
Is Mitochondrial Oxidative Stress a Viable Therapeutic Target in Preeclampsia?


Abstract
:1. Introduction
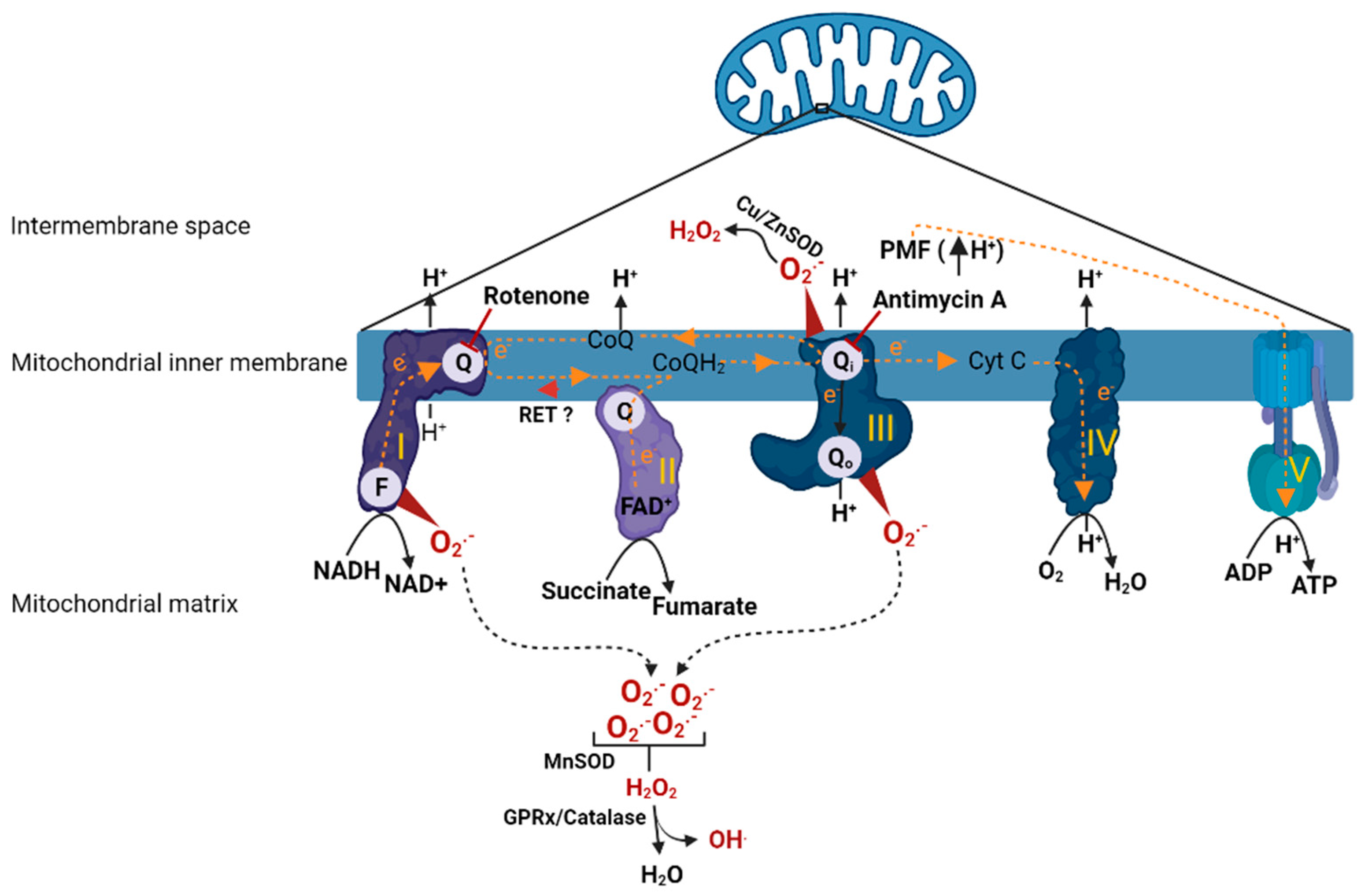
2. Mitochondrial Respiration and ROS Production
3. Mitochondrial Dysfunction in Preeclampsia
3.1. Clinical Studies
3.2. Animal Studies
3.3. In Vitro Cell-Based Studies
4. Mediators of Mitochondrial Dysfunction and Excessive ROS Production in Preeclampsia
5. Mitochondrial Targeted Antioxidants in Preeclampsia
5.1. MitoQ

{kind=link}
{kind=link}
| Antioxidant | Model | Treatment | Findings | Ref. | |||
|---|---|---|---|---|---|---|---|
| In Vitro | In Vivo | Dose | Duration | Route | |||
| MitoQ | L-NAME rat model a | 60 mg/kg/day | 5 days (GD 15–20) | Oral |
| [71] | |
| RUPP rat model | 500 µM/kg/day | 5 days (GD 14–19) | Oral |
| [7] | ||
| RUPP mouse model b | 100 µM/kg/day | 4 days (GD 13.5–17.5) | Oral |
| [43] | ||
| Hypoxia rat model | 125 µM | 5 days (GD 15–20) | I.V |
| [78] | ||
| Hypoxia rat model | 500 µM/kg/day | 14 days (GD6–20) | Oral |
| [79] | ||
| RUPP serum- treated HUVECs | 10% serum c | Overnight | Direct addition |
| [7] | ||
| H2O2 treated HTR8-S/Vneo trophoblast cells | 1 µM | Direct addition |
| [43] | |||
| MitoTempo | RUPP rat model | 1 mg/kg/day | 5 days (GD 14–19) | Oral |
| [7] | |
| PE serum treated HUVECs | 5 µM | 2 h | Direct addition |
| |||
| H2O2 treated HTR8-S/Vneo trophoblast cells | 1 µM | Direct addition |
| [43] | |||
| Ergothioneine | RUPP rat model | 25 mg/kg/day | 8 days (GD11–19) | Oral |
| [8] | |
| RUPP rat model | 25 mg/kg/day | 8 days (GD11–19) | Oral |
| [19] | ||
5.2. MitoTempo
5.3. Ergothioneine
6. The Potential of Mitochondrial Antioxidants in Treating Preeclampsia
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Catov, J.M.; Ouyang, P. Hypertensive Disorders of Pregnancy and Future Maternal Cardiovascular Risk. J. Am. Heart Assoc. 2018, 7, e009382. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L.; Cui, X. Oxidative stress in the placenta. Histochem. Cell Biol. 2004, 122, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.W.; Vaughan, J.E.; Wang, Y.; Roberts, L.J., II. Placental isoprostane is significantly increased in preeclampsia. FASEB J. 2000, 14, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Walsh, S.W. Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta 1998, 19, 581–586. [Google Scholar] [CrossRef]
- Wang, Y.; Walsh, S.W. Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in pre-eclampsia. Placenta 2001, 22, 206–212. [Google Scholar] [CrossRef]
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W., Jr.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; LaMarca, B. Role of Mitochondrial Dysfunction and Reactive Oxygen Species in Mediating Hypertension in the Reduced Uterine Perfusion Pressure Rat Model of Preeclampsia. Hypertension 2018, 72, 703–711. [Google Scholar] [CrossRef]
