
N-Acetyl-Cysteine: Modulating the Cysteine Redox Proteome in Neurodegenerative Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
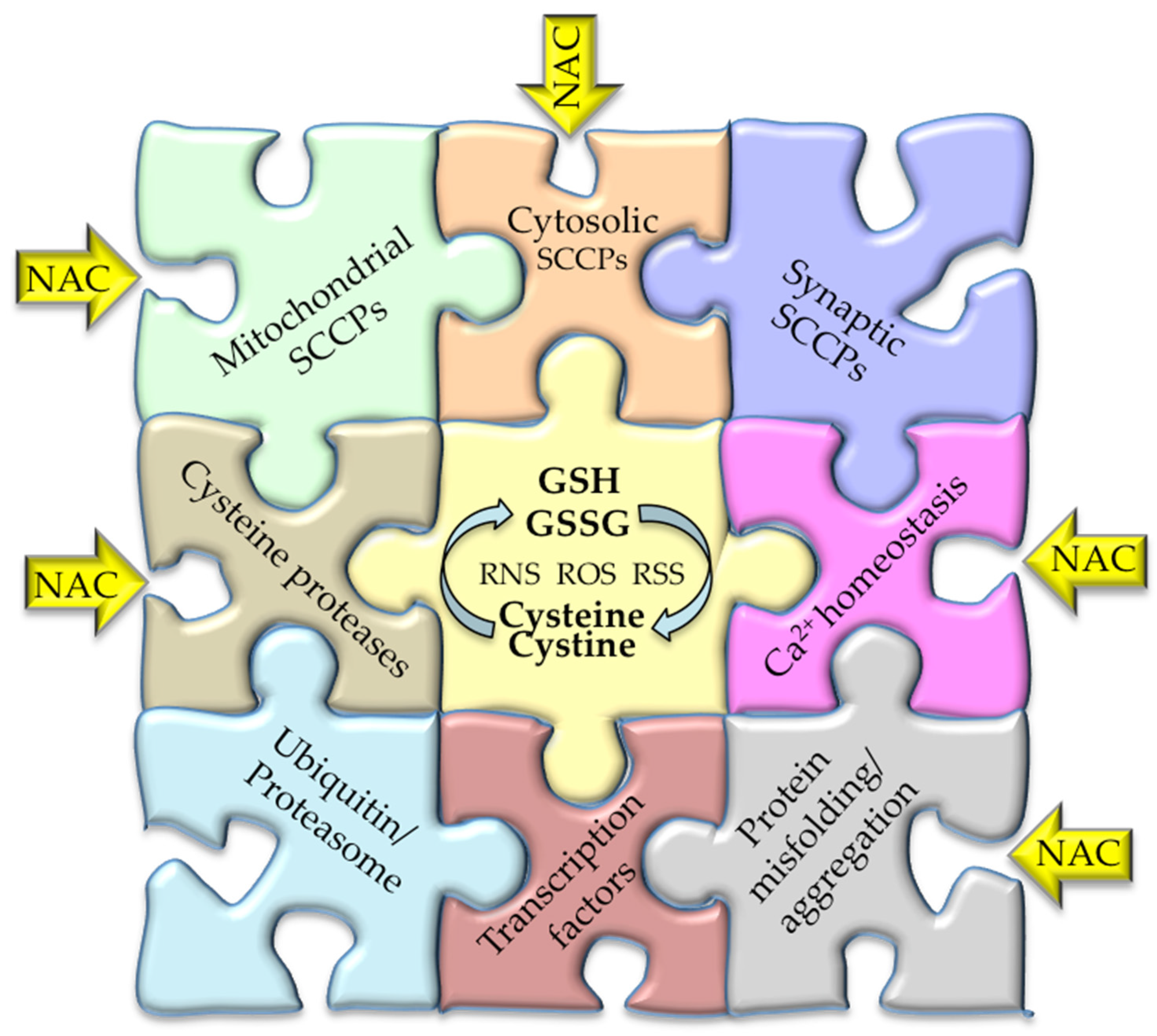
2. The Sensitive-Cysteine Redox Proteome (Cysteinet)
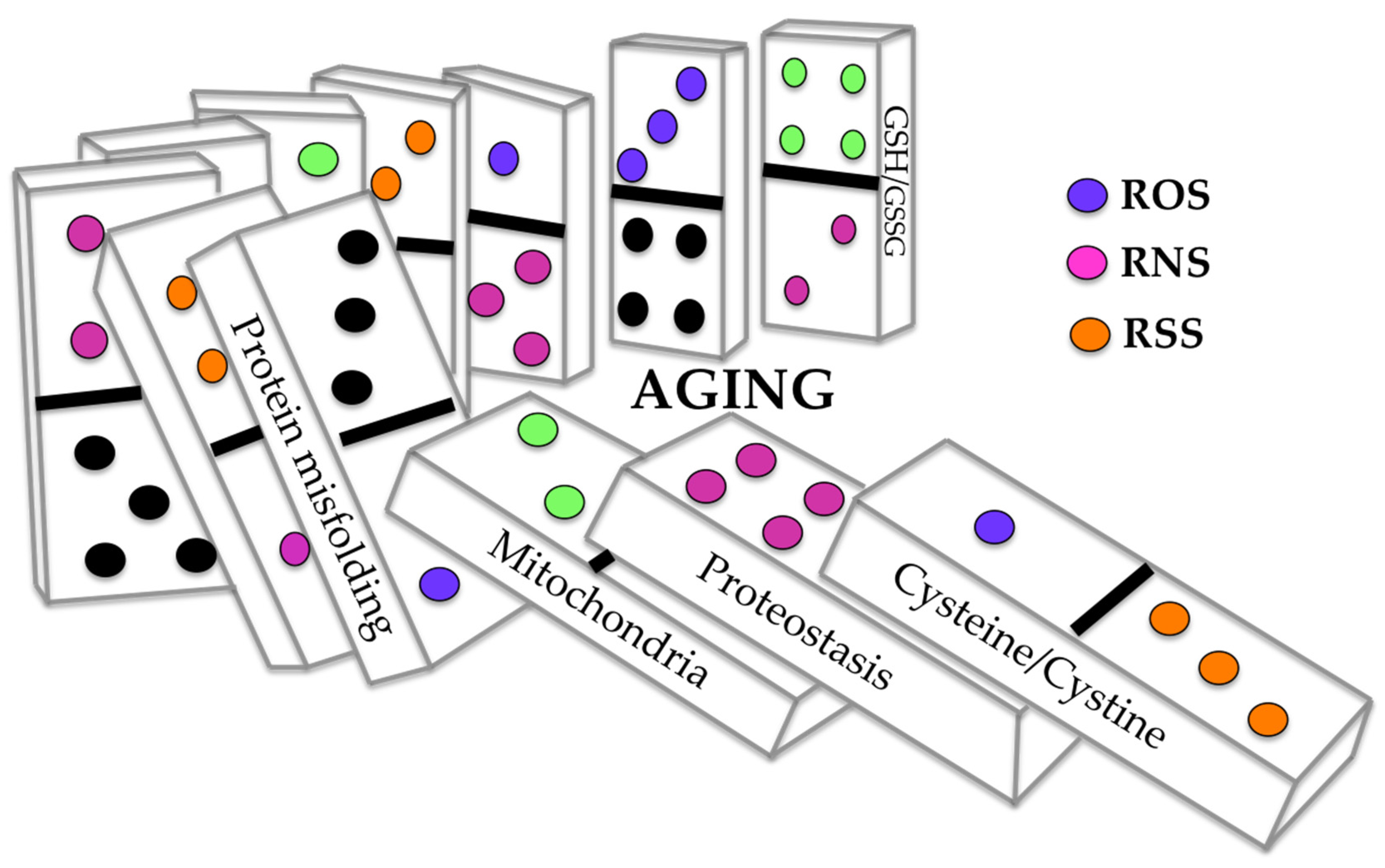
3. The Sensitive-Cysteine Redox Proteome (Cysteinet) in Aging
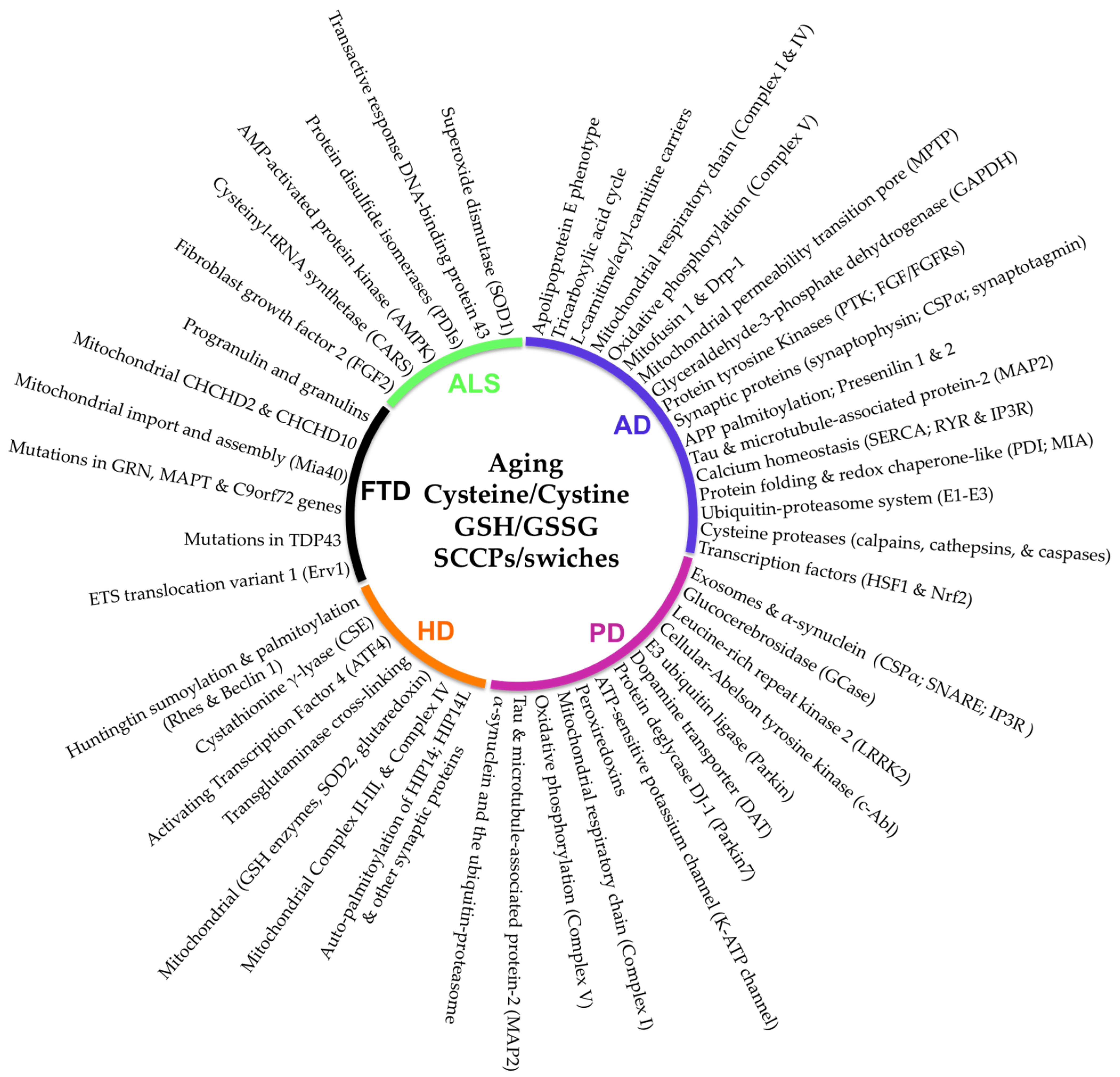
4. The Sensitive-Cysteine Redox Proteome (Cysteinet) in Neurodegenerative Diseases
5. Alzheimer’s Disease
5.1. Cysteinet Deregulation in AD
5.1.1. Apolipoprotein E (ApoE)
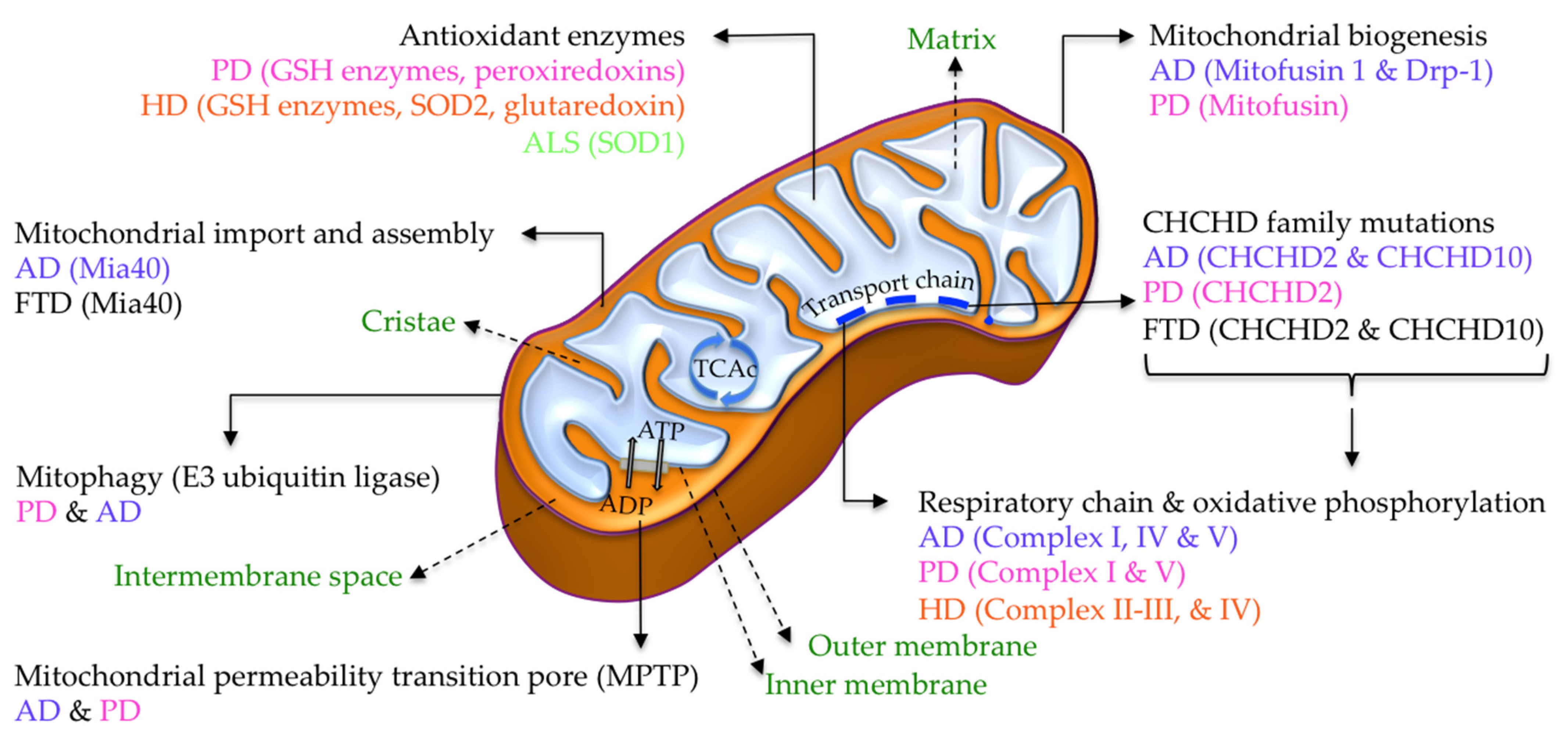
5.1.2. Mitochondrial SCCPs
5.1.3. Cytosolic SCCPs
5.1.4. Protein Tyrosine Kinases (PTK)
5.1.5. Synaptic SCCPs
5.1.6. APP Processing, Aβ Aggregation
5.1.7. Microtubule Associated Protein Tau (MAPT)
5.1.8. SCCPs in Calcium Homeostasis
5.1.9. SCCPs and Misfolding
5.1.10. SCCPs in the Ubiquitin-Proteasome
5.1.11. Cysteine Proteases
5.1.12. Transcription Factors as SCCPs
6. Parkinson’s Disease
6.1. Cysteinet Deregulation in PD
6.1.1. DnaJ Homolog C (DNAJC) Family
6.1.2. Glucocerebrosidase (GCase)
6.1.3. Leucine-Rich Repeat Kinase 2 (LRRK2)
6.1.4. Cellular-Abelson Tyrosine Kinase (c-Abl)
6.1.5. Parkin
6.1.6. Dopamine Transporter (DAT)
6.1.7. Protein Deglycase DJ-1 (Parkin7)
6.1.8. ATP-Sensitive Potassium Channel (K-ATP Channel)
6.1.9. Antioxidant Enzymatic System
6.1.10. Mitochondrial Respiratory Chain and Oxidative Phosphorylation
6.1.11. Microtubule-Associated Protein Tau and MAP2
6.1.12. α-Synuclein and the Ubiquitin-Proteasome
7. Amyotrophic Lateral Sclerosis
7.1. Cysteinet Deregulation in ALS
7.1.1. Superoxide Dismutase
7.1.2. Transactive Response DNA-Binding Protein 43 (TDP43)
7.1.3. Protein Disulfide Isomerases (PDIs)
7.1.4. AMP-Activated Protein Kinase (AMPK)
7.1.5. Fibroblast Growth Factor 2 (FGF2)
8. Huntington’s Disease
8.1. Cysteinet Deregulation in HD
8.1.1. Rhes and Beclin 1
8.1.2. Cystathionine γ-Lyase (CSE)
8.1.3. Activating Transcription Factor 4 (ATF4)
8.1.4. Transglutaminase Cross-Linking
8.1.5. Mitochondrial Antioxidant Enzymes
8.1.6. Mitochondrial Respiratory Enzymes
8.1.7. Palmitoylation
9. Frontotemporal Dementia
9.1. Cysteinet Deregulation in FTD
9.1.1. Progranulin and Granulins
9.1.2. Mitochondrial SCCPs
10. Modulation of Cysteinet by N-Acetyl-Cysteine (NAC)
10.1. NAC in Brain Aging and Neurodegenerative Diseases
10.1.1. Preclinical and Clinical Studies of NAC in AD
10.1.2. Preclinical and Clinical Studies of NAC in PD
10.1.3. Preclinical and Clinical Studies of NAC in HD
10.1.4. Preclinical and Clinical Studies of NAC in ALS
11. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Martinez-Banaclocha, M. Cellular cysteine network (Cysteinet): Pharmacological intervention in brain aging and neurodegenerative diseases. In Frontiers in Clinical Drug Research-Central Nervous System; Atta-ur-Rahman, Ed.; Bentham Science Publishers: Al Sharjah, United Arab Emirates, 2016; Volume 2, pp. 105–172. [Google Scholar]
- Martinez-Banaclocha, M. Cysteine network (CYSTEINET) dysregulation in Parkinson’s disease: Role of N-acetylcysteine. Curr. Drug Metab. 2016, 17, 368–385. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. N-acetylcysteine: A natural antidote for Alzheimer’s disease. Alzheimers Dis. Dement. 2016, 1, 4–15. [Google Scholar]
- Martinez-Banaclocha, M. Potential role of N-acetyl-cysteine in the cysteine proteome in Parkinson’s sisease? Clin. Pharmacol. Ther. 2020, 107, 1055. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. Proteomic complexity in Parkinson’s disease: A redox signaling perspective of the pathophysiology and progression. Neuroscience 2021, 453, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Feleciano, D.R.; Kirstein, J. Collapse of redox homeostasis during aging and stress. Mol. Cell. Oncol. 2015, 3, e1091060. [Google Scholar] [CrossRef] [Green Version]
- Radzinski, M.; Oppenheim, T.; Metanis, N.; Reichmann, D. The cys sense: Thiol redox switches mediate life cycles of celular proteins. Biomolecules 2021, 11, 469. [Google Scholar] [CrossRef]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Brandvold, K.R.; Morimoto, R.I. The chemical biology of molecular chaperones—Implications for modulation of proteostasis. J. Mol. Biol. 2015, 427, 2931–2947. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Fu, L.; Liu, K.; Tian, C.; Wu, Z.; Jung, Y.; Ferreira, R.B.; Carroll, K.S.; Blackwell, T.K.; Yang, J. Global profiling of distinct cysteine redox forms reveals wide-ranging redox regulation in C. elegans. Nat. Commun. 2021, 12, 1415. [Google Scholar] [CrossRef] [PubMed]
- Weerapana, E.; Wang, C.; Simon, G.M.; Richter, F.; Khare, S.; Dillon, M.B.; Bachovchin, D.A.; Mowen, K.; Baker, D.; Cravatt, B.F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef]
- Go, Y.-M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [Green Version]
- Benaroudj, N.; Zwickl, P.; Seemüller, E.; Baumeister, W.; Goldberg, A.L. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell 2003, 11, 69–78. [Google Scholar] [CrossRef]
- Haas, A.L.; Rose, I.A. The mechanism of ubiquitin activating enzyme. J. Biol. Chem. 1982, 257, 10329–10337. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine cesidues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular mechanism of cellular oxidative stress sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef] [Green Version]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernandez-Cardenas, L.P.; Blackwell, T.K. Cysteine sulfenylation directs IRE-1 to activate the SKN-1/Nrf2 antioxidant response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Knuesting, J.; Scheibe, R. Small molecules govern thiol redox switches. Trends Plant Sci. 2018, 23, 769–782. [Google Scholar] [CrossRef]
- Petrova, B.; Liu, K.; Tian, C.; Kitaoka, M.; Freinkman, E.; Yang, J.; Orr-Weaver, T.L. Dynamic redox balance directs the oocyte-to-embryo transition via developmentally controlled reactive cysteine changes. Proc. Natl. Acad. Sci. USA 2018, 115, E7978–E7986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Banaclocha, M. N-acetyl-cysteine in schizophrenia: Potential role on the sensitive cysteine proteome. Curr. Med. Chem. 2020, 27, 6424–6439. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. N-acetylcysteine in psychiatric disorders: Possible role of cysteinet deregulation. Inter. Neuropsy. Dis. J. 2018, 12, 1–6. [Google Scholar] [CrossRef]
- Barja, G. Endogenous oxidative stress: Relationship to aging, longevity and caloric restriction. Ageing Res. Rev. 2002, 1, 397–411. [Google Scholar] [CrossRef]
- Bokov, A.; Chaudhuri, A.; Richardson, A. The role of oxidative damage and stress in aging. Mech. Ageing Dev. 2004, 125, 811–826. [Google Scholar] [CrossRef]
- Perez, V.I.; Bokov, A.; Van Remmen, H.; Mele, J.; Ran, Q.; Ikeno, Y.; Richardson, A. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 2009, 1790, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.P.; Mody, V.C., Jr.; Carlson, J.L.; Lynn, M.J.; Sternberg, P., Jr. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic. Biol. Med. 2002, 33, 1290–1300. [Google Scholar] [CrossRef]
- Nuttall, S.L.; Martin, U.; Sinclair, A.J.; Kendall, M.J. Glutathione: In sickness and in health. Lancet 1998, 351, 645–646. [Google Scholar] [CrossRef]
- Liedhegner, E.A.S.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases—Focus on S-glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquel, J.; Ferrándiz, M.L.; De Juan, E.; Sevilla, I.; Martinez-Banaclocha, M. N-acetylcysteine protecs against age-related decline of oxidative phosphorylation in liver mitochondria. Eur. J. Pharmacol. 1995, 292, 333–335. [Google Scholar]
- McDonagh, B.; Sakellariou, G.K.; Smith, N.T.; Brownridge, P.; Jackson, M.J. Differential cysteine labelling and global label-free proteomics reveals an altered metabolic state in skeletal muscle aging. J. Proteome Res. 2014, 13, 5008–5021. [Google Scholar] [CrossRef] [PubMed]
- Freer, R.; Sormanni, P.; Vecchi, G.; Ciryam, P.; Dobson, C.M.; Vendruscolo, M. A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease. Sci. Adv. 2016, 2, e1600947. [Google Scholar] [CrossRef] [Green Version]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Ersoz, A.; Butterfield, D.A.; Mattson, M.P. Beneficial effects of dietary restriction on cerebral cortical synaptic terminals: Preservation of glucose transport and mitochondrial function after exposure to amyloid β-peptide and oxidative and metabolic insults. J. Neurochem. 2000, 75, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martell, J.; Seo, Y.; Bak, D.W.; Kingsley, S.F.; Tissenbaum, H.A.; Weerapana, E. Global cysteine-reactivity profiling during impaired insulin/IGF-1 signaling in C. elegans identifies uncharacterized mediators of longevity. Cell Chem. Biol. 2016, 23, 955–966. [Google Scholar] [CrossRef] [Green Version]
