
Combined Treatment with Bojungikgi-Tang and Riluzole Regulates Muscle Metabolism and Dysfunction in the hSOD1G93A Mouse Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. BJIGT and RZ Treatment
2.3. Tissue Preparation
2.4. Western Blotting
2.5. Nissl Staining and Immunohistochemistry
2.6. RNA Extraction and Real-Time Reverse Transcription Polymerase Chain Reaction
2.7. ATP Level, Creatine Level, and Creatine Kinase Assays
2.8. Statistical Analysis
3. Results
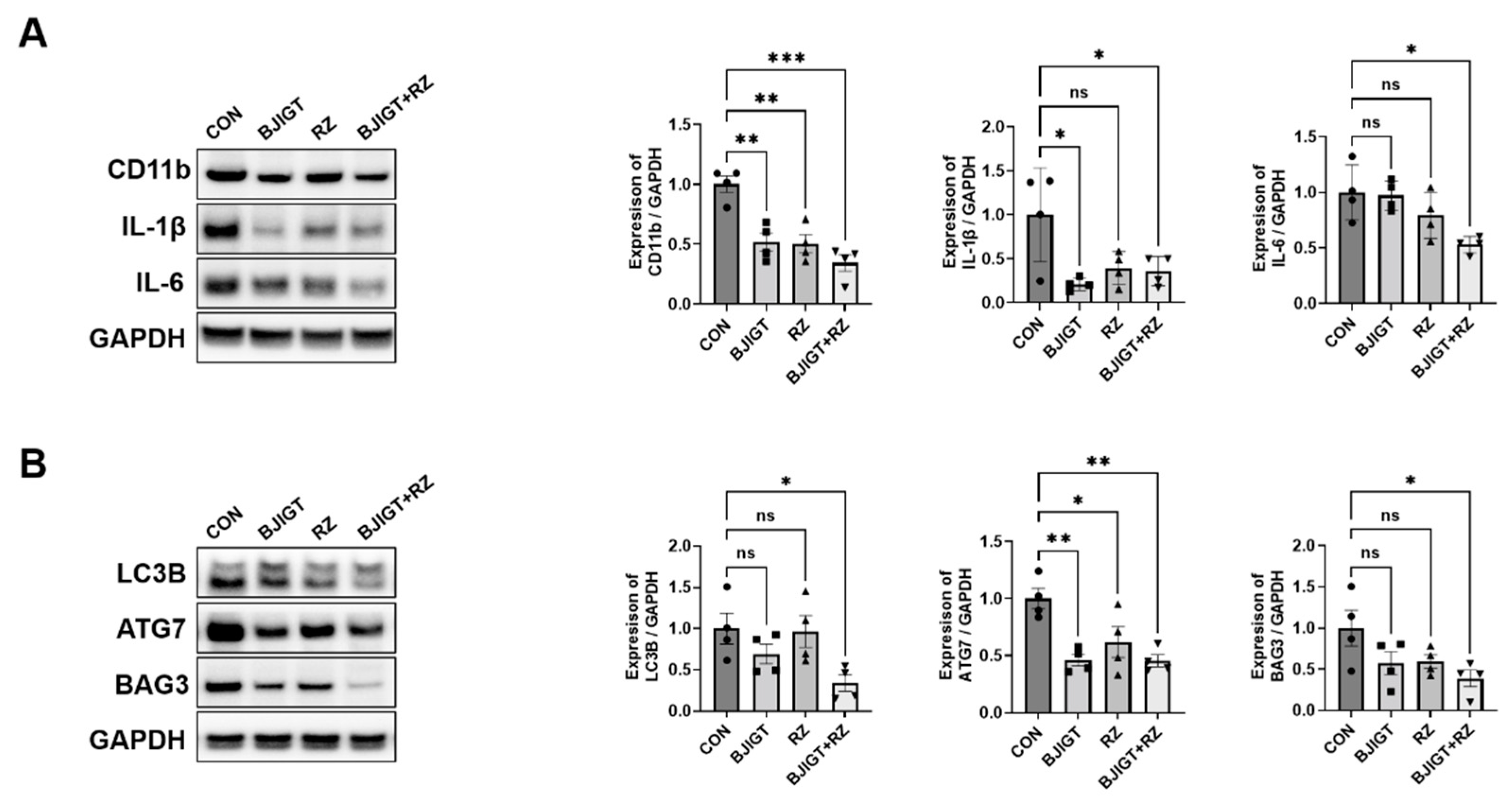
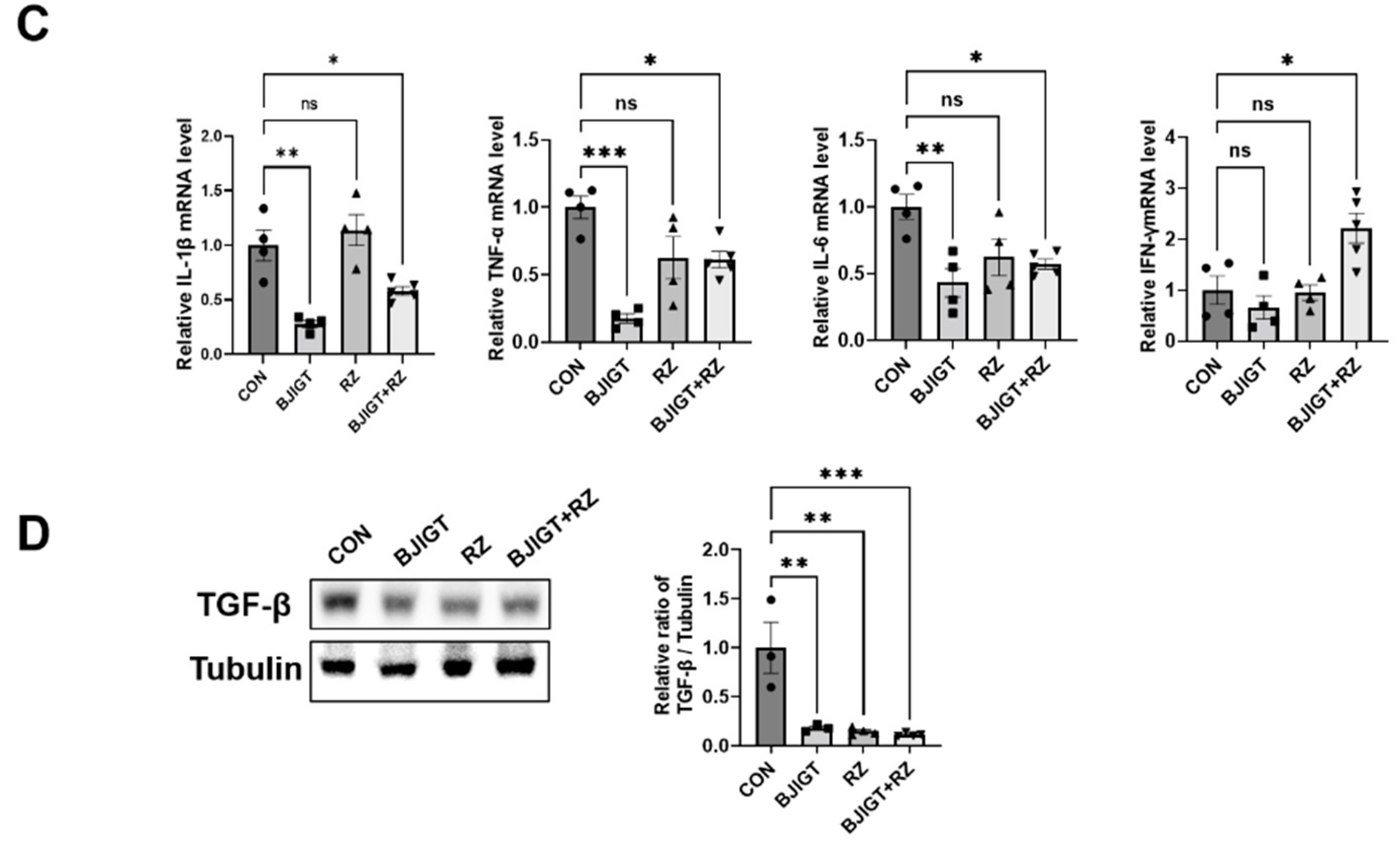
3.1. Combined BJIGT/RZ Treatment Affects Inflammation and Autophagy Dysfunction in the GC of the hSOD1G93A ALS Mouse Model