- Williamson, R.D.; McCarthy, F.P.; Manna, S.; Groarke, E.; Kell, D.B.; Kenny, L.C.; McCarthy, C.M. L-(+)-Ergothioneine Significantly Improves the Clinical Characteristics of Preeclampsia in the Reduced Uterine Perfusion Pressure Rat Model. Hypertension 2020, 75, 561–568. [Google Scholar] [CrossRef]
- McCarthy, C.; Kenny, L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016, 6, 32683. [Google Scholar] [CrossRef] [Green Version]
- Spinnato, J.A., II; Freire, S.; e Silva, J.L.P.; Rudge, M.V.C.; Martins-Costa, S.; Koch, M.A.; Goco, N.; de Barros Santos, C.; Cecatti, J.G.; Costa, R.; et al. Antioxidant therapy to prevent preeclampsia: A randomized controlled trial. Obstet. Gynecol. 2007, 110, 1311–1318. [Google Scholar] [CrossRef]
- Kiondo, P.; Wamuyu-Maina, G.; Wandabwa, J.; Bimenya, G.S.; Tumwesigye, N.M.; Okong, P. The effects of vitamin C supplementation on pre-eclampsia in Mulago Hospital, Kampala, Uganda: A randomized placebo controlled clinical trial. BMC Pregnancy Childbirth 2014, 14, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCance, D.R.; Holmes, V.A.; Maresh, M.J.; Patterson, C.C.; Walker, J.D.; Pearson, D.W.; Young, I.S. Diabetes and Pre-eclampsia Intervention Trial (DAPIT) Study Group. Vitamins C and E for prevention of pre-eclampsia in women with type 1 diabetes (DAPIT): A randomised placebo-controlled trial. Lancet 2010, 376, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Rolnik, D.L.; Wright, D.; Poon, L.C.Y.; Syngelaki, A.; O’Gorman, N.; de Paco Matallana, C.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. ASPRE trial: Performance of screening for preterm pre-eclampsia. Ultrasound Obstet. Gynecol. 2017, 50, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aranguren, L.C.; Espinosa-González, C.T.; González-Ortiz, L.M.; Sanabria-Barrera, S.M.; Riaño-Medina, C.E.; Nuñez, A.F.; Ahmed, A.; Vasquez-Vivar, J.; López, M. Soluble Fms-Like Tyrosine Kinase-1 Alters Cellular Metabolism and Mitochondrial Bioenergetics in Preeclampsia. Front. Physiol. 2018, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Vaka, V.R.; Cunningham, M.W.; Deer, E.; Franks, M.; Ibrahim, T.; Amaral, L.M.; Usry, N.; Cornelius, D.C.; Dechend, R.; Wallukat, G.; et al. Blockade of endogenous angiotensin II type I receptor agonistic autoantibody activity improves mitochondrial reactive oxygen species and hypertension in a rat model of preeclampsia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R256–R262. [Google Scholar] [CrossRef] [PubMed]