- Levine, C.G.; Mitra, D.; Sharma, A.; Smith, C.L.; Hegde, R.S. The efficiency of protein compartmentalization into the secretory pathway. Mol. Biol. Cell 2005, 16, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirstein, J.; Morito, D.; Kakihana, T.; Sugihara, M.; Minnen, A.; Hipp, M.S.; Nussbaum-Krammer, C.; Hartl, F.U.; Nagata, K.; Morimoto, R.I. Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 2015, 34, 2334–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Honer, W.G.; Masliah, E. Synapse alterations in the hippocampal-entorhinal formation in Alzheimer’s disease with and without Lewy body disease. Brain Res. 1994, 667, 24–32. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Abeta oligomer- induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. β-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef] [Green Version]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; Regan, C.M.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2006, 14, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddie, K.G.; Carroll, K.S. Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 2008, 12, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Fomenko, D.E.; Xing, W.; Adair, B.M.; Thomas, D.J.; Gladyshev, V.N. High-throughput identification of catalytic redox-active cysteine residues. Science 2007, 315, 387–389. [Google Scholar] [CrossRef] [Green Version]
- Pace, N.J.; Weerapana, E. Diverse functional roles of reactive cysteines. ACS Chem. Biol. 2013, 8, 283–296. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Kindy, M.; LaDu, M.J. Apolipoprotein E and Alzheimer’s disease: The protective effects of ApoE2 and E3. J. Alzheimer’s Dis. 2002, 4, 145–154. [Google Scholar] [CrossRef]
- Frieden, C.; Garai, K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8913–8918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, W.A.; Chan, S.L.; Mattson, M.P. A mechanism for the neuroprotective effect of apolipoprotein E: Isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 2000, 74, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Abrams, A.J.; Farooq, A.; Wang, G. S-nitrosylation of ApoE in Alzheimer’s disease. Biochemistry 2011, 50, 3405–3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Heijmsns, B.T.; Westendorp, R.G.; Slagboom, P.E. Common gene variants, mortality and extreme longevity in humans. Exp. Gerontol. 2000, 35, 865–877. [Google Scholar] [CrossRef]
- Katzman, R. Apolipoprotein E and Alzheimer’s disease. Curr. Opin. Neurobiol. 1994, 4, 703–707. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [Green Version]
- Aleshkov, S.B.; Li, X.; Lavrentiadou, S.N.; Zannis, V.I. Contribution of cysteine 158, the glycosylation site theonine 194, the amino-and carboxy-terminal domains of apolipoprotein E in the binding to amyloid peptide β (1–40). Biochemistry 1999, 38, 8918–8925. [Google Scholar] [CrossRef]
- Risør, M.W.; Poulsen, E.T.; Thomsen, L.R.; Dyrlund, T.F.; Nielsen, T.A.; Nielsen, N.C.; Sanggaard, K.W.; Enghild, J.J. The autolysis of human HtrA1 is governed by the redox state of its N-terminal domain. Biochemistry 2014, 53, 3851–3857. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.W.; Sunico, C.R.; Nakamura, T.; Lipton, S.A. Redox regulation of protein function via cysteine S-nitrosylation and its relevance to neurodegenerative diseases. Int. J. Cell Biol. 2012, 2012, 463756. [Google Scholar]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.X.; Yan, S.S. Role of mitochondrial amyloid-β in Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S569–S578. [Google Scholar] [CrossRef] [Green Version]
- García-Escudero, V.; Martín-Maestro, P.; Perry, G.; Avila, J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev. 2013, 2013, 162152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dua, H.; Guoa, L.; Yana, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Guo, L.; Lu, L.; Sun, H.; Shao, M.; Beck, S.J.; Li, L.; Ramachandran, J.; Du, Y.; Du, H. Synaptosomal mitochondrial dysfunction in 5xFAD mouse model of Alzheimer’s disease. PLoS ONE 2016, 11, e0150441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Quantitative proteomics of synaptic and nonsynaptic mitochondria: Insights for synaptic mitochondrial vulnerability. J. Proteome Res. 2014, 13, 2620–2636. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2013, 2, 123–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Kish, S.J. Mitochondria in Alzheimer’s disease. Int. Rev. Neurobiol. 2002, 53, 341–385. [Google Scholar]
- Cardoso, S.M.; Proenca, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Mahr, N.J.; Filley, C.M.; Parks, J.K.; Hughes, D.; Young, D.A.; Cullum, C.M. Reduced platelet cytochrome c oxidase activity in Alzheimer’s disease. Neurology 1994, 44, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragicevic, N.; Mamcarz, M.; Zhu, Y.; Buzzeo, R.; Tan, J.; Arendash, G.W.; Bradshaw, P.C. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimer’s Dis. 2010, 20, S535–S550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011, 18, 1478–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics fusion, fission, movement, and mitophagy in neurodegenerative diseases. Human Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Redpath, C.J.; Khalil, M.B.; Drozdzal, G.; Radisic, M.; McBride, H.M. Mitochondrial hyperfusion during oxidative stress is coupled to a dysregulation in calcium handling within a C2C12 cell model. PLoS ONE 2013, 8, e69165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroga, C.S.; Almeida, A.S.; Martel, C.; Queiroga, C.S.; Almeida, A.S.; Martel, C.; Brenner, C.; Alves, P.M.; Vieira, H.L. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 2010, 285, 17077–17088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonagh, B.; Martinez-Acedo, P.; Vazquez, J. Application of iTRAQ reagents to relatively quantify the reversible redox state of cysteine residues. Int. J. Proteom. 2012, 2012, 514847. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer disease: Many pathways to neurodegeneration. J. Alzheimer’s Dis. 2010, 20, 369–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell. Signal. 2011, 23, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 2012, 39, 674–679. [Google Scholar] [CrossRef]
- Li, S.; Whorton, A.R. Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch. Biochem. Biophys. 2003, 410, 269–279. [Google Scholar] [CrossRef]
- Kemble, D.J.; Sun, G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc. Natl. Acad. Sci. USA 2009, 106, 5070–5075. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.W.; Saitoh, T. Changes in protein kinases in brain aging and Alzheimer’s disease. Implications for drug therapy. Drugs Aging 1995, 6, 136–149. [Google Scholar] [CrossRef]
- Ferrer, I.; Martí, E. Distribution of fibroblast growth factor receptor-1 (FGFR-1) and FGFR-3 in the hippocampus of patients with Alzheimer’s disease. Neurosci. Lett. 1998, 240, 139–142. [Google Scholar] [CrossRef]
- Thorns, V.; Masliah, E. Evidence for neuroprotective effects of acidic fibroblast growth factor in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.A.; Eren-Kocak, E.; Inui, E.G.; Watson, S.J.; Akil, H. Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Semin. Cell Dev. Biol. 2015, 53, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimaschewski, L.; Claus, P. Fibroblast growth factor signalling in the diseased nervous system. Mol. Neurobiol. 2021, 58, 3884–3902. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.S.; d’Orange, M.; Troakes, C.; Shurovi, B.N.; Engmann, O.; Noble, W.; Hortobágyi, T.; Giese, K.P. Evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer inverted question marks disease. Mol. Brain 2015, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, R.D.; Morgan, A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 2015, 40, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, L.A.; Daniel, S.E.; Wilcock, G.K.; Love, S. Frontal cortical synaptophysin in Lewy body diseases: Relation to Alzheimer’s disease and dementia. J. Neurol. Neurosurg. Psychiatry 1998, 64, 653–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sze, C.; Troncoso, J.C.; Kawas, C.; Mouton, P.; Price, D.L.; Martin, L.J. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 933944. [Google Scholar] [CrossRef] [Green Version]
- Johnston, P.A.; Südhof, T.C. The multisubunit structure of synaptophysin. J. Biol. Chem. 1990, 265, 8869–8873. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Jao, C.C.; Der-Sarkissian, A.; Chen, J.; Langen, R. Structure of membrane-bound α-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA 2004, 101, 8331–8336. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Freed, C.R. Tyrosine-to-cysteine modification of human α-synuclein enhances protein aggregation and cellular toxicity. J. Biol. Chem. 2004, 279, 10128–10135. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. Synaptotagmins: Why so many? J. Biol. Chem. 2002, 277, 7629–7632. [Google Scholar] [CrossRef] [Green Version]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, M.; Alattia, J.-R.; Lemmin, T.; Lehal, R.; Fligier, A.; Houacine, J.; Hussain, I.; Radtke, F.; Dal Peraro, M.; Beher, D.; et al. Alzheimer’s disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat. Commun. 2013, 4, 2246. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Burdick, D.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. Neurodegeneration induced by β-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 1993, 13, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Laudon, H.; Hansson, E.M.; Melen, K.; Laudon, H.; Hansson, E.M.; Melén, K.; Bergman, A.; Farmery, M.R.; Winblad, B.; Lendahl, U.; et al. A nine transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005, 25, 25. [Google Scholar] [CrossRef] [Green Version]
