
Metabolism, Mitochondrial Dysfunction, and Redox Homeostasis in Pulmonary Hypertension

 ,
, 
{kind=link}
Abstract
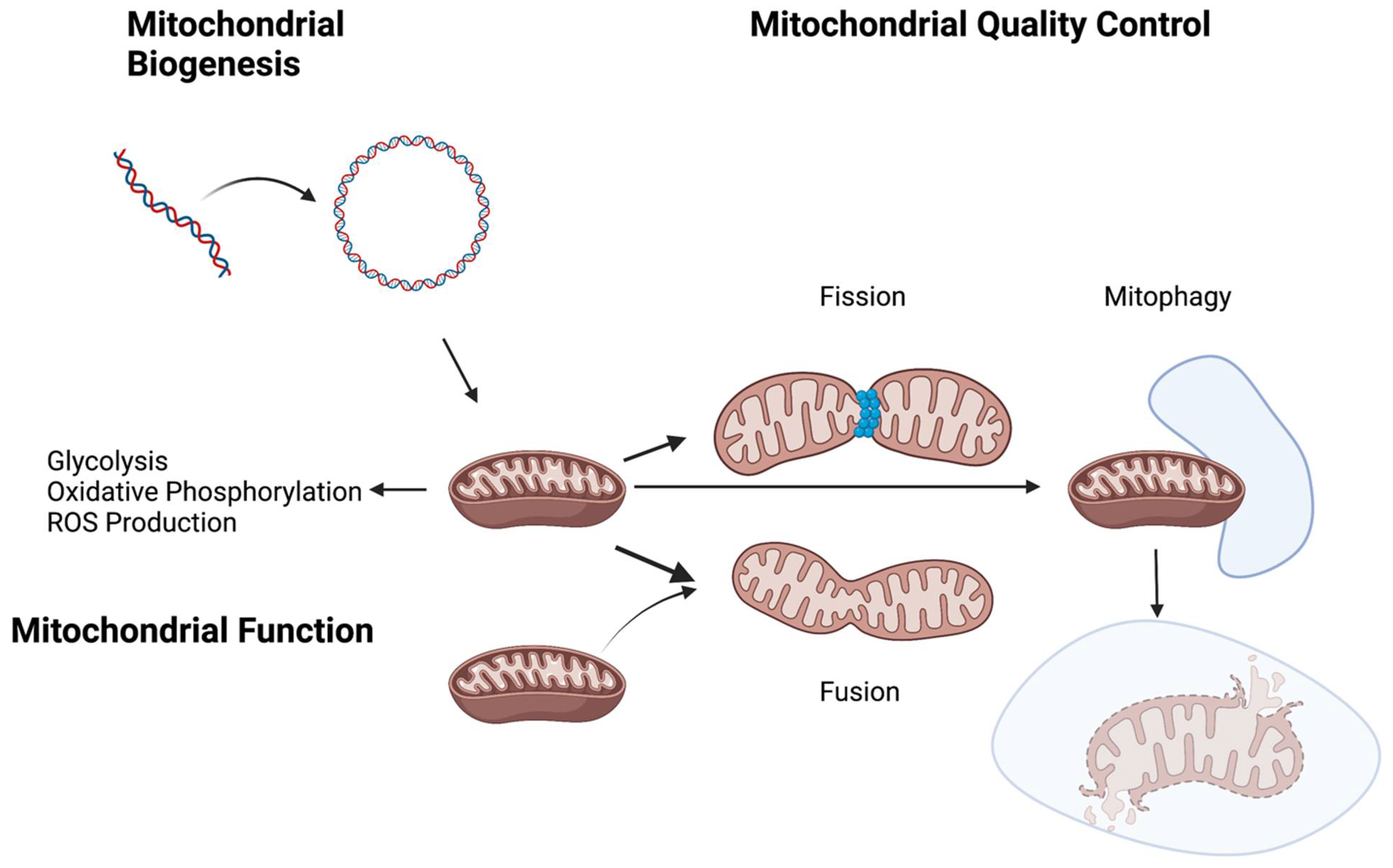
:1. Introduction
2. Glycolytic Switch and Energy Source in Pulmonary Hypertension
3. Mitochondrial Quality Control
4. Nuclear and Mitochondrial DNA Damage and Pulmonary Hypertension
5. ROS Production
6. Apoptosis Resistance
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Taichman, D.B.; Mandel, J. Epidemiology of Pulmonary Arterial Hypertension. Clin. Chest Med. 2013, 34, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Laurie, S.S. Vascular remodeling in pulmonary hypertension. J. Mol. Med. 2013, 91, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandras, S.A.; Mehta, H.S.; Vaidya, A. Pulmonary Hypertension: A Brief Guide for Clinicians. Mayo Clin. Proc. 2020, 95, 1978–1988. [Google Scholar] [CrossRef]
- Howard, L.S. Prognostic factors in pulmonary arterial hypertension: Assessing the course of the disease. Eur. Respir. Rev. 2011, 20, 236–242. [Google Scholar] [CrossRef]
- Veyssier-Belot, C.; Cacoub, P. Role of endothelial and smooth muscle cells in the physiopathology and treatment management of pulmonary hypertension. Cardiovasc. Res. 1999, 44, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Suliman, H.B.; Nozik-Grayck, E. Mitochondrial Dysfunction: Metabolic Drivers of Pulmonary Hypertension. Antioxid. Redox Signal. 2019, 31, 843–857. [Google Scholar] [CrossRef]
- Perros, F.; Sentenac, P.; Boulate, D.; Manaud, G.; Kotsimbos, T.; Lecerf, F.; Lamrani, L.; Fadel, E.; Mercier, O.; Londono-Vallejo, A.; et al. Smooth Muscle Phenotype in Idiopathic Pulmonary Hypertension: Hyper-Proliferative but not Cancerous. Int. J. Mol. Sci. 2019, 20, 3575. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Med. Cell. Longev. Publ. Online 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26, 170094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Zhou, Q.; Chen, J.; Rexius-Hall, M.L.; Rehman, J.; Zhou, G. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandro, A.; El Kasmi, K.C.; Plecitá-Hlavatá, L.; Ježek, P.; Li, M.; Zhang, H.; Gupte, S.A.; Stenmark, K.R. Hallmarks of Pulmonary Hypertension: Mesenchymal and Inflammatory Cell Metabolic Reprogramming. Antioxid. Redox Signal. 2018, 28, 230–250. [Google Scholar] [CrossRef]
- Chettimada, S.; Joshi, S.R.; Alzoubi, A.; Gebb, S.A.; McMurtry, I.F.; Gupte, R.; Gupte, S.A. Glucose-6-phosphate dehydrogenase plays a critical role in hypoxia-induced cd133+ progenitor cells self-renewal and stimulates their accumulation in the lungs of pulmonary hypertensive rats. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2014, 307, L545–L556. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, A.; Jacob, C.; Jordan, A.; Waddell, I.; McMurtry, I.F.; Gupte, S.A. Inhibition of Glucose-6-Phosphate Dehydrogenase Activity Attenuates Right Ventricle Pressure and Hypertrophy Elicited by VEGFR Inhibitor + Hypoxia. J. Pharmacol. Exp. Ther. 2021, 377, 284–292. [Google Scholar] [CrossRef]