- Vaka, V.R.; McMaster, K.M.; Cornelius, D.C.; Ibrahim, T.; Jayaram, A.; Usry, N.; Cunningham, M.W., Jr.; Amaral, L.M.; LaMarca, B. Natural killer cells contribute to mitochondrial dysfunction in response to placental ischemia in reduced uterine perfusion pressure rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R441–R447. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36 Pt 5, 976–980. [Google Scholar] [CrossRef]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Torbergsen, T.; Oian, P.; Mathiesen, E.; Borud, O. Pre-eclampsia—A mitochondrial disease? Acta Obstet. Gynecol. Scand. 1989, 68, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Minakami, H.; Sato, I.; Saito, T. Decrease in cytochrome c oxidase activity detected cytochemically in the placental trophoblast of patients with pre-eclampsia. Placenta 1997, 18, 255–259. [Google Scholar] [CrossRef]
- Rodríguez, J.C.M.; Mendoza, P.Y.; Bernal, M.P.; Acosta, J.L.A. Oxidative stress level and placental histological changes during preeclampsia. Ginecol. Obstet. Mex. 2008, 76, 319–326. [Google Scholar]
- Salgado, S.S.; Salgado, M.K.R. Structural changes in pre-eclamptic and eclamptic placentas—An ultrastructural study. J. Coll. Physicians Surg.—Pak. 2011, 21, 482–486. [Google Scholar]
- Zsengellér, Z.K.; Rajakumar, A.; Hunter, J.T.; Salahuddin, S.; Rana, S.; Stillman, I.E.; Karumanchi, S.A. Trophoblast mitochondrial function is impaired in preeclampsia and correlates negatively with the expression of soluble fms-like tyrosine kinase 1. Pregnancy Hypertens. 2016, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Muralimanoharan, S.; Maloyan, A.; Mele, J.; Guo, C.; Myatt, L.G.; Myatt, L. MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta 2012, 33, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaka, R.; Deer, E.; Cunningham, M.; McMaster, K.M.; Wallace, K.; Cornelius, D.C.; Amaral, L.M.; LaMarca, B. Characterization of Mitochondrial Bioenergetics in Preeclampsia. J. Clin. Med. 2021, 10, 5063. [Google Scholar] [CrossRef] [PubMed]
- Vishnyakova, P.A.; Volodina, M.A.; Tarasova, N.V.; Marey, M.V.; Tsvirkun, D.V.; Vavina, O.V.; Khodzhaeva, Z.S.; Kan, N.E.; Menon, R.; Vysokikh, M.Y.; et al. Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci. Rep. 2016, 6, 32410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, O.J.; Cuffe, J.S.M.; Nitert, M.D.; Callaway, L.; Cheung, K.A.K.; Radenkovic, F.; Perkins, A.V. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis. 2018, 9, 1150. [Google Scholar] [CrossRef] [Green Version]
- Mandò, C.; De Palma, C.; Stampalija, T.; Anelli, G.M.; Figus, M.; Novielli, C.; Parisi, F.; Clementi, E.; Ferrazzi, E.; Cetin, I. Placental mitochondrial content and function in intrauterine growth restriction and preeclampsia. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E404–E413. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.W.; Wang, Y. Deficient glutathione peroxidase activity in preeclampsia is associated with increased placental production of thromboxane and lipid peroxides. Am. J. Obstet. Gynecol. 1993, 169, 1456–1461. [Google Scholar] [CrossRef]
- Zusterzeel, P.L.; Steegers-Theunissen, R.P.; Harren, F.J.; Stekkinger, E.; Kateman, H.; Timmerman, B.H.; Berkelmans, R.; Nieuwenhuizen, A.; Peters, W.H.; Raijmakers, M.T.; et al. Ethene and other biomarkers of oxidative stress in hypertensive disorders of pregnancy. Hypertens. Pregnancy 2002, 21, 39–49. [Google Scholar] [CrossRef]