- Kornilova, A.Y.; Kim, J.; Laudon, H.; Wolfe, M.S. Deducing the transmembrane domain organization of presenilin-1 in γ-secretase by cysteine disulfide crosslinking. Biochemistry 2006, 45, 7598–7604. [Google Scholar] [CrossRef] [Green Version]
- Tedde, A.; Nacmias, B.; Ciantelli, M.; Forleo, P.; Cellini, E.; Bagnoli, S.; Piccini, C.; Caffarra, P.; Ghidoni, E.; Paganini, M.; et al. Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch. Neurol. 2003, 60, 1541–1544. [Google Scholar] [CrossRef]
- Tolia, A.; Chavez-Gutierrez, L.; Strooper, B.D. Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the γ-secretase complex. J. Biol. Chem. 2006, 281, 27633–27642. [Google Scholar] [CrossRef] [Green Version]
- Payne, A.J.; Gerdes, B.C.; Naumchuk, Y.; McCalley, A.E.; Kaja, S.; Coulen, P. Presenilins regulate the cellular activity of ryanodine receptors differentially through isotype-specific N-terminal cysteines. Exp. Neurol. 2013, 250, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef] [Green Version]
- Barnham, K.J.; McKinstry, W.J.; Multhaup, G.; Galatis, D.; Morton, C.J.; Curtain, C.C.; Williamson, N.A.; White, A.R.; Hinds, M.G.; Norton, R.S.; et al. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain: A regulator of neuronal copper homeostasis. J. Biol. Chem. 2003, 278, 17401–17407. [Google Scholar] [CrossRef] [Green Version]
- Schweers, O.; Mandelkow, E.; Biernat, J.; Mandelkow, E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein T controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. USA 1995, 92, 8463–8467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landino, L.M.; Skreslet, T.E.; Alston, J.A. Cysteine oxidation of Tau and microtubule-associated protein-2 by peroxynitrite. J. Biol. Chem. 2004, 279, 35101–35105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegmann, S.; Medalsy, I.D.; Mandelkow, E.; Müller, D.J. The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectric brush. Proc. Natl. Acad. Sci. USA 2012, 109, E313–E321. [Google Scholar]
- Soeda, Y.; Yoshikawa, M.; Almeida, O.F.; Sumioka, A.; Maeda, S.; Osada, H.; Kondoh, Y.; Saito, A.; Miyasaka, T.; Kimura, T.; et al. Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat. Commun. 2015, 6, 10216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, O.; Gant, J.C.; Landfield, P.W. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: Minding the store. Aging Cell 2007, 6, 307–317. [Google Scholar] [CrossRef]
- Berridge, M.J. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013, 7, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Laferla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Xu, L.; Eu, J.P.; Stamler, J.S.; Meissner, G. Nitric oxide, NOC-12, and S-nitrosoglutathione modulate the skeletal muscle calcium release channel/ryanodine receptor by different mechanisms. An allosteric function for O2 in S-nitrosylation of the channel. J. Biol. Chem. 2003, 278, 8184–8189. [Google Scholar] [CrossRef] [Green Version]
- Bánsághi, S.; Golenár, T.; Madesh, M.; Csordás, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnóczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Rossi, A.M.; Riley, A.M.; Potter, B.V.; Taylor, C.W. Subtype-selective regulation of IP3 receptors by thimerosal via cysteine residues within the IP3-binding core and suppressor domain. Biochem. J. 2013, 451, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurumatani, T.; Fastboma, J.; Bonkalea, W.L.; Bogdanovic, N.; Winblad, B.; Ohm, T.G.; Cowburn, R.F. Loss of inositol 1,4,5-trisphosphate receptor sites and decreased PKC levels correlate with staging of Alzheimer’s disease neurofibrillary pathology. Brain Res. 1998, 796, 209–221. [Google Scholar] [CrossRef]
- Tonga, X.Y.; Yinga, J.; Pimentel, D.R.; Trucillo, M.; Adachi, T.; Cohen, R.A. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J. Mol. Cell. Cardiol. 2008, 44, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Sharov, V.S.; Dremina, E.S.; Galeva, N.A. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC–electrospray-tandem MS: Selective protein oxidation during biological aging. Biochem. J. 2006, 394, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Qina, M.; Wanga, W.; Thirumalai, D. Protein folding guides disulfide bond formation. Proc. Natl. Acad. Sci. USA 2015, 112, 11241–11246. [Google Scholar] [CrossRef] [Green Version]
- Haase-Pettingell, C.; Betts, S.; Raso, S.W.; Stuart, L.; Robinson, A.; King, J. Role for cysteine residues in the in vivo folding and assembly of the phage P22 tail spike. Protein Sci. 2001, 10, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Winklhofer, K.F.; Tatzeltand, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef]
- Shin, H.-C. Protein folding, misfolding, and refolding of therapeutic proteins. Biotechnol. Bioprocess Eng. 2001, 6, 237–243. [Google Scholar] [CrossRef]
- Creighton, T.E. Disulfide bonds as probes of protein folding pathways. Methods Enzymol. 1986, 131, 83–106. [Google Scholar]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-nitrosylated protein disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef]
- Riemer, J.; Bulleid, N.; Herrmann, J.M. Disulfide formation in the ER and mitochondria: Two solutions to a common process. Science 2009, 324, 1284–1287. [Google Scholar] [CrossRef]
- Koch, J.R.; Schmid, F.X. Mia40 targets cysteines in a hydrophobic environment to direct oxidative protein folding in the mitochondria. Nat. Commun. 2014, 5, 3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Mata, R.; Gao, Y.S.; Sztul, E. Hassles with taking out the garbage: Aggravating aggresomes. Traffic 2002, 3, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Lecke, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Jahngen-Hodge, J.; Obin, M.S.; Gong, X.; Shang, F.; Nowell, T.R., Jr.; Gong, J.; Abasi, H.; Blumberg, J.; Taylor, A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J. Biol. Chem. 1997, 272, 28218–28226. [Google Scholar] [CrossRef] [Green Version]
- Doris, K.S.; Rumsby, E.L.; Gong, B.A. Oxidative stress responses involve oxidation of a conserved ubiquitin pathway enzyme. Mol. Cell. Biol. 2012, 32, 4472–4481. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Huang, H.C.; Jiang, Z.F. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef]
- Upadhya, S.C.; Hegde, A.N. Role of the ubiquitin proteasome system in Alzheimer’s disease. BMC Biochem. 2007, 8, S12. [Google Scholar] [CrossRef] [Green Version]
- Hegde, A.N.; Smith, S.G.; Duke, L.M.; Pourquoi, A.; Vaz, S. Perturbations of ubiquitin-proteasome-mediated proteolysis in aging and Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 324. [Google Scholar] [CrossRef]
- Harris, L.D.; Jasem, S.; Licchesi, J.D.F. The ubiquitin system in Alzheimer’s disease. Adv. Exp. Med. Biol. 2020, 1233, 195–221. [Google Scholar]
- Fischer, D.F.; van Dijk, R.; van Tijn, P.; Hobo, B.; Verhage, M.C.; van der Schors, R.C.; Li, K.W.; van Minnen, J.; Hol, E.M.; van Leeuwen, F.W. Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol. Aging 2009, 30, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Hasanbasic, S.; Jahic, A.; Karahmet, E. The role of cysteine proteases in Alzheimer disease. Mater Sociomed. 2016, 28, 235–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinchese, F.; Liu, S.; Zhang, H.; Hidalgo, A.; Schmidt, S.D.; Yamaguchi, H.; Yoshii, N.; Mathews, P.M.; Nixon, R.A.; Arancio, O. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Investig. 2008, 1, 2796–2807. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, T.; Hosfield, C.M.; Lim, D.; Elce, J.S.; Jia, Z.; Davies, P.L. A Ca2+ switch aligns the active site of calpain. Cell 2002, 108, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Sorimachi, H. Calpains-An elaborate proteolytic system. Biochim. Biophys. Acta 2012, 1824, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Melino, G.; Bernassola, F.; Knight, R.A.; Corasaniti, M.T.; Nistico, G.; Finazzi-Agro, A. S-nitrosylation regulates apoptosis. Nature 1997, 388, 432–433. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef]
