
Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of SOD1 in Normal Physiology and Diseases
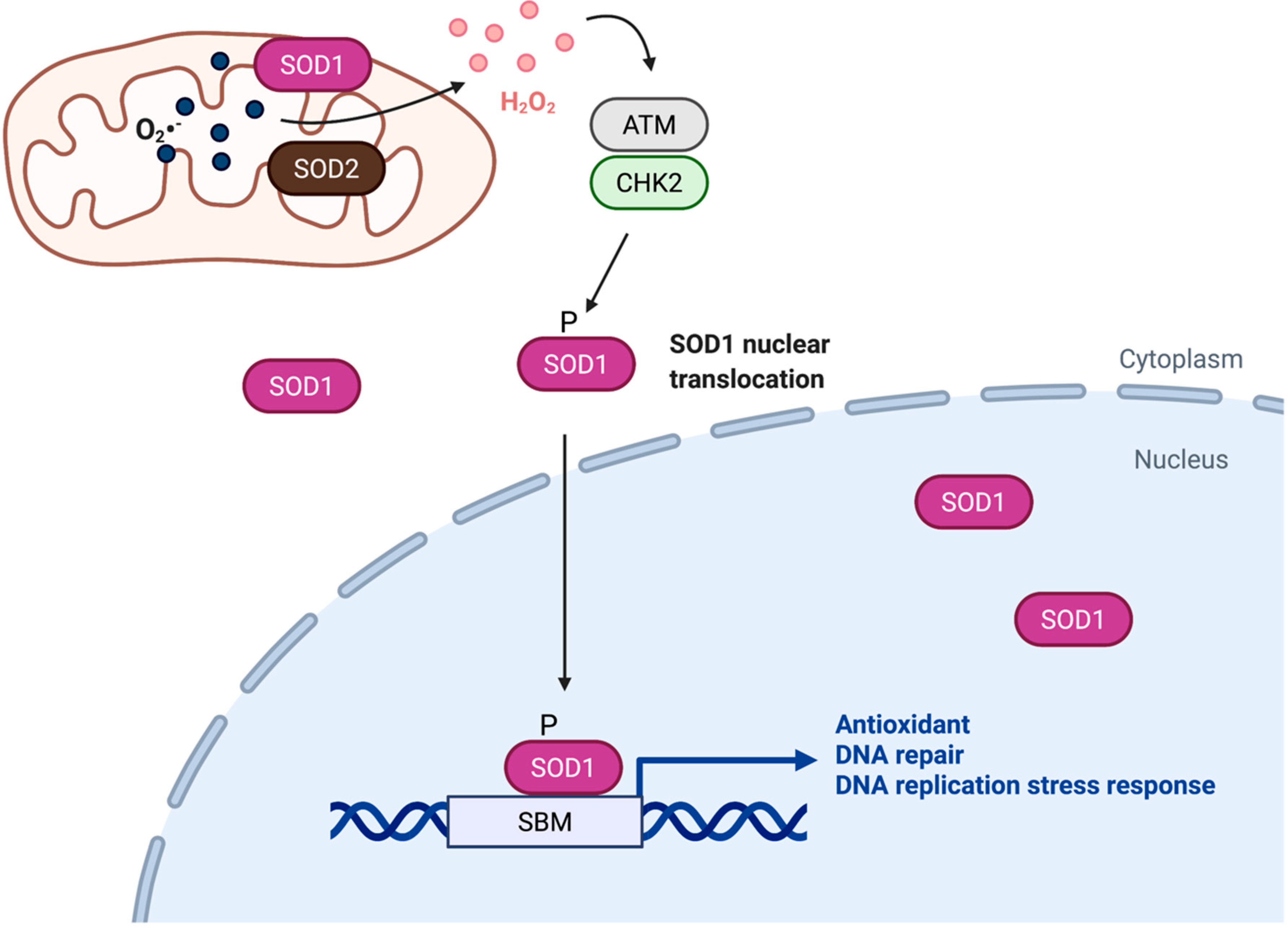
3. Localization and Regulation of SOD1 Protein in the Nucleus
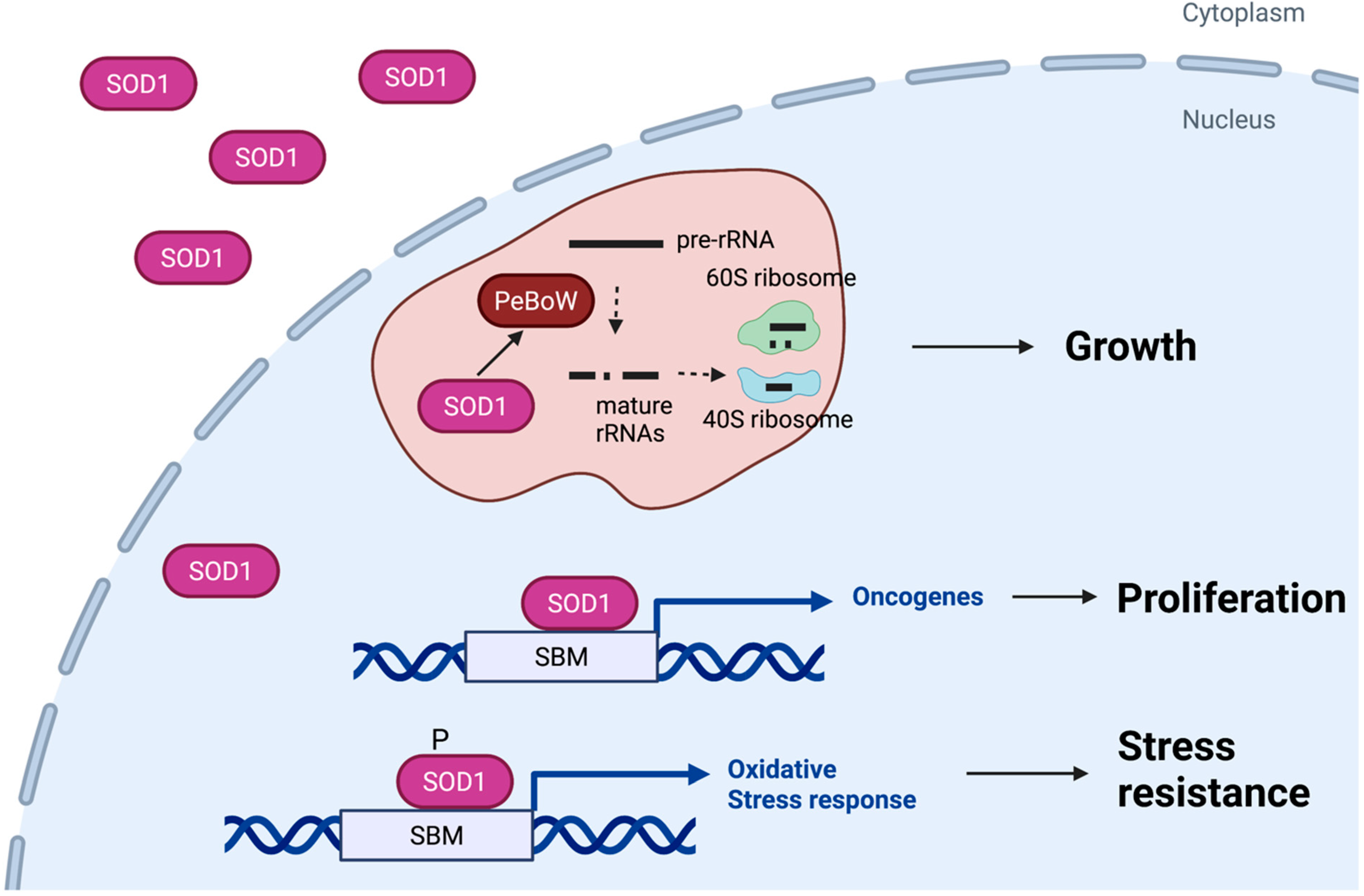
4. Nuclear SOD1 Acts as a Transcription Factor during the Oxidative Stress Response
5. Roles of Nuclear SOD1 in Cancer
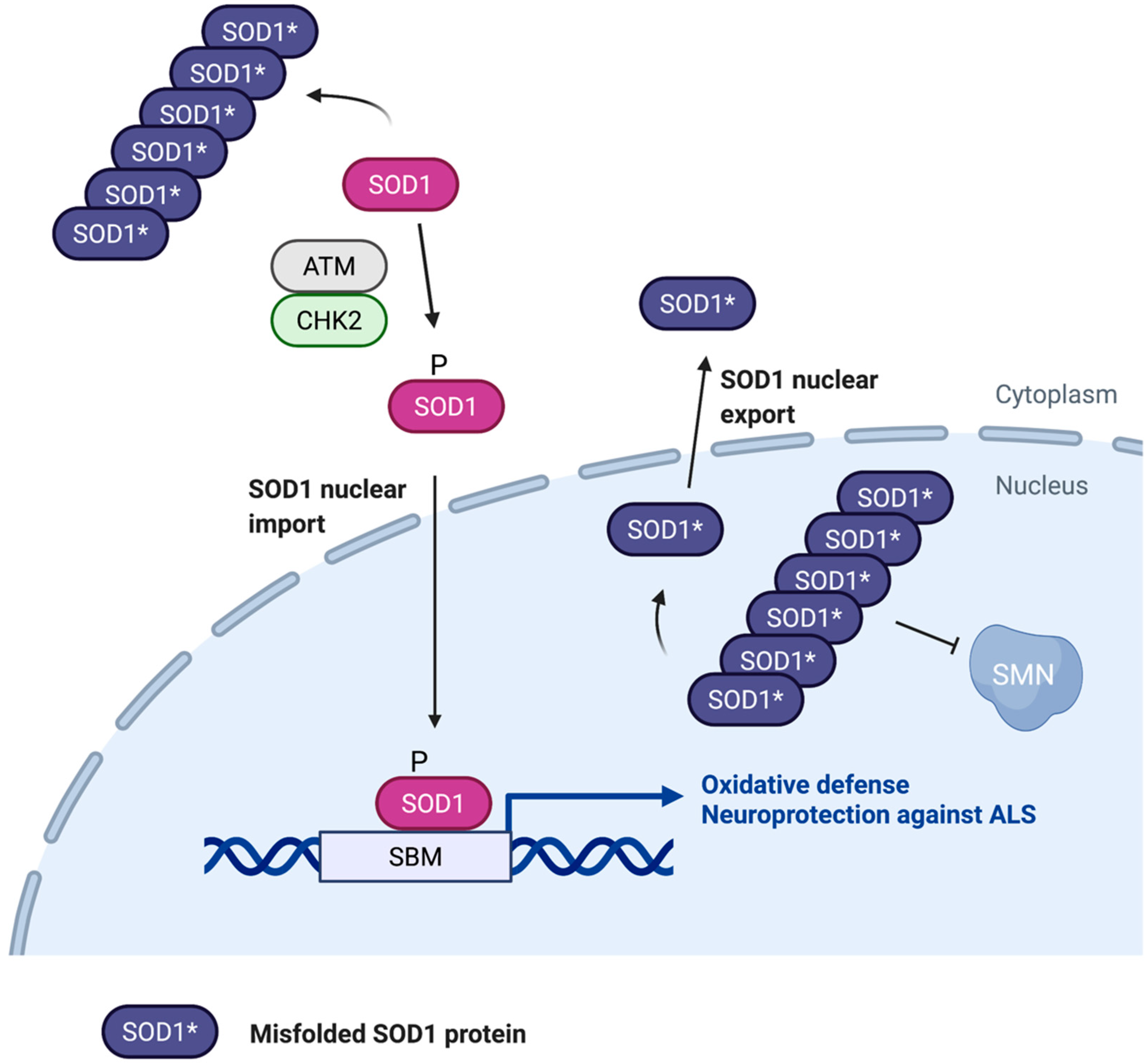
6. Role of Nuclear SOD1 in ALS
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stowe, D.F.; Camara, A.K.S. Mitochondrial Reactive Oxygen Species Production in Excitable Cells: Modulators of Mitochondrial and Cell Function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Sallam, R.M. Reactive Oxygen Species in Health and Disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Barber, S.C.; Mead, R.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.M.; He, W.; Liou, Y.-C. The redox language in neurodegenerative diseases: Oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 2021, 12, 58. [Google Scholar] [CrossRef]
- Che, M.; Wang, R.; Li, X.; Wang, H.-Y.; Zheng, X.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.R.; Carroll, R.S.; Steers, G.J.; Hrabe, J.; Domann, F.E.; Cullen, J.J. Impact of EcSOD Perturbations in Cancer Progression. Antioxidants 2021, 10, 1219. [Google Scholar] [CrossRef]
- Mccord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; Van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1−/− mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Huai, J.; Zhang, Z. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front. Neurol. 2019, 10, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabuta, T.; Suzuki, Y.; Wada, K. Degradation of Amyotrophic Lateral Sclerosis-linked Mutant Cu,Zn-Superoxide Dismutase Proteins by Macroautophagy and the Proteasome. J. Biol. Chem. 2006, 281, 30524–30533. [Google Scholar] [CrossRef] [Green Version]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non–small-cell