- Jin, X.; Xu, Z.; Cao, J.; Shao, P.; Zhou, M.; Qin, Z.; Liu, Y.; Yu, F.; Zhou, X.; Ji, W.; et al. Proteomics analysis of human placenta reveals glutathione metabolism dysfunction as the underlying pathogenesis for preeclampsia. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1207–1214. [Google Scholar] [CrossRef]
- Knapen, M.F.; Peters, W.H.; Mulder, T.P.; Merkus, H.M.; Jansen, J.B.; Steegers, E.A. Glutathione and glutathione-related enzymes in decidua and placenta of controls and women with pre-eclampsia. Placenta 1999, 20, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aranguren, L.C.; Prada, C.E.; Riaño-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Hevner, K.; Enquobahrie, D.A.; Williams, M.A. A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int. J. Mol. Epidemiol. Genet. 2012, 3, 237–244. [Google Scholar] [PubMed]
- Marschalek, J.; Wohlrab, P.; Ott, J.; Wojta, J.; Speidl, W.; Klein, K.U.; Kiss, H.; Pateisky, P.; Zeisler, H.; Kuessel, L. Maternal serum mitochondrial DNA (mtDNA) levels are elevated in preeclampsia—A matched case-control study. Pregnancy Hypertens. 2018, 14, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhao, X.; Zhou, W.; Qi, H.; Zhang, H.; Han, T.L.; Baker, P. Impaired placental mitophagy and oxidative stress are associated with dysregulated BNIP3 in preeclampsia. Sci. Rep. 2021, 11, 20469. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, P.; Zhu, F.; Liao, J.; Wu, Y.; Hu, M.; Fu, H.; Qiao, J.; Lin, L.; Huang, B.; et al. The Potent Antioxidant MitoQ Protects Against Preeclampsia During Late Gestation but Increases the Risk of Preeclampsia When Administered in Early Pregnancy. Antioxid. Redox Signal. 2021, 34, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Booz, G.W.; Kennedy, D.; Bowling, M.; Robinson, T.; Azubuike, D.; Fisher, B.; Brooks, K.; Chinthakuntla, P.; Hoang, N.H.; Hosler, J.P.; et al. Angiotensin II type 1 receptor agonistic autoantibody blockade improves postpartum hypertension and cardiac mitochondrial function in rat model of preeclampsia. Biol. Sex Differ. 2021, 12, 58. [Google Scholar] [CrossRef]
- Sanchez-Aranguren, L.C.; Rezai, H.; Ahmad, S.; Alzahrani, F.A.; Sparatore, A.; Wang, K.; Ahmed, A. MZe786 Rescues Cardiac Mitochondrial Activity in High sFlt-1 and Low HO-1 Environment. Antioxidants 2020, 9, 598. [Google Scholar] [CrossRef] [PubMed]
- Okatani, Y.; Wakatsuki, A.; Shinohara, K.; Taniguchi, K.; Fukaya, T. Melatonin protects against oxidative mitochondrial damage induced in rat placenta by ischemia and reperfusion. J. Pineal Res. 2001, 31, 173–178. [Google Scholar] [CrossRef]
- Chen, G.; Lin, Y.; Chen, L.; Zeng, F.; Zhang, L.; Huang, Y.; Huang, P.; Liao, L.; Yu, Y. Role of DRAM1 in mitophagy contributes to preeclampsia regulation in mice. Mol. Med. Rep. 2020, 22, 1847–1858. [Google Scholar] [CrossRef]