- Tsang, A.H.K.; Lee, Y.I.L.; Ko, H.S.; Savitt, J.M.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M.; Chung, K.K. S-nitrosylation of XIAP compromises neuronal survival in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 4900–4905. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Kim, H.E.; Li, C.R.; Kim, S.; Kwak, I.J.; Lee, Y.J.; Kim, S.S.; Moon, J.Y.; Kim, C.H.; Kim, D.K.; et al. Two distinct disulfide bonds formed in human heat shock transcription factor 1 actin opposition to regulate its DNA binding activity. Biochemistry 2008, 47, 6007–6015. [Google Scholar] [CrossRef]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Yan, X.X.; Freeman, M.L. Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene 2002, 21, 6829–6834. [Google Scholar] [CrossRef] [Green Version]
- Piccirillo, S.; Filomeni, G.; Brune, B.; Rotilio, G.; Ciriolo, M.R. Redox mechanisms involved in the selective activation of Nrf2-mediated resistance versus p53-dependent apoptosis in adenocarcinoma gastric cells. J. Biol. Chem. 2009, 284, 27721–27733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, D.W. Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Trinkaus, V.A.; Riera-Tur, I.; Martínez-Sánchez, A.; Bäuerlein, F.; Guo, Q.; Arzberger, T.; Baumeister, W.; Dudanova, I.; Hipp, M.S.; Hartl, F.U.; et al. In situ architecture of neuronal α-Synuclein inclusions. Nat. Commun. 2021, 12, 2110. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. A critical reappraisal of current staging of Lewy related pathology in human brain. Acta Neuropathol. 2008, 116, 1. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Schaeffer, W.J. Is cell death primary or secondary in the pathophysiology of idiopathic Parkinson’s disease? Biomolecules 2015, 5, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S. Lewy-body formation is an aggresome-related process: A hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef]
- Veldman, B.A.; Wijn, A.M.; Knoers, N.; Praamstra, P.; Horstinka, M.W.I.M. Genetic and environmental risk factors in Parkinson’s Disease. Clin. Neurol. Neurosurg. 1998, 100, 15–26. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, X.; Bandrés-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol. Dis. 2019, 124, 230–239. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Fahn, S.; Cohen, G. The oxidant stress hypothesis in Parkinson’s disease: Evidence supporting it. Ann. Neurol. 1992, 32, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–924. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett. 1983, 33, 305–310. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Bowling, A.C.; Mutisya, E.M.; Walker, L.C.; Price, D.L.; Cork, L.C.; Beal, M.F. Age-dependent impairment of mitochondrial function in primate brain. J. Neurochem. 1993, 60, 1964–1967. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Kanski, J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 2001, 122, 945–962. [Google Scholar] [CrossRef]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montine, T.J.; Neely, M.D.; Quinn, J.F.; Beal, M.F.; Markesbery, W.R.; Roberts, L.J.; Morrow, J.D. Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radic. Biol. Med. 2002, 33, 620–626. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martinez, N.; Ferrandiz, M.L. Age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1996, 731, 246–248. [Google Scholar] [CrossRef]
- Medina, S.; Martinez-Banaclocha, M.; Hernanz, A. Antioxidants inhibit the human cortical neuron apoptosis induced by hydrogen peroxide, tumor necrosis factor alpha, dopamine and beta-amyloid peptide 1–42. Free Radic. Res. 2002, 36, 1179–1184. [Google Scholar] [CrossRef]
- Li, Y.I.; Wong, G.; Humphrey, J.; Raj, T. Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat. Commun. 2019, 10, 994. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, M.P.; Milanese, C.; Di Maio, R.; Hu, X.; Montero, L.M.; Sanders, L.H.; Tapias, V.; Sepe, S.; van Cappellen, W.A.; Burton, E.A.; et al. Single-cell redox imaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults. Antioxid. Redox Signal. 2011, 15, 855–871. [Google Scholar] [CrossRef] [Green Version]
- Chinta, S.; Andersen, J. Redox imbalance in Parkinson’s disease. Biochim. Biophys. Acta 2008, 1780, 1362–1367. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Garcia, A.; Zavala-Flores, L.; Rodriguez-Rocha, H.; Franco, R. Thiol-redox signaling, dopaminergic cell death, and Parkinson’s disease. Antioxid. Redox Signal. 2012, 17, 1764–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox mechanisms in neurodegeneration: From disease outcomes to therapeutic opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef]
- Milanese, C.; Payán-Gómez, C.; Mastroberardino, P.G. Cysteine oxidation and redox signaling in dopaminergic neurons physiology and in Parkinson’s disease. Curr. Opin. Physiol. 2019, 9, 73–78. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-dependent thiol modifications: Implications for the release of extracellular vesicles. Cell. Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef]
- Pasquet, J.M.; Dachary-Prigent, J.; Nurden, A.T. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur. J. Biochem. 1996, 239, 647–654. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [Green Version]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosen, D.A.; Blauwendraat, C.; Cookson, M.R.; Lewis, P.A. DNAJC proteins and pathways to parkinsonism. FEBS J. 2019, 286, 3080–3094. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Yoshida, S.; Sugeno, N.; Kobayashi, J.; Aoki, M. DnaJ/Hsp40 family and Parkinson’s disease. Front. Neurosci. 2018, 11, 743. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernández-Chacón, R.; Schlüter, O.M.; Südhof, T.C. α-synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160534. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Wang, B.; Li, X.; Fu, C.; Wang, C.; Kang, X. α-Synuclein: A multifunctional player in exocytosis, endocytosis, and vesicle recycling. Front. Neurosci. 2019, 13, 28. [Google Scholar] [CrossRef]
- Deng, J.; Koutras, C.; Donnelier, J.; Alshehri, M.; Fotouhi, M.; Girard, M.; Casha, S.; McPherson, P.S.; Robbins, S.M.; Braun, J.E.A. Neurons export extracellular vesicles enriched in cysteine string protein and misfolded protein cargo. Sci. Rep. 2017, 7, 956. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.; Garcia-Garcia, E.; Royo, F.; Falcon-Perez, J.M.; Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med. 2019, 11, 514. [Google Scholar] [CrossRef]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-β-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Kang, S.; Ray, S.; Jackson, J.; Zaitsev, A.D.; Gerber, S.A.; Cuny, G.D.; Glicksman, M.A. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry 2011, 50, 9399–9408. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.H.; Knape, M.J.; Boassa, D.; Mumdey, N.; Kornev, A.P.; Ellisman, M.H.; Taylor, S.S.; Herberg, F.W. The dynamic switch mechanism that leads to activation of LRRK2 is embedded in the DFGψ motif in the kinase domain. Proc. Natl. Acad. Sci. USA 2019, 116, 14979–14988. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Aasly, J.O.; Sæther, O.; Johansen, K.K.; Bathen, T.F.; Giskeødegård, G.F.; White, L.R. Changes to intermediary metabolites in sporadic and LRRK2 Parkinson’s disease demonstrated by proton magnetic resonance spectroscopy. Parkinson Dis. 2015, 2015, 264896. [Google Scholar]
- Brahmachari, S.; Karuppagounder, S.S.; Ge, P.; Lee, S.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. c-Abl and Parkinson’s disease: Mechanisms and therapeutic potential. J. Parkinson Dis. 2017, 7, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Leonberg, A.K.; Chai, Y.C. The functional role of cysteine residues for c-Abl kinase activity. Mol. Cell. Biochem. 2007, 304, 207–212. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.; Park, Y.J.; Yun, S.P.; Kwon, S.H.; Kim, D.; Kim, D.Y.; Shin, J.S.; Cho, D.J.; Lee, G.Y.; et al. The c-Abl inhibitor, Radotinib HCl, is neuroprotective in a preclinical Parkinson’s disease mouse model. Hum. Mol. Genet. 2018, 27, 2344–2356. [Google Scholar] [CrossRef] [Green Version]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-dependent regulation of parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [Green Version]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, K.; Komatsubara, A.T.; Nishimura, Y.; Sawada, T.; Kawafune, H.; Tsumoto, H.; Tsuji, Y.; Zhao, J.; Kyotani, Y.; Tanaka, T.; et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 2013, 3, 2202. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2014, 25, S32–S39. [Google Scholar] [CrossRef]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitocondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Ferrer, J.V.; Javitch, J.A.; Justice, J.B., Jr. Transport-dependent accessibility of a cytoplasmic loop cysteine in the human dopamine transporter. J. Biol. Chem. 2000, 275, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Rastedt, D.E.; Vaughan, R.A.; Foster, J.D. Palmitoylation mechanisms in dopamine transporter regulation. J. Chem. Neuroanat. 2017, 83–84, 3–9. [Google Scholar] [CrossRef]