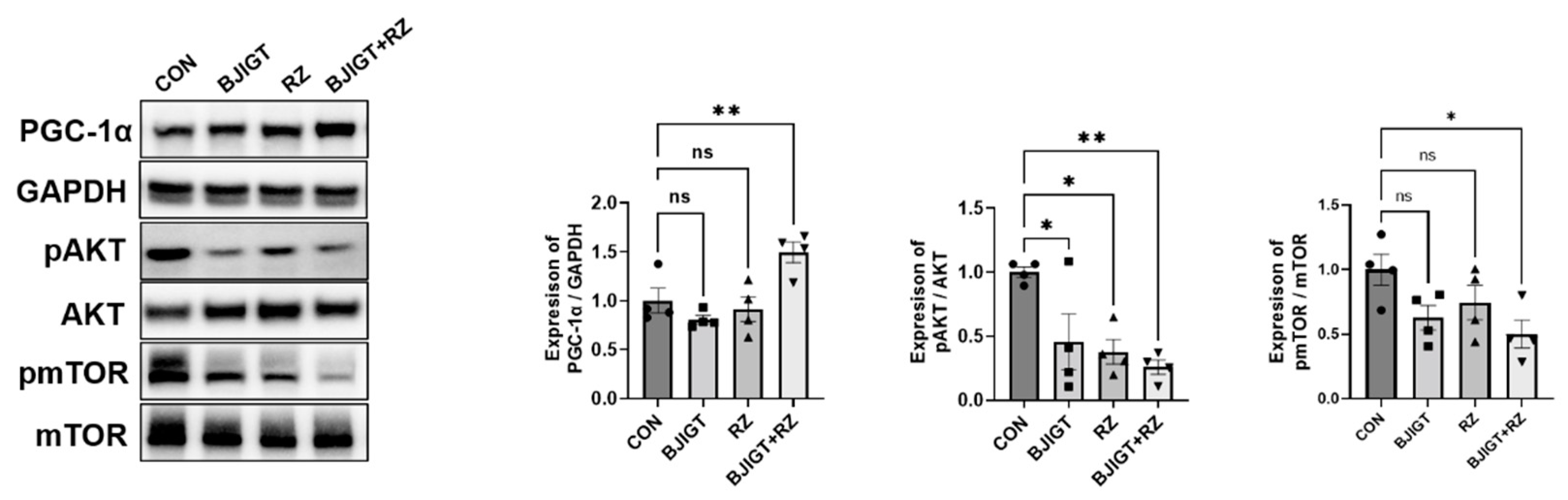
3.2. Combined BJIGT/RZ Treatment Ameliorates Metabolic Dysfunction in the GC of the hSOD1G93A ALS Mouse Model
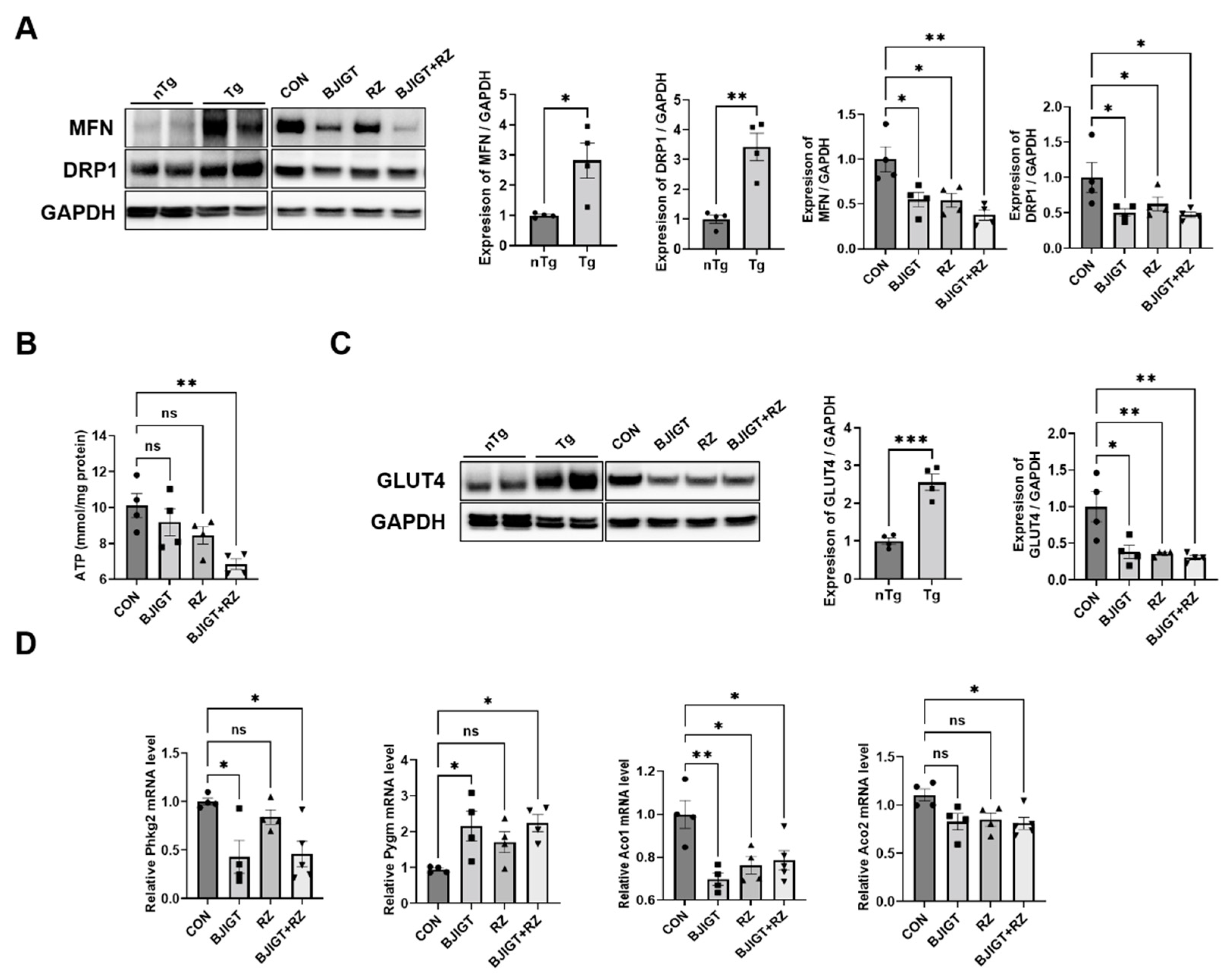