- Varghese, M.V.; James, J.; Rafikova, O.; Rafikov, R. Glucose-6-phosphate dehydrogenase deficiency contributes to metabolic abnormality and pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2021, 320, L508–L521. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Eccles, C.A.; Desai, A.; Gonzalez-Garay, M.; Yuan, J.X.-J.; Garcia, J.G.N.; Rafikova, O.; Rafikov, R. New cases of Glucose-6-Phosphate Dehydrogenase Deficiency in Pulmonary Arterial Hypertension. PLoS ONE 2018, 13, e0203493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Zhang, R.; Shen, Y.-F.; Huang, K.-Y.; He, Y.-Y.; Zhao, J.-H.; Jing, Z.-C. 3-Bromopyruvate Attenuates Experimental Pulmonary Hypertension via Inhibition of Glycolysis. Am. J. Hypertens. 2018, 32, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-H.; Qiu, M.-H.; Wang, X.-J.; Sun, K.; Zheng, Y.; Jing, Z.-C. Up-regulation of hexokinase1 in the right ventricle of monocrotaline induced pulmonary hypertension. Respir. Res. 2014, 15, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malenfant, S.; Potus, F.; Fournier, F.; Breuils-Bonnet, S.; Pflieger, A.; Bourassa, S.; Tremblay, E.; Nehmé, B.; Droit, A.; Bonnet, S.; et al. Skeletal muscle proteomic signature and metabolic impairment in pulmonary hypertension. J. Mol. Med. 2014, 93, 573–584. [Google Scholar] [CrossRef]
- Archer, S.L.; Fang, Y.; Ryan, J.J.; Piao, L. Metabolism and Bioenergetics in the Right Ventricle and Pulmonary Vasculature in Pulmonary Hypertension. Pulm. Circ. 2013, 3, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.L.; Corey, C.; White, P.; Watson, A.; Gladwin, M.T.; Simon, M.; Shiva, S. Platelets from pulmonary hypertension patients show increased mitochondrial reserve capacity. JCI Insight 2017, 2, e91415. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.B.; Kumar, R.; Mickael, C.; Sanders, L.; Gebreab, L.; Huber, K.M.; Perez, M.; Smith-Jones, P.; Serkova, N.J.; Tuder, R.M. Severe pulmonary hypertension is associated with altered right ventricle metabolic substrate uptake. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L435–L440. [Google Scholar] [CrossRef] [Green Version]
- Prisco, S.Z.; Rose, L.; Potus, F.; Tian, L.; Wu, D.; Hartweck, L.; Al-Qazazi, R.; Neuber-Hess, M.; Eklund, M.; Hsu, S.; et al. Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 7278. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Futur. Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.B.; Michelakis, E.D. Fatty Acid Oxidation and Malonyl-CoA Decarboxylase in the Vascular Remodeling of Pulmonary Hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Manhas, A.; Kaur, G.; Jagavelu, K.; Hanif, K. Inhibition of fatty acid synthase is protective in pulmonary hypertension. J. Cereb. Blood Flow Metab. 2016, 173, 2030–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakao, S.; Kawakami, E.; Shoji, H.; Naito, A.; Miwa, H.; Suda, R.; Sanada, T.J.; Tanabe, N.; Tatsumi, K. Metabolic remodeling in the right ventricle of rats with severe pulmonary arterial hypertension. Mol. Med. Rep. 2021, 23, 1. [Google Scholar] [CrossRef]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef]