- Torres, G.E.; Gainetdinov, R.R.; Caron, M.G. Plasma membrane monoamine transporters: Structure, regulation and function. Nat. Rev. Neurosci. 2003, 4, 13–25. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Ito, G.; Ariga, H.; Nakagawa, Y.; Iwatsubo, T. Roles of distinct cysteine residues in S-nitrosylation and dimerization of DJ-1. Biochem. Biophys. Res. Commun. 2006, 339, 667–672. [Google Scholar] [CrossRef]
- Choi, M.S.; Nakamura, T.; Cho, S.J.; Han, X.; Holland, E.A.; Qu, J.; Petsko, G.A.; Yates, J.R., 3rd; Liddington, R.C.; Lipton, S.A. Transnitrosylation from DJ-1 to PTEN attenuates neuronal cell death in Parkinson’s disease models. J. Neurosci. 2014, 34, 15123–15131. [Google Scholar] [CrossRef]
- Coetzee, W.A.; Nakamura, T.Y.; Faivre, J.F. Effects of thiol-modifying agents on KATP channels in guinea pig ventricular cells. Am. J. Physiol. 1995, 269, H1625–H1633. [Google Scholar] [CrossRef]
- Jiang, B.; Tang, G.; Cao, K.; Wu, L.; Wang, R. Molecular mechanism for H2S-induced activation of KATP channels. Antioxid. Redox Signal. 2010, 12, 1167–1178. [Google Scholar] [CrossRef]
- Avshalumov, M.V.; Chen, B.T.; Koós, T.; Tepper, J.M.; Rice, M.E. Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP-sensitive potassium channels. J. Neurosci. 2005, 25, 4222–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lim, S.; Haque, M.M.; Ryoo, N.; Hong, H.S.; Rhim, H.; Lee, D.E.; Chang, Y.T.; Lee, J.S.; Cheong, E.; et al. Identification of disulfide cross-linked tau dimer responsable for tau propagation. Sci. Rep. 2015, 5, 15231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiee, S.; Asadollahi, K.; Riazi, G.; Ahmadian, S.; Saboury, A.A. Vitamin B12 inhibits Tau fibrillization via binding to cysteine residues of Tau. ACS Chem. Neurosci. 2017, 8, 2676–2682. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Recchia, A.; Debetto, P.; Negro, A.; Guidolin, D.; Skaper, S.D.; Giusti, P. α-Synuclein and Parkinson’s disease. FASEB J. 2004, 18, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Jha, N.; Kumar, M.J.; Boonplueang, R.; Andersen, J.K. Glutathione decreases in dopaminergic PC12 cells interfere with the ubiquitin protein degradation pathway: Relevance for Parkinson’s disease? J. Neurochem. 2002, 80, 555–561. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, A.; Bubacco, L.; Longhena, F.; Parrella, E.; Faustini, G.; Porrini, V.; Bono, F.; Missale, C.; Pizzi, M. Nuclear Factor-κB dysregulation and α-synuclein pathology: Critical interplay in the pathogenesis of Parkinson’s disease. Front. Aging Neurosci. 2020, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 2548. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Maruyama, S. Synapse loss in anterior horn neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1994, 88, 222–227. [Google Scholar] [CrossRef]
- Lee, E.B.; Lee, V.M.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2011, 13, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Valle, C.; Carrì, M.T. Cysteine modifications in the pathogenesis of ALS. Front. Mol. Neurosci. 2017, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.; Antonyuk, S.; Hasnain, S. The biophysics of superoxide dismutase-1 and amyotrophic lateral sclerosis. Q. Rev. Biophys. 2019, 52, E12. [Google Scholar] [CrossRef]
- Sala, F.A.; Wright, G.S.A.; Antonyuk, S.V.; Garratt, R.C.; Hasnain, S.S. Molecular recognition and maturation of SOD1 by its evolutionarily destabilized cognate chaperone hCCS. PLoS Biol. 2019, 17, e3000141. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Ferri, A.; Carrì, M.T. Amyotrophic lateral sclerosis: From current developments in the laboratory to clinical implications. Antioxid. Redox Signal. 2008, 10, 405–444. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antinone, S.E.; Ghadge, G.D.; Lam, T.T.; Wang, L.; Roos, R.P.; Green, W.N. Palmitoylation of superoxide dismutase 1 (SOD1) is increased for familial ALS-linked SOD1 mutants. J. Biol. Chem. 2013, 288, 21606–21617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antinone, S.E.; Ghadge, G.D.; Ostrow, L.W.; Roos, R.P.; Green, W.N. S-acylation of SOD1, CCS, and a stable SOD1-CCS heterodimer in human spinal cords from ALS and non-ALS subjects. Sci. Rep. 2017, 7, 41141. [Google Scholar] [CrossRef] [Green Version]
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 proteinopathy and ALS: Insights into disease mechanisms and therapeutic targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M. Redox signaling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzo, F.; Salvatori, I.; Iacovelli, F.; Mirra, A.; Rossi, S.; Cozzolino, M.; Falconi, M.; Valle, C.; Carrì, M.T. Structural insights into the multi-determinant aggregation of TDP-43 in motor neuron-like cells. Neurobiol. Dis. 2016, 94, 63–72. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Li, C.; Guan, T.; Shang, H.; Cui, L.; Li, X.M.; Kong, J. S-nitrosylated protein disulfide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J. Neurochem. 2013, 124, 45–58. [Google Scholar] [CrossRef]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K. Protein disulfide isomerase and the endoplasmic reticulum in amyotrophic lateral sclerosis. J. Neurosci. 2010, 30, 3865–3867. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Farg, M.A.; Bye, C.R.; McLean, C.A.; Horne, M.K.; Atkin, J.D. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 2010, 133, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Nitrosative stress in the ER: A new role for S-nitrosylation in neurodegenerative diseases. ACS Chem. Biol. 2006, 1, 355–358. [Google Scholar] [PubMed] [Green Version]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Soo, K.Y.; Walker, A.K.; Pham, H.; Orian, J.; Horne, M.K.; Warraich, S.T.; Williams, K.L.; Blair, I.P.; Atkin, J.D. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 2012, 33, 2855–2868. [Google Scholar] [CrossRef]
- Zhao, G.; Lu, H.; Li, C. Proapoptotic activities of protein disulfide isomerase (PDI) and PDIA3 protein, a role of the Bcl-2 protein Bak. J. Biol. Chem. 2015, 290, 8949–8963. [Google Scholar] [CrossRef] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Rodríguez, C.; Muñoz, M.; Contreras, C.; Prieto, D. AMPK, metabolism, and vascular function. FEBS J. 2021, 288, 3746–3771. [Google Scholar] [CrossRef]
- Yuan, M.; Yan, R.; Zhang, Y.; Qiu, Y.; Jiang, Z.; Liu, H.; Wang, Y.; Sun, L.; Zhang, H.; Gao, P. CARS senses cysteine deprivation to activate AMPK for cell survival. EMBO J. 2021, 40, e108028. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, M.J.; Hendershot, L.M. Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 2011, 23, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, J.E.; Marciniak, S.J. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 2. Protein misfolding and ER stress. Am. J. Physiol. Cell Physiol. 2014, 307, C657–C670. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [Green Version]
- Thau, N.; Jungnickel, J.; Knippenberg, S.; Ratzka, A.; Dengler, R.; Petri, S.; Grothe, C. Prolonged survival and milder impairment of motor function in the SOD1 ALS mouse model devoid of fibroblast growth factor 2. Neurobiol. Dis. 2012, 47, 248–257. [Google Scholar] [CrossRef]
- Lee, J.; Blaber, M. Structural basis of conserved cysteine in the fibroblast growth factor family: Evidence for a vestigial halfcystine. J. Mol. Biol. 2009, 393, 128–139. [Google Scholar] [CrossRef]
- Liu, X.; Resch, J.; Rush, T.; Lobner, D. Functional upregulation of system xc- by fibroblast growth factor-2. Neuropharmacology 2012, 62, 901–906. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Estrada Sanchez, A.M.; Mejia-Toiber, J.; Massieu, L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch. Med. Res. 2008, 39, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Lemarié, F.L.; Caron, N.S.; Sanders, S.S.; Schmidt, M.E.; Nguyen, Y.T.N.; Ko, S.; Xu, X.; Pouladi, M.A.; Martin, D.D.O.; Hayden, M.R. Rescue of aberrant huntingtin palmitoylation ameliorates mutant huntingtin-induced toxicity. Neurobiol. Dis. 2021, 158, 105479. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef] [Green Version]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Sanchez, M.; Thomson, F.; Zavodszky, E.; Rubinsztein, D.C. Autophagy and polyglutamine diseases. Prog. Neurobiol. 2012, 97, 67–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Tran, S.; Fairlie, W.D.; Lee, E.F. BECLIN1: Protein Structure, Function and Regulation. Cells 2021, 10, 1522. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Neurodegeneration in Huntington’s disease involves loss of cystathionine γ-lyase. Cell Cycle 2014, 13, 2491–2493. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Guo, C.; Lao, Y.; Yang, J.; Chen, F.; Zhao, Y.; Yang, Y.; Yang, J.; Yi, J. A fine-tuning mechanism underlying self-control for autophagy: deSUMOylation of BECN1 by SENP3. Autophagy 2020, 16, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 8843–8848. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Kahlem, P.; Green, H.; Djian, P. Transglutaminase action imitates Huntington’s disease: Selective polymerization of Huntingtin containing expanded polyglutamine. Mol. Cell 1998, 1, 595–601. [Google Scholar] [CrossRef]
