstein, P.; deKemp, R.; da Silva, J.; et al. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 2014, 289, 14812–14828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diotte, N.M.; Xiong, Y.; Gao, J.; Chua, B.H.; Ho, Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta 2009, 1793, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschenson, J.; Mailloux, R.J. The glutathionylation agent disulfiram augments superoxide/hydrogen peroxide production when liver mitochondria are oxidizing ubiquinone pool-linked and branched chain amino acid substrates. Free Radic. Biol. Med. 2021, 172, 1–8. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Chalker, J.; Slade, L.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic. Biol. Med. 2017, 106, 302–314. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Rothschild, D.E.; Quinlan, C.L.; Scott, G.K.; Benz, C.C.; Brand, M.D. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biol. 2014, 2, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Shabalina, I.G.; Vrbacky, M.; Pecinova, A.; Kalinovich, A.V.; Drahota, Z.; Houstek, J.; Mracek, T.; Cannon, B.; Nedergaard, J. ROS production in brown adipose tissue mitochondria: The question of UCP1-dependence. Biochim. Biophys. Acta 2014, 1837, 2017–2030. [Google Scholar] [CrossRef] [Green Version]
- Oldford, C.; Kuksal, N.; Gill, R.; Young, A.; Mailloux, R.J. Estimation of the hydrogen peroxide producing capacities of liver and cardiac mitochondria isolated from C57BL/6N and C57BL/6J mice. Free Radic. Biol. Med. 2019, 135, 15–27. [Google Scholar] [CrossRef]
- Orr, A.L.; Ashok, D.; Sarantos, M.R.; Ng, R.; Shi, T.; Gerencser, A.A.; Hughes, R.E.; Brand, M.D. Novel inhibitors of mitochondrial sn-glycerol 3-phosphate dehydrogenase. PLoS ONE 2014, 9, e89938. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Benoit, B.; Brand, M.D. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic. Biol. Med. 2019, 130, 140–150. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; David, L.; Ho, Y.S.; Lou, M.F. Glutaredoxin 2 (Grx2) gene deletion induces early onset of age-dependent cataracts in mice. J. Biol. Chem. 2014, 289, 36125–36139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Gardiner, D.; O’Brien, M. 2-Oxoglutarate dehydrogenase is a more significant source of O2·−/H2O2 than pyruvate dehydrogenase in cardiac and liver tissue. Free Radic. Biol. Med. 2016, 97, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.; Gardiner, D.; Kuksal, N.; Mailloux, R.J. Characterization of the impact of glutaredoxin-2 (GRX2) deficiency on superoxide/hydrogen peroxide release from cardiac and liver mitochondria. Redox Biol. 2018, 15, 216–227. [Google Scholar] [CrossRef] [PubMed]
- McLain, A.L.; Cormier, P.J.; Kinter, M.; Szweda, L.I. Glutathionylation of alpha-ketoglutarate dehydrogenase: The chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 2013, 61, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Merrihew, G.E.; Egertson, J.D.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef]
- Whitson, J.A.; Martin-Perez, M.; Zhang, T.; Gaffrey, M.J.; Merrihew, G.E.; Huang, E.; White, C.C.; Kavanagh, T.J.; Qian, W.J.; Campbell, M.D.; et al. Elamipretide (SS-31) treatment attenuates age-associated post-translational modifications of heart proteins. Geroscience 2021, 43, 2395–2412. [Google Scholar] [CrossRef]
- Young, A.; Gardiner, D.; Kuksal, N.; Gill, R.; O’Brien, M.; Mailloux, R.J. Deletion of the Glutaredoxin-2 Gene Protects Mice from Diet-Induced Weight Gain, Which Correlates with Increased Mitochondrial Respiration and Proton Leaks in Skeletal Muscle. Antioxid. Redox Signal. 2019, 31, 1272–1288. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- VanHecke, G.C.; Abeywardana, M.Y.; Ahn, Y.H. Proteomic Identification of Protein Glutathionylation in Cardiomyocytes. J. Proteome Res. 2019, 18, 1806–1818. [Google Scholar] [CrossRef]
- Wang, S.B.; Foster, D.B.; Rucker, J.; O’Rourke, B.; Kass, D.A.; van Eyk, J.E. Redox regulation of mitochondrial ATP synthase: Implications for cardiac resynchronization therapy. Circ. Res. 2011, 109, 750–757. [Google Scholar] [CrossRef]
- Randi, E.B.; Zuhra, K.; Pecze, L.; Panagaki, T.; Szabo, C. Physiological concentrations of cyanide stimulate mitochondrial Complex IV and enhance cellular bioenergetics. Proc. Natl. Acad. Sci. USA 2021, 118, e2026245118. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Hirschenson, J.; Moore, A.; Mailloux, R.J. Conditions Conducive to the Glutathionylation of Complex I Subunit NDUFS1 Augment ROS Production following the Oxidation of Ubiquinone Linked Substrates, Glycerol-3-Phosphate and Proline. Antioxidants 2022, 11, 2043. https://doi.org/10.3390/antiox11102043
Wang K, Hirschenson J, Moore A, Mailloux RJ. Conditions Conducive to the Glutathionylation of Complex I Subunit NDUFS1 Augment ROS Production following the Oxidation of Ubiquinone Linked Substrates, Glycerol-3-Phosphate and Proline. Antioxidants. 2022; 11(10):2043. https://doi.org/10.3390/antiox11102043
Chicago/Turabian StyleWang, Kevin, Jonathan Hirschenson, Amanda Moore, and Ryan J. Mailloux. 2022. "Conditions Conducive to the Glutathionylation of Complex I Subunit NDUFS1 Augment ROS Production following the Oxidation of Ubiquinone Linked Substrates, Glycerol-3-Phosphate and Proline" Antioxidants 11, no. 10: 2043. https://doi.org/10.3390/antiox11102043
APA StyleWang, K., Hirschenson, J., Moore, A., & Mailloux, R. J. (2022). Conditions Conducive to the Glutathionylation of Complex I Subunit NDUFS1 Augment ROS Production following the Oxidation of Ubiquinone Linked Substrates, Glycerol-3-Phosphate and Proline. Antioxidants, 11(10), 2043. https://doi.org/10.3390/antiox11102043