- Matsushita, T.; Ikeda, S.; Miyahara, Y.; Yakabe, K.; Yamaguchi, K.; Furukawa, K.; Iwasaki, T.; Shikuwa, M.; Fukui, J.; Kohno, S. Use of [123I]-BMIPP Myocardial Scintigraphy for the Clinical Evaluation of a Fatty-Acid Metabolism Disorder of the Right Ventricle in Chronic Respiratory and Pulmonary Vascular Disease. J. Int. Med Res. 2000, 28, 111–123. [Google Scholar] [CrossRef]
- Koop, A.C.; Bossers, G.P.L.; Ploegstra, M.; Hagdorn, Q.A.J.; Berger, R.M.F.; Silljé, H.H.W.; Bartelds, B. Metabolic Remodeling in the Pressure-Loaded Right Ventricle: Shifts in Glucose and Fatty Acid Metabolism—A Systematic Review and Meta-Analysis. J. Am. Hear. Assoc. 2019, 8, e012086. [Google Scholar] [CrossRef]
- Harvey, L.D.; Chan, S.Y. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. J. Clin. Med. 2017, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; McIntyre, R.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Chen, X.; Chen, X.; Li, J.; Cheng, B.; Xia, J. Mitochondrial Dynamics: Fission and Fusion in Fate Determination of Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2020, 8, 580070. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [Green Version]
- James, J.; Varghese, M.V.; Vasilyev, M.; Langlais, P.R.; Tofovic, S.P.; Rafikova, O.; Rafikov, R. Complex III Inhibition-Induced Pulmonary Hypertension Affects the Mitochondrial Proteomic Landscape. Int. J. Mol. Sci. 2020, 21, 5683. [Google Scholar] [CrossRef]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Bárcena, C.; López-Otín, C.; Yehia, G.; et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Alvira, C.M.; Umesh, A.; Husted, C.; Ying, L.; Hou, Y.; Lyu, S.-C.; Nowak, J.; Cornfield, D.N. Voltage-Dependent Anion Channel-2 Interaction with Nitric Oxide Synthase Enhances Pulmonary Artery Endothelial Cell Nitric Oxide Production. Am. J. Respir. Cell Mol. Biol. 2012, 47, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Corssac, G.B.; Bonetto, J.P.; Campos-Carraro, C.; Cechinel, L.R.; Zimmer, A.; Parmeggiani, B.; Grings, M.; Carregal, V.M.; Massensini, A.R.; Siqueira, I.; et al. Pulmonary arterial hypertension induces the release of circulating extracellular vesicles with oxidative content and alters redox and mitochondrial homeostasis in the brains of rats. Hypertens. Res. 2021, 44, 918–931. [Google Scholar] [CrossRef]
- Yeligar, S.M.; Kang, B.-Y.; Bijli, K.M.; Kleinhenz, J.M.; Murphy, T.C.; Torres, G.; Martin, A.S.; Sutliff, R.L.; Hart, C.M. PPARγ Regulates Mitochondrial Structure and Function and Human Pulmonary Artery Smooth Muscle Cell Proliferation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 648–657. [Google Scholar] [CrossRef]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef] [Green Version]
- Zurlo, G.; Piquereau, J.; Moulin, M.; Da Silva, J.P.; Gressette, M.; Ranchoux, B.; Garnier, A.; Ventura-Clapier, R.; Fadel, E.; Humbert, M.; et al. Sirtuin 1 regulates pulmonary artery smooth muscle cell proliferation: Role in pulmonary arterial hypertension. J. Hypertens. 2018, 36, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Afolayan, A.J.; Eis, A.; Alexander, M.; Michalkiewicz, T.; Teng, R.