lung cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Gomez, M.; Shah, N.; Kenny, T.C.; Jenkins, E.; Germain, D. SOD1 is essential for oncogene-driven mammary tumor formation but dispensable for normal development and proliferation. Oncogene 2019, 38, 5751–5765. [Google Scholar] [CrossRef]
- Papa, L.; Hahn, M.; Marsh, E.L.; Evans, B.S.; Germain, D. SOD2 to SOD1 Switch in Breast Cancer. J. Biol. Chem. 2014, 289, 5412–5416. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Liu, T.; Chen, J.; Ni, H.; Li, W. Survivin in breast cancer–derived exosomes activates fibroblasts by up-regulating SOD1, whose feedback promotes cancer proliferation and metastasis. J. Biol. Chem. 2020, 295, 13737–13752. [Google Scholar] [CrossRef]
- Salem, K.; McCormick, M.L.; Wendlandt, E.; Zhan, F.; Goel, A. Copper–zinc superoxide dismutase-mediated redox regulation of bortezomib resistance in multiple myeloma. Redox Biol. 2015, 4, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Fu, L.; Tian, T.; Deng, L.; Li, H.; Xia, W.; Gong, Q. Disrupting SOD1 activity inhibits cell growth and enhances lipid accumulation in nasopharyngeal carcinoma. Cell Commun. Signal. 2018, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, H.; Sapio, R.; Yang, J.; Wong, J.; Zhang, X.; Guo, J.Y.; Pine, S.; Van Remmen, H.; Li, H.; et al. SOD1 regulates ribosome biogenesis in KRAS mutant non-small cell lung cancer. Nat. Commun. 2021, 12, 2259. [Google Scholar] [CrossRef] [PubMed]
- Lowndes, S.A.; Adams, A.; Timms, A.; Fisher, N.; Smythe, J.; Watt, S.M.; Joel, S.; Donate, F.; Hayward, C.; Reich, S.; et al. Phase I Study of Copper-Binding Agent ATN-224 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2008, 14, 7526–7534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Zahurak, M.; Beer, T.M.; Ryan, C.J.; Wilding, G.; Mathew, P.; Morris, M.; Callahan, J.A.; Gordon, G.; Reich, S.D.; et al. A non-comparative randomized phase II study of 2 doses of ATN-224, a