- Popova, T.A.; Perfilova, V.N.; Zhakupova, G.A.; Verovsky, V.E.; Ostrovskij, O.V.; Tyurenkov, I.N. The effect of sulodexide on placental mitochondria function in rats with experimental preeclampsia. Biomeditsinskaia Khimiia 2016, 62, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.; Vaka, V.R.; McMaster, K.M.; Wallace, K.; Cornelius, D.C.; Amaral, L.M.; Cunningham, M.W.; LaMarca, B. Vascular endothelial mitochondrial oxidative stress in response to preeclampsia: A role for angiotension II type 1 autoantibodies. Am. J. Obstet. Gynecol. MFM 2021, 3, 100275. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W., Jr.; Vaka, V.R.; McMaster, K.; Ibrahim, T.; Cornelius, D.C.; Amaral, L.; Campbell, N.; Wallukat, G.; McDuffy, S.; Usry, N.; et al. Renal natural killer cell activation and mitochondrial oxidative stress; new mechanisms in AT1-AA mediated hypertensive pregnancy. Pregnancy Hypertens. 2019, 15, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinvalet, D.; Zhu, P.; Lieberman, J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 2005, 22, 355–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiusolo, V.; Jacquemin, G.; Bassoy, E.Y.; Vinet, L.; Liguori, L.; Walch, M.; Kozjak-Pavlovic, V.; Martinvalet, D. Granzyme B enters the mitochondria in a Sam50-, Tim22- and mtHsp70-dependent manner to induce apoptosis. Cell Death Differ. 2017, 24, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Martinvalet, D.; Dykxhoorn, D.M.; Ferrini, R.; Lieberman, J. Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 2008, 133, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, F.; Roy, S.; Nicholson, D.; Thornberry, N.; Rosen, A.; Casciola-Rosen, L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: Implications for CTL-induced apoptosis. Immunity 1998, 8, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Jayaram, A.; Deer, E.; Amaral, L.M.; Campbell, N.; Vaka, V.R.; Cunningham, M.; Ibrahim, T.; Cornelius, D.C.; LaMarca, B.B. The role of tumor necrosis factor in triggering activation of natural killer cell, multi-organ mitochondrial dysfunction and hypertension during pregnancy. Pregnancy Hypertens. 2021, 24, 65–72. [Google Scholar] [CrossRef]
- Cunningham, M.W.; Jayaram, A.; Deer, E.; Amaral, L.M.; Vaka, V.R.; Ibrahim, T.; Cornelius, D.C.; LaMarca, B. Tumor necrosis factor alpha (TNF-α) blockade improves natural killer cell (NK) activation, hypertension, and mitochondrial oxidative stress in a preclinical rat model of preeclampsia. Hypertens. Pregnancy 2020, 39, 399–404. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Wallukat, G.; Llinas, M.; Herse, F.; Dechend, R.; Granger, J.P. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 2008, 52, 1168–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deer, E.; Reeve, K.E.; Amaral, L.; Vaka, V.R.; Franks, M.; Campbell, N.; Fitzgerald, S.; Herrock, O.; Ibrahim, T.; Cornelius, D.; et al. CD4+ T cells cause renal and placental mitochondrial oxidative stress as mechanisms of hypertension in response to placental ischemia. Am. J. Physiol. Ren. Physiol. 2021, 320, F47–F54. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.; Amaral, L.M.; Campbell, N.; Fitzgerald, S.; Herrock, O.; Ibrahim, T.; LaMarca, B. Low Dose of IL-2 Normalizes Hypertension and Mitochondrial Function in the RUPP Rat Model of Placental Ischemia. Cells 2021, 10, 2797. [Google Scholar] [CrossRef]
- Deer, E.; Jones, J.; Cornelius, D.C.; Comley, K.; Herrock, O.; Campbell, N.; Fitzgerald, S.; Ibrahim, T.; LaMarca, B.; Amaral, L.M. Progesterone Induced Blocking Factor Reduces Hypertension and Placental Mitochondrial Dysfunction with in Response to sFlt-1 during Pregnancy. Cells 2021, 10, 2817. [Google Scholar] [CrossRef]
- Jiang, Z.; Zou, Y.; Ge, Z.; Zuo, Q.; Huang, S.Y.; Sun, L. A Role of sFlt-1 in Oxidative Stress and Apoptosis in Human and Mouse Pre-Eclamptic Trophoblasts. Biol. Reprod. 2015, 93, 73. [Google Scholar] [CrossRef] [PubMed]
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Tuohey, L.; Parry, L.J.; Senadheera, S.; Illanes, S.E.; Kaitu’u-Lino, T.J.; Tong, S. Metformin as a prevention and treatment for preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am. J. Obstet. Gynecol. 2016, 214, 356.e1–356.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastie, R.; Brownfoot, F.C.; Pritchard, N.; Hannan, N.J.; Cannon, P.; Nguyen, V.; Palmer, K.; Beard, S.; Tong, S.; Kaitu’u-Lino, T.J. EGFR (Epidermal Growth Factor Receptor) Signaling and the Mitochondria Regulate sFlt-1 (Soluble FMS-Like Tyrosine Kinase-1) Secretion. Hypertension 2019, 73, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Parrish, M.R.; Murphy, S.R.; Rutland, S.; Wallace, K.; Wenzel, K.; Wallukat, G.; Keiser, S.; Ray, L.F.; Dechend, R.; Martin, J.N.; et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am. J. Hypertens. 2010, 23, 911–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cochemé, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [Green Version]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Teran, E.; Hernández, I.; Tana, L.; Teran, S.; Galaviz-Hernandez, C.; Sosa-Macías, M.; Molina, G.; Calle, A. Mitochondria and Coenzyme Q10 in the Pathogenesis of Preeclampsia. Front. Physiol. 2018, 9, 1561. [Google Scholar] [CrossRef]
- Xu, X.; Pan, J.R.; Zhang, Y.Z. CoQ10 alleviate preeclampsia symptoms by enhancing the function of mitochondria in the placenta of pregnant rats with preeclampsia. Hypertens. Pregnancy 2019, 38, 217–222. [Google Scholar] [CrossRef]