-J.; Lakshminrusimha, S.; Konduri, G.G. Decreased endothelial nitric oxide synthase expression and function contribute to impaired mitochondrial biogenesis and oxidative stress in fetal lambs with persistent pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L40–L49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Sud, N.; Wiseman, D.A.; Carter, A.L.; Kumar, S.; Hou, Y.; Rau, T.; Wilham, J.; Harmon, C.; Oishi, P.; et al. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L46–L56. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 240, pp. 159–188. [Google Scholar] [CrossRef]
- Ferguson, S.M.; De Camilli, P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 2012, 13, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pan, Z.; Ju, J.; Xing, C.; Li, X.; Shan, M.; Sun, S. DRP1 deficiency induces mitochondrial dysfunction and oxidative stress-mediated apoptosis during porcine oocyte maturation. J. Anim. Sci. Biotechnol. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Cho, B.; Cho, H.M.; Jo, Y.; Kim, H.D.; Song, M.; Moon, C.; Kim, H.; Kim, K.; Sesaki, H.; Rhyu, I.J.; et al. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat. Commun. 2017, 8, 15754. [Google Scholar] [CrossRef] [Green Version]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.; Hinshaw, J.E.; Green, D.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Potus, F.; Wu, D.; Dasgupta, A.; Chen, K.-H.; Mewburn, J.; Lima, P.; Archer, S.L. Increased Drp1-Mediated Mitochondrial Fission Promotes Proliferation and Collagen Production by Right Ventricular Fibroblasts in Experimental Pulmonary Arterial Hypertension. Front. Physiol. 2018, 9, 828. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D.; et al. Continuous Subcutaneous Infusion of Treprostinil, a Prostacyclin Analogue, in Patients with Pulmonary Arterial Hypertension: A Double-Blind, Randomized, Placebo-Controlled Trial. Am. J. Respir. Crit. Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef]
- Abu-Hanna, J.; Taanman, J.-W.; Abraham, D.; Clapp, L. Impact of treprostinil on dynamin-related protein 1 (DRP1) and mitochondrial fragmentation in pulmonary arterial hypertension (PAH). Eur. Respir. J. 2018, 52, PA3059. [Google Scholar] [CrossRef]
- Goldenberg, N.M.; Hu, Y.; Hu, X.; Volchuk, A.; Zhao, Y.D.; Kucherenko, M.M.; Knosalla, C.; De Perrot, M.; Tracey, K.J.; Al-Abed, Y.; et al. Therapeutic Targeting of High-Mobility Group Box-1 in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Wang, J.; Yan, X.; Zhang, Q.; Chai, L.; Wang, Q.; Shi, W.; Chen, Y.; Liu, J.; Qu, Z.; et al. ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. 2021, 54, e13048. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Guo, X.; Ma, D.; Guo, Y.; Li, Q.; Yang, D.; Li, P.; Qiu, X.; Wen, S.; Xiao, R.-P.; et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 2004, 6, 872–883. [Google Scholar] [CrossRef]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.J.; Marsboom, G.; Fang, Y.-H.; Toth, P.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J.; et al. PGC1α-mediated Mitofusin-2 Deficiency in Female Rats and Humans with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Mair, K.M.; Johansen, A.K.Z.; Wright, A.F.; Wallace, E.; MacLean, M.R. Pulmonary arterial hypertension: Basis of sex differences in incidence and treatment response. Br. J. Pharmacol. 