copper/zinc superoxide dismutase inhibitor, in patients with biochemically recurrent hormone-naïve prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez, J.C.; Betancourt, O.; Pirie-Shepherd, S.R.; Guan, X.; Price, M.L.; Shaw, D.E.; Mazar, A.P.; Doñate, F. Copper Binding by Tetrathiomolybdate Attenuates Angiogenesis and Tumor Cell Proliferation through the Inhibition of Superoxide Dismutase 1. Clin. Cancer Res. 2006, 12, 4974–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Roberts, L.J.; Van Remmen, H.; Epstein, C.J.; Huang, T.-T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2004, 24, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.K.; Chen, M.; Cheng, X.; Qi, Y.; Chen, Y.; Das, I.; Li, X.; Vallat, B.; Fu, L.-W.; Qian, C.-N.; et al. SOD1 Phosphorylation by mTORC1 Couples Nutrient Sensing and Redox Regulation. Mol. Cell 2018, 70, 502–515.e8. [Google Scholar] [CrossRef] [Green Version]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver: Cu,Zn-SOD IN MITOCHONDRIA*. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Wang, J.; Henderson, M.; Yang, P.; Hagen, B.M.; Siddique, T.; Vogel, B.E.; Deng, H.-X.; Fang, S. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. ELife 2017, 6, e23759. [Google Scholar] [CrossRef]
- Bordoni, M.; Pansarasa, O.; Dell’Orco, M.; Crippa, V.; Gagliardi, S.; Sproviero, D.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Tedeschi, G.; et al. Nuclear Phospho-SOD1 Protects DNA from Oxidative Stress Damage in Amyotrophic Lateral Sclerosis. J. Clin. Med. 2019, 8, 729. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM Activation by Oxidative Stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Qiu, S.; Shi, J.; Wang, S.; Wang, M.; Xu, Y.; Nie, Z.; Liu, C.; Liu, C. A new function of copper zinc superoxide dismutase: As a regulatory DNA-binding protein in gene expression in response to intracellular hydrogen peroxide. Nucleic Acids Res. 2019, 47, 5074–5085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattaj, I.W.; Englmeier, L. Nucleocytoplasmic Transport: The Soluble Phase. Annu. Rev. Biochem. 1998, 67, 265–306. [Google Scholar] [CrossRef]