- Fox, J.H.; Barber, D.S.; Singh, B.; Zucker, B.; Swindell, M.K.; Norflus, F.; Buzescu, R.; Chopra, R.; Ferrante, R.J.; Kazantsev, A.; et al. Cystamine increases L-cysteine levels in Huntington’s disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J. Neurochem. 2004, 91, 413–422. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Pinto, J.T.; Cooper, A.J.L. Cystamine and cysteamine as inhibitors of transglutaminase activity in vivo. Biosci. Rep. 2018, 38, BSR20180691. [Google Scholar] [CrossRef] [Green Version]
- Arbez, N.; Roby, E.; Akimov, S.; Eddings, C.; Ren, M.; Wang, X.; Ross, C.A. Cysteamine protects neurons from mutant Huntingtin toxicity. J. Huntingt. Dis. 2019, 8, 129–143. [Google Scholar] [CrossRef]
- Fox, J.H.; Connor, T.; Stiles, M.; Kama, J.; Lu, Z.; Dorsey, K.; Lieberman, G.; Sapp, E.; Cherny, R.A.; Banks, M.; et al. Cysteine oxidation within N-terminal mutant huntingtin promotes oligomerization and delays clearance of soluble protein. J. Biol. Chem. 2011, 286, 18320–18330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, W.A.; Bird, E.D.; Aprille, J.R. Regional mitochondria respiratory activity in Huntington’s disease brain. J. Neurochem. 1985, 44, 1948–1950. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H.V. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Mann, V.M.; Copper, J.M.; Jvoy-Agid, Y.; Jenner, Y.; Schapira, A.H.V. Mitochondrial function and parental sex effect in Huntington’s disease. Lancet 1990, 336, 749. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Koutouzis, T.K.; Freeman, T.B.; Cahill, D.W.; Sanberg, P.R. Behavioral pathology induced by repeated systemic injection of 3-nitropropionic acid mimics the motor symptoms of Huntington’s disease. Brain Res. 1995, 697, 254–257. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Workman, J.; Hart, P.E.; Mangiarini, L.; Mahal, A.; Bates, G.; Cooper, J.M.; Schapira, A.H.V. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann. Neurol. 2000, 47, 80–86. [Google Scholar] [CrossRef]
- Burke, J.R.; Enghild, J.J.; Martin, M.E.; Jou, Y.; Myers, R.; Roses, A.; Vance, J.; Strittmatter, W. Huntington and DRPLA proteins selectively interact with the enzyme GAPDH. Nat. Med. 1996, 2, 347–350. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Smotrys, J.E.; Linder, M.E. Palmitoylation of intracellular signaling proteins: Regulation and function. Ann. Rev. Biochem. 2004, 73, 559–587. [Google Scholar] [CrossRef]
- Fukata, Y.; Fukata, M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 2010, 11, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Globa, A.K.; Bamji, S.X. Protein palmitoylation in the development and plasticity of neuronal connections. Curr. Opin. Neurobiol. 2017, 45, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Matt, L.; Kim, K.; Chowdhury, D.; Hell, J.W. Role of palmitoylation of postsynaptic proteins in promoting synaptic plasticity. Front. Mol. Neurosci. 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.A.; et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Zamzow, D.R.; Elias, V.; Acosta, V.A.; Escobedo, E.; Magnusson, K.R. Higher levels of protein palmitoylation in the frontal cortex across aging were associated with reference memory and executive function declines. eNeuro 2019, 6, ENEURO.0310-18.2019. [Google Scholar] [CrossRef]
- Arvanitakis, Z. Update on frontotemporal dementia. Neurologist 2010, 16, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Kao, A.W.; McKay, A.; Singh, P.P.; Brunet, A.; Huang, E.J. Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci. 2017, 18, 325–333. [Google Scholar] [CrossRef]
- Mann, D.M.A.; Snowden, J.S. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017, 27, 723–736. [Google Scholar] [CrossRef]
- Sieben, A.; Van Langenhove, T.; Engelborghs, S.; Martin, J.J.; Boon, P.; Cras, P.; De Deyn, P.P.; Santens, P.; Van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef] [Green Version]
- Chitramuthu, B.P.; Bennett, H.P.J.; Bateman, A. Progranulin: A new avenue towards the understanding and treatment of neurodegenerative disease. Brain 2017, 140, 3081–3104. [Google Scholar] [CrossRef]
- Wang, J.; Van Damme, P.; Cruchaga, C.; Gitcho, M.A.; Vidal, J.M.; Seijo-Martínez, M.; Wang, L.; Wu, J.Y.; Robberecht, W.; Goate, A. Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J. Neurochem. 2010, 112, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holler, C.J.; Taylor, G.; Deng, Q.; Kukar, T. Intracellular proteolysis of progranulin generates stable, lysosomal granulins that are haploinsufficient in patients with frontotemporal dementia caused by GRN mutations. eNeuro 2017, 4, ENEURO.0100-17.2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef]
- Fischer, M.; Horn, S.; Belkacemi, A.; Kojer, K.; Petrungaro, C.; Habich, M.; Ali, M.; Kuttner, V.; Bien, M.; Kauff, F.; et al. Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol. Biol. Cell 2013, 24, 2160–2170. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Meng, H.; Shiba-Fukushima, K.; Hattori, N. Twin CHCH Proteins, CHCHD2, and CHCHD10: Key molecules of Parkinson’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Int. J. Mol. Sci. 2019, 20, 908. [Google Scholar] [CrossRef] [Green Version]
- Darshi, M.; Trinh, K.N.; Murphy, A.N.; Taylor, S.S. Targeting and import mechanism of coiled-coil helix coiled-coil helix domain-containing protein 3 (ChChd3) into the mitochondrial intermembrane space. J. Biol. Chem. 2012, 287, 39480–39491. [Google Scholar] [CrossRef] [Green Version]
- Oka, O.B.; Bulleid, N.J. Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2425–2429. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Riemer, J. The mitochondrial disulfide relay system: Roles in oxidative protein folding and beyond. Int. J. Cell Biol. 2013, 2013, 742923. [Google Scholar] [CrossRef] [Green Version]
- Carrie, C.; Soll, J. To Mia or not to Mia: Stepwise evolution of the mitochondrial intermembrane space disulfide relay. BMC Biol. 2017, 15, 119. [Google Scholar] [CrossRef] [Green Version]
- Ang, S.K.; Zhang, M.; Lodi, T.; Lu, H. Mitochondrial thiol oxidase Erv1: Both shuttle cysteine residues are required for its function with distinct roles. Biochem. J. 2014, 460, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Banaclocha, M. N-acetylcysteine elicited increase in complex I activity in synaptic mitochondria from aged mice: Implications for treatment of Parkinson’s disease. Brain Res. 2000, 859, 173–175. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. Therapeutic potential of N-acetylcysteine in age-related mitochondrial neurodegenerative diseases. Med. Hypotheses 2001, 56, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. N-acetyl-cysteine in the treatment of Parkinson’s disease. What are we waiting for? Med. Hypotheses 2012, 79, 8–12. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Martínez, N. N-acetylcysteine elicited increase in cytochrome c oxidase activity in mice synaptic mitochondria. Brain Res 1999, 842, 249–251. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Martinez, N.; Hernandez, A.I.; Ferrandiz, M.L. Hypothesis: Can N-acetylcysteine be beneficial in Parkinson’s disease? Life Sci. 1999, 64, 1253–1257. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. Cysteinet dysregulation in muscular dystrophies: A pathogenic network susceptible to therapy. Curr. Med. Chem. 2017, 24, 312–330. [Google Scholar] [CrossRef]
- Rossell, S.L.; Francis, P.S.; Galletly, C.; Harris, A.; Siskind, D.; Berk, M.; Bozaoglu, K.; Dark, F.; Dean, O.; Liu, D.; et al. N-acetylcysteine (NAC) in schizophrenia resistant to clozapine: A double blind randomised placebo controlled trial targeting negative symptoms. BMC Psychiatry 2016, 16, 320. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martinez, N. N-Acetylcysteine delays age-associated memory impairment in mice: Role in synaptic mitochondria. Brain Res. 2000, 855, 100–106. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martínez, N.; Ferrándiz, M.L. N-acetylcysteine protects against age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1997, 762, 256–258. [Google Scholar] [CrossRef]
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170. [Google Scholar] [CrossRef]
- Van Zandwijk, N. N-Acetylcysteine for lung cancer prevention. Chest 1995, 107, 1437–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Flora, S.; Astengo, M.; Serra, D.; Bennicelli, C. Inhibition of urethan-induced lung tumors in mice by dietary N-acetylcysteine. Cancer Lett. 1986, 32, 235–241. [Google Scholar] [CrossRef]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Schwalfenberg, G.K. N-Acetylcysteine: A review of clinical usefulness (an old drug with new tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef]