2014, 171, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Soriano, F.X.; Liesa, M.; Bach, D.; Chan, D.C.; Palacín, M.; Zorzano, A. Evidence for a Mitochondrial Regulatory Pathway Defined by Peroxisome Proliferator–Activated Receptor-γ Coactivator-1α, Estrogen-Related Receptor-α, and Mitofusin 2. Diabetes 2006, 55, 1783–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased Plasma Endothelin-1 in Pulmonary Hypertension: Marker or Mediator of Disease? Ann. Intern. Med. 1991, 114, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, K.-P.; Hsu, C.-L.; Fan, L.-C.; Huang, Z.; Bhatia, D.; Chen, Y.-J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Szabo, A.; Sumegi, K.; Fekete, K.; Hocsak, E.; Debreceni, B.; Setalo, G.; Kovacs, K.; Deres, L.; Kengyel, A.; Kovacs, D.; et al. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem. Pharmacol. 2018, 150, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186, 111207. [Google Scholar] [CrossRef]
- Um, J.-H.; Yun, A.J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopitz, J.; Kisen, G.O.; Gordon, P.B.; Bohley, P.; Seglen, P.O. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 1990, 111, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Hibshman, J.D.; Leuthner, T.C.; Shoben, C.; Mello, D.F.; Sherwood, D.R.; Meyer, J.; Baugh, L.R. Nonselective autophagy reduces mitochondrial content during starvation in Caenorhabditis elegans. Am. J. Physiol. Physiol. 2018, 315, C781–C792. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef]
- Hoshino, A.; Wang, W.-J.; Wada, S.; McDermott-Roe, C.; Evans, C.; Gosis, B.; Morley, M.P.; Rathi, K.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef]
- Ordureau, A.; Sarraf, S.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Sarraf, S.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Mannam, P.; Zhang, J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L433–L452. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zuo, Q.; Zhou, Y.; Li, X. Mitophagy plays an important role in hypoxia-induced pulmonary artery smooth muscle cell proliferation. Eur. Respir. J. 2020, 56, 1485. [Google Scholar] [CrossRef]
- Klingenberg, M. Uncoupling Protein—A Useful Energy Dissepator. J. Bioenerg. Biomembr. 1999, 31, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Dromparis, P.; Paulin, R.; Sutendra, G.; Qi, A.C.; Bonnet, S.; Michelakis, E.D. Uncoupling Protein 2 Deficiency Mimics the Effects of Hypoxia and Endoplasmic Reticulum Stress on Mitochondria and Triggers Pseudohypoxic Pulmonary Vascular Remodeling and Pulmonary Hypertension. Circ. Res. 2013, 113, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haslip, M.; Dostanic, I.; Huang, Y.; Zhang, Y.; Russell, K.S.; Jurczak, M.J.; Mannam, P.; Giordano, F.; Erzurum, S.C.; Lee, P.J. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arter. Thromb. Vasc. Biol. 2015, 35, 1166–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsboom, G.; Toth, P.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1–Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Clin. Trans. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Sharma, S.; Aldred, M.A. DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes 2020, 11, 1224. [Google Scholar] [CrossRef]