- Schulz, C.; Obermüller-Jevic, U.C.; Hasselwander, O.; Bernhardt, J.; Biesalski, H.K. Comparison of the relative bioavailability of different coenzyme Q10 formulations with a novel solubilizate (Solu Q10). Int. J. Food Sci. Nutr. 2006, 57, 546–555. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Beyer, R.E. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem. Cell Biol. 1992, 70, 390–403. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [Green Version]
- Junior, R.F.R.; Dabkowski, E.R.; Shekar, K.C.; KA, O.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free. Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Gottwald, E.M.; Duss, M.; Bugarski, M.; Haenni, D.; Schuh, C.D.; Landau, E.M.; Hall, A.M. The targeted anti-oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue. Physiol. Rep. 2018, 6, e13667. [Google Scholar] [CrossRef]
- Rehman, H.; Liu, Q.; Krishnasamy, Y.; Shi, Z.; Ramshesh, V.K.; Haque, K.; Schnellmann, R.G.; Murphy, M.P.; Lemasters, J.J.; Rockey, D.C.; et al. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 14–27. [Google Scholar]
- Phillips, T.J.; Scott, H.; Menassa, D.A.; Bignell, A.L.; Sood, A.; Morton, J.S.; Akagi, T.; Azuma, K.; Rogers, M.F.; Gilmore, C.E.; et al. Treating the placenta to prevent adverse effects of gestational hypoxia on fetal brain development. Sci. Rep. 2017, 7, 9079. [Google Scholar] [CrossRef] [Green Version]
- Nuzzo, A.M.; Camm, E.J.; Sferruzzi-Perri, A.N.; Ashmore, T.J.; Yung, H.W.; Cindrova-Davies, T.; Spiroski, A.M.; Sutherland, M.R.; Logan, A.; Austin-Williams, S.; et al. Placental Adaptation to Early-Onset Hypoxic Pregnancy and Mitochondria-Targeted Antioxidant Therapy in a Rodent Model. Am. J. Pathol. 2018, 188, 2704–2716. [Google Scholar] [CrossRef] [Green Version]
- Sedeek, M.; Gilbert, J.S.; LaMarca, B.B.; Sholook, M.; Chandler, D.L.; Wang, Y.; Granger, J.P. Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. Am. J. Hypertens. 2008, 21, 1152–1156. [Google Scholar] [CrossRef] [Green Version]
- Dhillion, P.; Wallace, K.; Herse, F.; Scott, J.; Wallukat, G.; Heath, J.; Mosely, J.; Martin, J.N., Jr.; Dechend, R.; LaMarca, B. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R353–R358. [Google Scholar] [CrossRef] [Green Version]
- Parrish, M.R.; Wallace, K.; Tam, K.B.T.; Herse, F.; Weimer, A.; Wenzel, K.; Wallukat, G.; Ray, L.F.; Arany, M.; Cockrell, K.; et al. Hypertension in response to AT1-AA: Role of reactive oxygen species in pregnancy-induced hypertension. Am. J. Hypertens. 2011, 24, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.S.; Weydert, C.J.; Lazartigues, E.; Kutschke, W.J.; Kienzle, M.F.; Leach, J.E.; Sharma, J.A.; Sharma, R.V.; Davisson, R.L. Chronic tempol prevents hypertension, proteinuria, and poor feto-placental outcomes in BPH/5 mouse model of preeclampsia. Hypertension 2008, 51, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, C.; Bauer, T.; Surek, B.; Schömig, E.; Gründemann, D. Cyanobacteria produce high levels of ergothioneine. Food Chem. 2011, 129, 1766–1769. [Google Scholar] [CrossRef]