- Inoue, E.; Tano, K.; Yoshii, H.; Nakamura, J.; Tada, S.; Watanabe, M.; Seki, M.; Enomoto, T. SOD1 Is Essential for the Viability of DT40 Cells and Nuclear SOD1 Functions as a Guardian of Genomic DNA. J. Nucleic Acids 2010, 2010, 795946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Xie, Z.; Onishi, A.; Yu, X.; Jiang, L.; Lin, J.; Rho, H.-S.; Woodard, C.; Wang, H.; Jeong, J.-S.; et al. Profiling the Human Protein-DNA Interactome Reveals ERK2 as a Transcriptional Repressor of Interferon Signaling. Cell 2009, 139, 610–622. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Li, H.; Tsang, C.K.; Watkins, M.; Bertram, P.G.; Zheng, X.F.S. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature 2006, 442, 1058–1061. [Google Scholar] [CrossRef]
- Tsang, C.K.; Zheng, X.S. TOR-in(g) the Nucleus. Cell Cycle 2007, 6, 25–29. [Google Scholar] [CrossRef]
- Tsang, C.K.; Liu, H.; Zheng, X.F.S. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle 2010, 9, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Hölzel, M.; Rohrmoser, M.; Schlee, M.; Grimm, T.; Harasim, T.; Malamoussi, A.; Gruber-Eber, A.; Kremmer, E.; Hiddemann, W.; Bornkamm, G.W.; et al. Mammalian WDR12 is a novel member of the Pes1–Bop1 complex and is required for ribosome biogenesis and cell proliferation. J. Cell Biol. 2005, 170, 367–378. [Google Scholar] [CrossRef]
- Lapik, Y.R.; Fernandes, C.J.; Lau, L.F.; Pestov, D.G. Physical and Functional Interaction between Pes1 and Bop1 in Mammalian Ribosome Biogenesis. Mol. Cell 2004, 15, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, L.; Treré, D.; Derenzini, M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2014, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.D.; Kitchen, L.E.; Au, W.