- Ranchoux, B.; Meloche, J.; Paulin, R.; Boucherat, O.; Provencher, S.; Bonnet, S. DNA Damage and Pulmonary Hypertension. Int. J. Mol. Sci. 2016, 17, 990. [Google Scholar] [CrossRef] [Green Version]
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta- Gene Regul. Mech. 2012, 1819, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2020, 2019, 6175804. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Chi, A.Y.; Waypa, G.B.; Mungai, P.T.; Schumacker, P.T. Prolonged Hypoxia Increases ROS Signaling and RhoA Activation in Pulmonary Artery Smooth Muscle and Endothelial Cells. Antioxid. Redox Signal. 2010, 12, 603–610. [Google Scholar] [CrossRef]
- Waypa, G.B.; Marks, J.D.; Guzy, R.; Mungai, P.T.; Schriewer, J.; Dokic, D.; Schumacker, P.T. Hypoxia Triggers Subcellular Compartmental Redox Signaling in Vascular Smooth Muscle Cells. Circ. Res. 2010, 106, 526–535. [Google Scholar] [CrossRef]
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide Generated at Mitochondrial Complex III Triggers Acute Responses to Hypoxia in the Pulmonary Circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Rafikov, R.; Sun, X.; Rafikova, O.; Meadows, M.L.; Desai, A.; Khalpey, Z.; Yuan, J.X.-J.; Fineman, J.R.; Black, S.M. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol. 2015, 6, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A Fatal Mitochondrial Disease Is Associated with Defective NFU1 Function in the Maturation of a Subset of Mitochondrial Fe-S Proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Ahting, U.; Mayr, J.; Vanlander, A.V.; Hardy, S.A.; Santra, S.; Makowski, C.; Alston, C.; Zimmermann, F.A.; Abela, L.; Plecko, B.; et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front. Genet. 2015, 6, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, J.; Zemskova, M.; Eccles, C.A.; Varghese, M.V.; Niihori, M.; Barker, N.K.; Luo, M.; Mandarino, L.J.; Langlais, P.R.; Rafikova, O.; et al. Single Mutation in the NFU1 Gene Metabolically Reprograms Pulmonary Artery Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2021, 41, 734–754. [Google Scholar] [CrossRef] [PubMed]
- Niihori, M.; Eccles, C.A.; Kurdyukov, S.; Zemskova, M.; Varghese, M.V.; Stepanova, A.A.; Galkin, A.; Rafikov, R.; Rafikova, O. Rats with a Human Mutation of NFU1 Develop Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 62, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Michelakis, E.D.; Porter, C.; Andrade, M.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An Abnormal Mitochondrial–Hypoxia Inducible Factor-1α–Kv Channel Pathway Disrupts Oxygen Sensing and Triggers Pulmonary Arterial Hypertension in Fawn Hooded Rats: Similarities to Human Pulmonary Arterial Hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.N.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham-Snary, K.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Culley, M.K.; Chan, S.Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Investig. 