- De Flora, S.; Izzotti, A.; D’Agostini, F.; Balansky, R.M. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 2001, 22, 999–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekhuijzen, P.N.; van Beurden, W.J. The role for N-acetylcysteine in the management of COPD. Int. J. Chron. Obstruct. Pulmon Dis. 2006, 1, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Caro, L.; Ghizzi, A.; Costa, R.; Longo, A.; Ventresca, G.P.; Lodola, E. Pharmacokinetics and bioavailability of oral acetylcysteine in healthy volunteers. Arzneimittelforschung 1989, 39, 382–386. [Google Scholar]
- Harman, D. The biological clock: The mitochondria. J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar] [CrossRef] [Green Version]
- Stadtman, E.R. Protein oxidation and aging. Science 1992, 257, 1220–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Banaclocha, M.; Ferrandiz, M.L.; Díez, A.; Miquel, J. Depletion of cytosolic GSH decreases the ATP levels and viability of synaptosomes from aged mice but not from young mice. Mech. Ageing Dev. 1995, 84, 77–81. [Google Scholar] [CrossRef]
- Ferrandiz, M.L.; Martinez-Banaclocha, M.; De Juan, E.; Díez, A.; Bustos, G.; Miquel, J. Impairment of mitochondrial oxidative phosphorylation in the brain of aged mice. Brain Res. 1994, 644, 335–338. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M.; Ferrandiz, M.L.; De Juan, E.; Miquel, J. Age-related changes in glutathione and lipid peroxide content in mouse synaptic mitochondria: Relationship to cytochrome c oxidase decline. Neurosci. Lett. 1994, 170, 121–124. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L.; Duan, W. Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. Physiol. Rev. 2002, 82, 637–672. [Google Scholar] [CrossRef] [Green Version]
- Miquel, J.; Martinez-Banaclocha, M.; Díez, A.; De Juan, E.; Soler, A.; Ramirez, A.; Laborda, J.; Carrión, M. Effects of turmeric on blood and liver lipoperoxide levels of mice: Lack of toxicity. Age 1995, 18, 171–174. [Google Scholar] [CrossRef]
- Yu, Z.F.; Mattson, M.P. Dietary restriction and 2-deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: Evidence for a preconditioning mechanism. J. Neurosci. Res. 1999, 57, 830–839. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, Z.; Yu, Q.S.; Chan, S.L.; Camandola, S.; Guo, Z.; Greig, N.H.; Mattson, M.P. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 2001, 77, 220–228. [Google Scholar] [CrossRef]
- Duan, W.; Zhu, X.; Ladenheim, B.; Yu, Q.S.; Guo, Z.; Oyler, J.; Cutler, R.G.; Cadet, J.L.; Greig, N.H.; Mattson, M.P. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol. 2002, 52, 597–606. [Google Scholar] [CrossRef]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease α-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc. Nat. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Banaclocha, M. Interfering with the reactive cysteine proteome in COVID-19. Curr. Med. Chem. 2021, 28, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, R.V.; Vykhovanets, O.; Kondratova, A.A.; Antoch, M.P. Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging 2009, 1, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Mullen, L.; Mengozzi, M.; Hanschmann, E.M.; Alberts, B.; Ghezzi, P. How the redox state regulates immunity. Free Radic. Biol. Med. 2020, 157, 3–14. [Google Scholar] [CrossRef]
- García-Piñeres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in p65/NF-κB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 2001, 276, 39713–39720. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-κB by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992, 20, 3821–3830. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Machius, M.; Nestler, E.J.; Rudenko, G. Activator protein-1: Redox switch controlling structure and DNA-binding. Nucleic Acids Res. 2017, 45, 11425–11436. [Google Scholar] [CrossRef] [Green Version]
- Del Sorbo, L.; Zhang, H. Is there a place for N-acetylcysteine in the treatment of septic shock? Crit. Care 2004, 8, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Studer, R.; Baysang, G.; Brack, C. N-acetyl-L-cystein downregulates beta-amyloid precursor protein gene transcription in human neuroblastoma cells. Biogerontology 2001, 2, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, E. Protein misfolding and degenerative diseases. Nat. Educ. 2010, 3, 28. [Google Scholar]
- Takai, E.; Uda, K.; Yoshida, T.; Zako, T.; Maeda, M.; Shiraki, K. Cysteine inhibits the fibrillisation and cytotoxicity of amyloid-β 40 and 42: Implications for the contribution of the thiophilic interaction. Phys. Chem. Chem. Phys. 2014, 16, 3566–3572. [Google Scholar] [CrossRef]
- Yan, C.Y.; Greene, L.A. Prevention of PC12 cell death by N-Acetylcysteine requires activation of the Ras pathway. J. Neurosci. 1998, 18, 4042–4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, G.A.; Gunawardena, H.P.; Campbell, S.L. Biophysical and proteomic characterization strategies for cysteine modifications in Ras GTPases. Meth. Mol. Biol. 2014, 1120, 75–96. [Google Scholar]
- Tsai, L.H.; Delalle, I.; Caviness, V.S., Jr.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.; Huang, Q.Q.; Qi, Z.; Winkfein, R.J.; Aebersold, R.; Hunt, T.; Wang, J.H. A brain-specific activator of cyclin-dependent kinase 5. Nature 1994, 371, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Ward, J.M.; Huh, C.G.; Longenecker, G.; Pant, H.C.; Brady, R.O.; Martin, L.J.; Kulkarni, A.B. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 1996, 93, 11173–11178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yu, P.C.; Tsang, A.H.; Chen, Y.; Fu, A.K.; Fu, W.Y.; Chung, K.K.; Ip, N.Y. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J. Neurosci. 2010, 30, 14366–14370. [Google Scholar] [CrossRef]
- Hu, S.Q.; Ye, J.S.; Zong, Y.Y.; Sun, C.C.; Liu, D.H.; Wu, Y.P.; Song, T.; Zhang, G.Y. S-nitrosylation of mixed lineage kinase 3 contributes to its activation after cerebral ischemia. J. Biol. Chem. 2011, 287, 2364–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Zhang, Q.; Li, H.; Zhang, G. Antioxidant N-acetylcysteine and AMPA/KA receptor antagonist DNQX inhibited mixed lineage kinase-3 activation following cerebral ischemia in rat hippocampus. Neurosci. Res. 2003, 47, 47–53. [Google Scholar] [CrossRef]
- Chen, C.H.; Li, W.; Sultana, R.; You, M.H.; Kondo, A.; Shahpasand, K.; Kim, B.M.; Luo, M.L.; Nechama, M.; Lin, Y.M.; et al. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol. Dis. 2015, 76, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, V.; Mishra, R.; Sondarva, G.; Das, S.; Lee, T.H.; Bakowska, J.C.; Tzivion, G.; Malter, J.S.; Rana, B.; Lu, K.P.; et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc. Natl. Acad. Sci. USA 2012, 109, 8149–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchantchou, F.; Graves, M.; Rogers, E.; Ortiz, D.; Shea, T.B. N-acteylcysteine alleviates oxidative damage to central nervous system of ApoE-deficient mice following folate and vitamin E-deficiency. J. Alzheimer’s Dis. 2005, 7, 135–138; discussion 173–180. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Ahl, M.; Bush, A.; Westaway, D.; Huang, X.; Rogers, J.T. Pilot study of the reducing effect on amyloidosis in vivo by three FDA pre-approved drugs via the Alzheimer’s APP 5′ untranslated region. Curr. Alzheimer Res. 2005, 2, 249–254. [Google Scholar] [CrossRef]
- Tchantchou, F.; Graves, M.; Ortiz, D.; Rogers, E.; Shea, T.B. Dietary supplementation with 3-deaza adenosine, N-acetyl-cysteine, and S-adenosyl methionine provide neuroprotection against multiple consequences of vitamin deficiency and oxidative challenge. Neuromol. Med. 2004, 6, 93–103. [Google Scholar] [CrossRef]
- Tucker, S.; Ahl, M.; Cho, H.H.; Bandyopadhyay, S.; Cuny, G.D.; Bush, A.I.; Goldstein, L.E.; Westaway, D.; Huang, X.; Rogers, J.T. RNA therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin, and N-acetyl-cysteine. Curr. Alzheimer Res. 2006, 3, 221–227. [Google Scholar] [CrossRef]
- Farr, S.A.; Poon, H.F.; Dogrukol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.; Butterfield, D.A.; Morley, J.E. The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Aluise, C.D.; Joshi, G.; Sultana, R.; St Clair, D.K.; Markesbery, W.R.; Butterfield, D.A. Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: Toward therapeutic modulation of mild cognitive impairment. J. Neurosci. Res. 2010, 88, 2618–2629. [Google Scholar] [CrossRef]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R.; Zhou, X.Z.; Lu, K.P.; Butterfield, D.A. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol. Aging 2006, 27, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, G.; Baysang, G.; Meier, F.; Müller-Spahn, F.; Stähelin, H.B.; Brockhaus, M.; Brack, C. N-acetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: Effects on beta-amyloid secretion and tau phosphorylation. J. Neurochem. 2001, 76, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, B.J.; Parks, J.K.; Cassarino, S.W.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [CrossRef]