- Deiana, M.; Rosa, A.; Casu, V.; Piga, R.; Dessì, M.A.; Aruoma, O.I. L-ergothioneine modulates oxidative damage in the kidney and liver of rats in vivo: Studies upon the profile of polyunsaturated fatty acids. Clin. Nutr. 2004, 23, 183–193. [Google Scholar] [CrossRef]
- Bedirli, A.; Sakrak, O.; Muhtaroglu, S.; Soyuer, I.; Guler, I.; Erdogan, A.R.; Sozuer, E.M. Ergothioneine pretreatment protects the liver from ischemia-reperfusion injury caused by increasing hepatic heat shock protein 70. J. Surg. Res. 2004, 122, 96–102. [Google Scholar] [CrossRef]
- Lamhonwah, A.M.; Tein, I. Novel localization of OCTN1, an organic cation/carnitine transporter, to mammalian mitochondria. Biochem. Biophys. Res. Commun. 2006, 345, 1315–1325. [Google Scholar] [CrossRef]
- Morillon, A.C.; Williamson, R.D.; Baker, P.N.; Kell, D.B.; Kenny, L.C.; English, J.A.; McCarthy, F.P.; McCarthy, C. Effect of L-Ergothioneine on the metabolic plasma profile of the RUPP rat model of pre-eclampsia. PLoS ONE 2020, 15, e0230977. [Google Scholar] [CrossRef]
- Cheah, I.K.; Tang, R.M.; Yew, T.S.; Lim, K.H.; Halliwell, B. Administration of Pure Ergothioneine to Healthy Human Subjects: Uptake, Metabolism, and Effects on Biomarkers of Oxidative Damage and Inflammation. Antioxid. Redox Signal. 2017, 26, 193–206. [Google Scholar] [CrossRef]
- Marone, P.A.; Trampota, J.; Weisman, S. A Safety Evaluation of a Nature-Identical l-Ergothioneine in Sprague Dawley Rats. Int. J. Toxicol. 2016, 35, 568–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, R.; Spézia, F.; Papineau, D.; Sabadie, C.; Erdelmeier, I.; Moutet, M.; Yadan, J.C. Reproductive safety evaluation of L-Ergothioneine. Food Chem. Toxicol. 2015, 80, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, M.V.; Indart, A.; Aruoma, O.I.; Viana, M.; Bonet, B. Effects of ergothioneine on diabetic embryopathy in pregnant rats. Food Chem. Toxicol. 2002, 40, 1751–1755. [Google Scholar] [CrossRef]
- Turner, E.; Brewster, J.A.; Simpson, N.A.; Walker, J.J.; Fisher, J. Imidazole-based erythrocyte markers of oxidative stress in preeclampsia—An NMR investigation. Reprod. Sci. 2009, 16, 1040–1051. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaka, R.; Deer, E.; LaMarca, B. Is Mitochondrial Oxidative Stress a Viable Therapeutic Target in Preeclampsia? Antioxidants 2022, 11, 210. https://doi.org/10.3390/antiox11020210
Vaka R, Deer E, LaMarca B. Is Mitochondrial Oxidative Stress a Viable Therapeutic Target in Preeclampsia? Antioxidants. 2022; 11(2):210. https://doi.org/10.3390/antiox11020210
Chicago/Turabian StyleVaka, Ramana, Evangeline Deer, and Babbette LaMarca. 2022. "Is Mitochondrial Oxidative Stress a Viable Therapeutic Target in Preeclampsia?" Antioxidants 11, no. 2: 210. https://doi.org/10.3390/antiox11020210
APA StyleVaka, R., Deer, E., & LaMarca, B. (2022). Is Mitochondrial Oxidative Stress a Viable Therapeutic Target in Preeclampsia? Antioxidants, 11(2), 210. https://doi.org/10.3390/antiox11020210