-C.; Babic, C.M.; Basrai, M.A. Loss of SOD1 and LYS7 Sensitizes Saccharomyces cerevisiae to Hydroxyurea and DNA Damage Agents and Downregulates MEC1 Pathway Effectors. Mol. Cell. Biol. 2005, 25, 10273–10285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Sadowska-Bartosz, I.; Königstorfer, A.; Kettle, T.; Winterbourn, C.C. Superoxide dismutase protects ribonucleotide reductase from inactivation in yeast. Free Radic. Biol. Med. 2018, 116, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Valentine, J.S.; Doucette, P.A.; Potter, S.Z. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 2005, 74, 563–593. [Google Scholar] [CrossRef] [Green Version]
- Cereda, C.; Leoni, E.; Milani, P.; Pansarasa, O.; Mazzini, G.; Guareschi, S.; Alvisi, E.; Ghiroldi, A.; Diamanti, L.; Bernuzzi, S.; et al. Altered Intracellular Localization of SOD1 in Leukocytes from Patients with Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e75916. [Google Scholar] [CrossRef] [Green Version]
- Gertz, B.; Wong, M.; Martin, L.J. Nuclear Localization of Human SOD1 and Mutant SOD1-Specific Disruption of Survival Motor Neuron Protein Complex in Transgenic Amyotrophic Lateral Sclerosis Mice. J. Neuropathol. Exp. Neurol. 2012, 71, 162–177. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Su, X.; Burley, S.K.; Zheng, X.F.S. Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer. Antioxidants 2022, 11, 427. https://doi.org/10.3390/antiox11020427
Xu J, Su X, Burley SK, Zheng XFS. Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer. Antioxidants. 2022; 11(2):427. https://doi.org/10.3390/antiox11020427
Chicago/Turabian StyleXu, Joyce, Xiaoyang Su, Stephen K. Burley, and X. F. Steven Zheng. 2022. "Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer" Antioxidants 11, no. 2: 427. https://doi.org/10.3390/antiox11020427
APA StyleXu, J., Su, X., Burley, S. K., & Zheng, X. F. S. (2022). Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer. Antioxidants, 11(2), 427. https://doi.org/10.3390/antiox11020427