- Blanchard, B.J.; Park, T.; Fripp, W.J.; Lerman, L.S.; Ingram, V.M. A mitochondrial DNA deletion in normaly aging and in Alzheimer brain tissue. Neuroreport 1993, 4, 799–802. [Google Scholar] [CrossRef]
- Hutchin, T.; Cortopassi, G. A mitochondrial DNA clone is associated with increased risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1995, 92, 6892–6895. [Google Scholar] [CrossRef] [Green Version]
- Soffner, J.M.; Brawn, M.D.; Torroni, A.; Lott, M.T.; Cabell, M.F.; Mirra, S.S.; Beal, M.F.; Yang, C.-C.; Gearing, M.; Salvo, R.; et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 1993, 17, 171–184. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondria, free radicals, and neurodegeneration. Curr. Opin. Neurobiol. 1996, 6, 661–666. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Giordano, T.; Brady, D.R.; Stoll, J.; Martin, L.J.; Rapoport, S.I. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer’s disease. Brain Res. Mol. Brain Res. 1994, 24, 336–340. [Google Scholar] [CrossRef]
- Simonian, N.A.; Hyman, B.T. Functional alterations in Alzheimer’s disease: Selective loss of mitochondrial-encoded cytochrome oxidase mRNA in the hyppocampal formation. J. Neuropathol. Exp. Neurol. 1994, 53, 508–512. [Google Scholar] [CrossRef]
- Moreira, P.I.; Harris, P.L.; Zhu, X.; Santos, M.S.; Oliveira, C.R.; Smith, M.A.; Perry, G. Lipoic acid and N-acetyl-cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimer’s Dis. 2007, 12, 195–206. [Google Scholar] [CrossRef]
- Adair, J.C.; Knoefel, J.E.; Morgan, N. Controlled trial of N-acetylcysteine for patients with probable Alzheimer’s disease. Neurology 2001, 57, 1515–1517. [Google Scholar] [CrossRef] [PubMed]
- Tanel, A.; Averill-Bates, D.A. Inhibition of acrolein-induced apoptosis by the antioxidant N-acetylcysteine. J. Pharmacol. Exp. Ther. 2007, 321, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Chan, A.; Paskavitz, J.; Shea, T.B. Efficacy of a vitamin/nutriceutical formulation for moderate-stage to later-stage Alzheimer’s disease: A placebo-controlled pilot study. Am. J. Alzheimers Dis. Other Dement. 2009, 24, 27–33. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A.; Davies, G. Co-administration of N-acetylcysteine, vitamin B12 and folate in cognitively impaired hyperhomocysteinaemic patients. Int. J. Geriatr. Psychiatry 2005, 20, 998–1000. [Google Scholar] [CrossRef]
- Henze, A.; Raila, J.; Scholze, A.; Zidek, W.; Tepel, M.; Schweigert, F.J. Does N-acetylcysteine modulate post-translational modifications of transthyretin in hemodialysis patients? Antioxid. Redox Signal. 2013, 19, 1166–1672. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Ladiwala, A.R.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J. Neurosci. 2013, 33, 19423–19433. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Reixach, N. Transthyretin: The servant of many masters. Cell. Mol. Life Sci. 2009, 66, 3095–3101. [Google Scholar] [CrossRef] [Green Version]
- Gamba, P.; Guglielmotto, M.; Testa, G.; Monteleone, D.; Zerbinati, C.; Gargiulo, S.; Biasi, F.; Iuliano, L.; Giaccone, G.; Mauro, A.; et al. Up-regulation of β-amyloidogenesis in neuron-like human cells by both 24- and 27-hydroxycholesterol: Protective effect of N-acetyl-cysteine. Aging Cell 2014, 13, 561–572. [Google Scholar] [CrossRef]
- Ghosh, S.; Won, S.J.; Wang, J.; Fong, R.; Butler, N.J.M.; Moss, A.; Wong, C.; Pan, J.; Sanchez, J.; Huynh, A.; et al. α-synuclein aggregates induce c-Abl activation and dopaminergic neuronal loss by a feed-forward redox stress mechanism. Prog. Neurobiol. 2021, 202, 102070. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Cai, J.; Wei, X.; Bazzan, A.J.; Zhong, L.; Bowen, B.; et al. N-Acetyl-cysteine may support dopamine neurons in Parkinson’s disease: Preliminary clinical and cell line data. PLoS ONE 2016, 11, e0157602. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-acetyl-cysteine is associated with dopaminergic improvement in Parkinson’s disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef]
- Lee, Y.M.; Park, S.H.; Shin, D.I.; Hwang, J.Y.; Park, B.; Park, Y.J.; Lee, T.H.; Chae, H.Z.; Jin, B.K.; Oh, T.H.; et al. Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J. Biol. Chem. 2008, 283, 9986–9998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; et al. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1–/– mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Sandhir, R.; Sood, A.; Mehrotra, A.; Kamboj, S.S. N-acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener. Dis. 2012, 9, 145–157. [Google Scholar] [CrossRef]
- La Fontaine, M.A.; Geddes, J.W.; Banks, A.; Butterfield, D.A. Effect of exogenous and endogenous antioxidants on 3-nitropropionic acid-induced in vivo oxidative stress and striatal lesions: Insights into Huntington’s disease. J. Neurochem. 2000, 75, 1709–1715. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.J.; Gray, L.J.; Finkelstein, D.I.; Crouch, P.J.; Pow, D.; Pang, T.Y.; Li, S.; Smith, Z.M.; Francis, P.S.; Renoir, T.; et al. N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 2923–2933. [Google Scholar] [CrossRef] [Green Version]
- Beretta, S.; Sala, G.; Mattavelli, L.; Ceresa, C.; Casciati, A.; Ferri, A.; Carrì, M.T.; Ferrarese, C. Mitochondrial dysfunction due to mutant copper/zinc superoxide dismutase associated with amyotrophic lateral sclerosis is reversed by N-acetylcysteine. Neurobiol. Dis. 2003, 13, 213–221. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Klivenyi, P.; Beal, M.F.; Bush, A.I. N-acetyl-L-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport 2000, 11, 2491–2493. [Google Scholar] [CrossRef] [PubMed]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.; Meyjes, F.; de Jong, J. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.E.; Odlaug, B.L.; Kim, S.W. N-acetylcysteine, a glutamate modulator, in the treatment of trichotillomania: A double-blind, placebo-controlled study. Arch. Gen. Psychiatry 2009, 66, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the effects of N-acetylcysteine in neurodegenerative diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-H.; Lane, H.-Y. Plasma glutathione levels decreased with cognitive decline among people with mild cognitive impairment (MCI): A two-year prospective study. Antioxidants 2021, 10, 1839. [Google Scholar] [CrossRef] [PubMed]
- Nagumo, K.; Tanaka, M.; Chuang, V.T.; Setoyama, H.; Watanabe, H.; Yamada, N.; Kubota, K.; Tanaka, M.; Matsushita, K.; Yoshida, A.; et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS ONE 2014, 9, e85216. [Google Scholar] [CrossRef] [PubMed]
- Paramasivan, S.; Adav, S.S.; Ngan, S.C.; Dalan, R.; Leow, M.K.; Ho, H.H.; Sze, S.K. Serum albumin cysteine trioxidation is a potential oxidative stress biomarker of type 2 diabetes mellitus. Sci. Rep. 2020, 10, 6475. [Google Scholar] [CrossRef] [Green Version]
- Bocedi, A.; Cattani, G.; Stella, L.; Massoud, R.; Ricci, G. Thiol disulfide exchange reactions in human serum albumin: The apparent paradox of the redox transitions of Cys34. FEBS J. 2018, 285, 3225–3237. [Google Scholar] [CrossRef] [Green Version]
- Tunold, J.A.; Geut, H.; Rozemuller, J.M.A.; Henriksen, S.P.; Toft, M.; van de Berg, W.D.J.; Pihlstrøm, L. APOE and MAPT are associated with dementia in neuropathologically confirmed Parkinson’s disease. Front. Neurol. 2021, 12, 631145. [Google Scholar] [CrossRef]
- Saito, T.; Chiku, T.; Oka, M.; Wada-Kakuda, S.; Nobuhara, M.; Oba, T.; Shinno, K.; Abe, S.; Asada, A.; Sumioka, A.; et al. Disulfide bond formation in microtubule-associated tau protein promotes tau accumulation and toxicity in vivo. Hum. Mol. Genet. 2021, 30, 1955–1967. [Google Scholar] [CrossRef]
- Toshikawa, H.; Ikenaka, A.; Li, L.; Nishinaka-Arai, Y.; Niwa, A.; Ashida, A.; Kazuki, Y.; Nakahata, T.; Tamai, H.; Russell, D.W.; et al. N-acetylcysteine prevents amyloid-β secretion in neurons derived from human pluripotent stem cells with trisomy 21. Sci. Rep. 2021, 11, 17377. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Banaclocha, M. N-Acetyl-Cysteine: Modulating the Cysteine Redox Proteome in Neurodegenerative Diseases. Antioxidants 2022, 11, 416. https://doi.org/10.3390/antiox11020416
Martinez-Banaclocha M. N-Acetyl-Cysteine: Modulating the Cysteine Redox Proteome in Neurodegenerative Diseases. Antioxidants. 2022; 11(2):416. https://doi.org/10.3390/antiox11020416
Chicago/Turabian StyleMartinez-Banaclocha, Marcos. 2022. "N-Acetyl-Cysteine: Modulating the Cysteine Redox Proteome in Neurodegenerative Diseases" Antioxidants 11, no. 2: 416. https://doi.org/10.3390/antiox11020416
APA StyleMartinez-Banaclocha, M. (2022). N-Acetyl-Cysteine: Modulating the Cysteine Redox Proteome in Neurodegenerative Diseases. Antioxidants, 11(2), 416. https://doi.org/10.3390/antiox11020416