2018, 128, 3704–3715. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic Targeting of Mitochondrial Superoxide in Hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Hartney, J.M.; Stidham, T.; Goldstrohm, D.A.; Oberley-Deegan, R.E.; Weaver, M.R.; Valnickova-Hansen, Z.; Scavenius, C.; Benninger, R.K.; Leahy, K.F.; Johnson, R.; et al. A Common Polymorphism in Extracellular Superoxide Dismutase Affects Cardiopulmonary Disease Risk by Altering Protein Distribution. Circ. Cardiovasc. Genet. 2014, 7, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Nozik-Grayck, E.; Woods, C.; Taylor, J.M.; Benninger, R.K.P.; Johnson, R.D.; Villegas, L.R.; Stenmark, K.R.; Harrison, D.G.; Majka, S.M.; Irwin, D.; et al. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L868–L876. [Google Scholar] [CrossRef] [Green Version]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial Fibroblasts Induce a Distinct Proinflammatory/Profibrotic Macrophage Phenotype in Pulmonary Hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, L.; Zhao, Q.-H.; Jiang, R.; Gong, S.-G.; Jiang, X.; Xu, X.-Q.; He, Y.-Y.; Li, Y.; Jing, Z.-C. Alteration of Extracellular Superoxide Dismutase in Idiopathic Pulmonary Arterial Hypertension. Front. Med. 2020, 7, 509. [Google Scholar] [CrossRef] [PubMed]
- Kusuyama, J.; Alves-Wagner, A.B.; Conlin, R.H.; Makarewicz, N.S.; Albertson, B.G.; Prince, N.B.; Kobayashi, S.; Kozuka, C.; Møller, M.; Bjerre, M.; et al. Placental superoxide dismutase 3 mediates benefits of maternal exercise on offspring health. Cell Metab. 2021, 33, 939–956.e8. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Bazan, I.; Zhang, Y.; Fares, W.H.; Lee, P.J. Mitochondrial dysfunction and pulmonary hypertension: Cause, effect, or both. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L782–L796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurtry, M.S.; Archer, S.L.; Altieri, D.C.; Bonnet, S.; Haromy, A.; Harry, G.; Bonnet, S.; Puttagunta, L.; Michelakis, E.D. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J. Clin. Investig. 2005, 115, 1479–1491. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colon Hidalgo, D.; Elajaili, H.; Suliman, H.; George, M.P.; Delaney, C.; Nozik, E. Metabolism, Mitochondrial Dysfunction, and Redox Homeostasis in Pulmonary Hypertension. Antioxidants 2022, 11, 428. https://doi.org/10.3390/antiox11020428
Colon Hidalgo D, Elajaili H, Suliman H, George MP, Delaney C, Nozik E. Metabolism, Mitochondrial Dysfunction, and Redox Homeostasis in Pulmonary Hypertension. Antioxidants. 2022; 11(2):428. https://doi.org/10.3390/antiox11020428
Chicago/Turabian StyleColon Hidalgo, Daniel, Hanan Elajaili, Hagir Suliman, Marjorie Patricia George, Cassidy Delaney, and Eva Nozik. 2022. "Metabolism, Mitochondrial Dysfunction, and Redox Homeostasis in Pulmonary Hypertension" Antioxidants 11, no. 2: 428. https://doi.org/10.3390/antiox11020428
APA StyleColon Hidalgo, D., Elajaili, H., Suliman, H., George, M. P., Delaney, C., & Nozik, E. (2022). Metabolism, Mitochondrial Dysfunction, and Redox Homeostasis in Pulmonary Hypertension. Antioxidants, 11(2), 428. https://doi.org/10.3390/antiox11020428