3.3. Combined BJIGT/RZ Treatment Regulates Mitochondrial Dysfunction in the GC of hSOD1G93A ALS Mouse Model
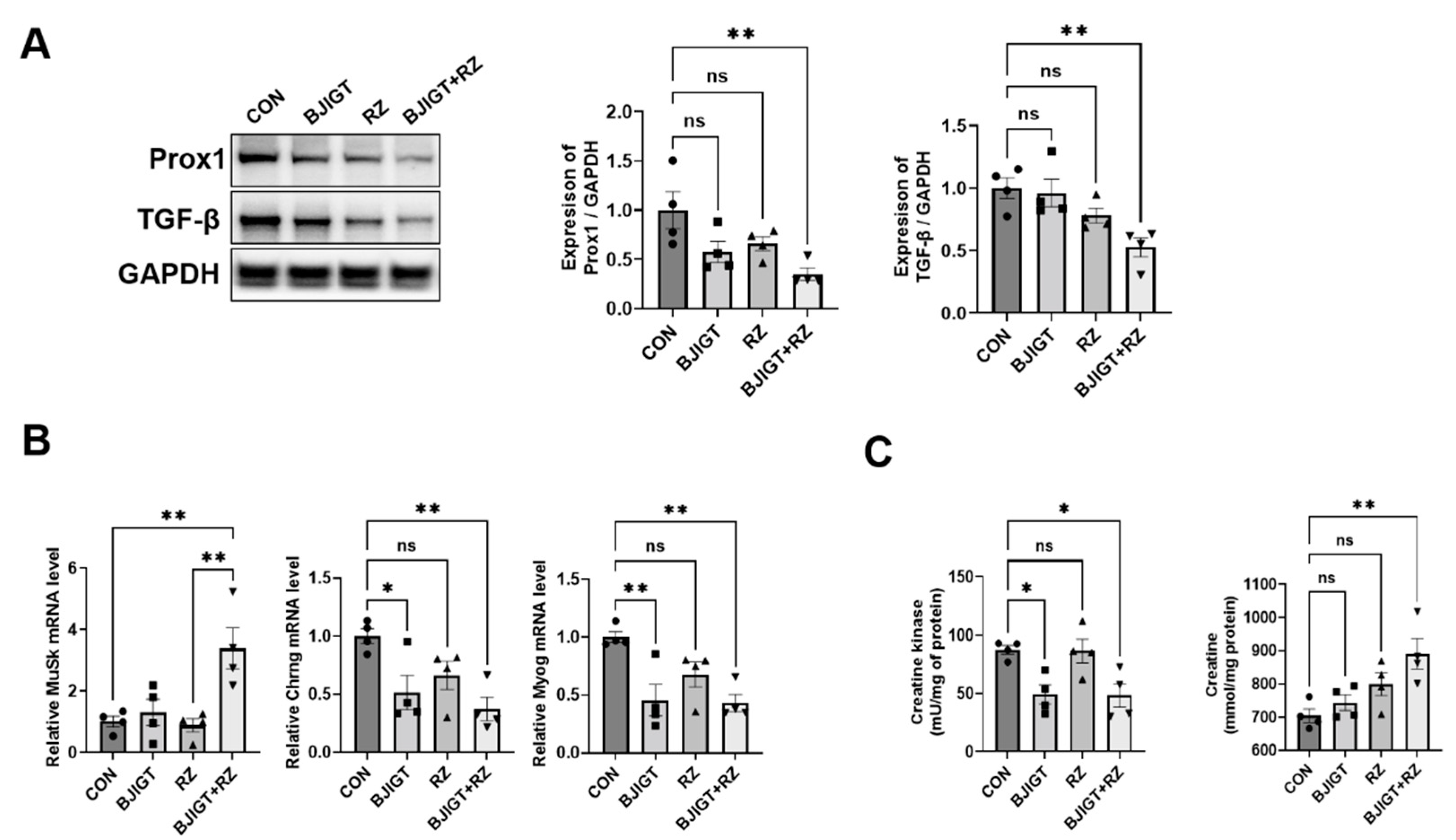
3.4. Combined BJIGT/RZ Treatment Attenuates Muscle Denervation in the GC of the hSOD1G93A ALS Mouse Model
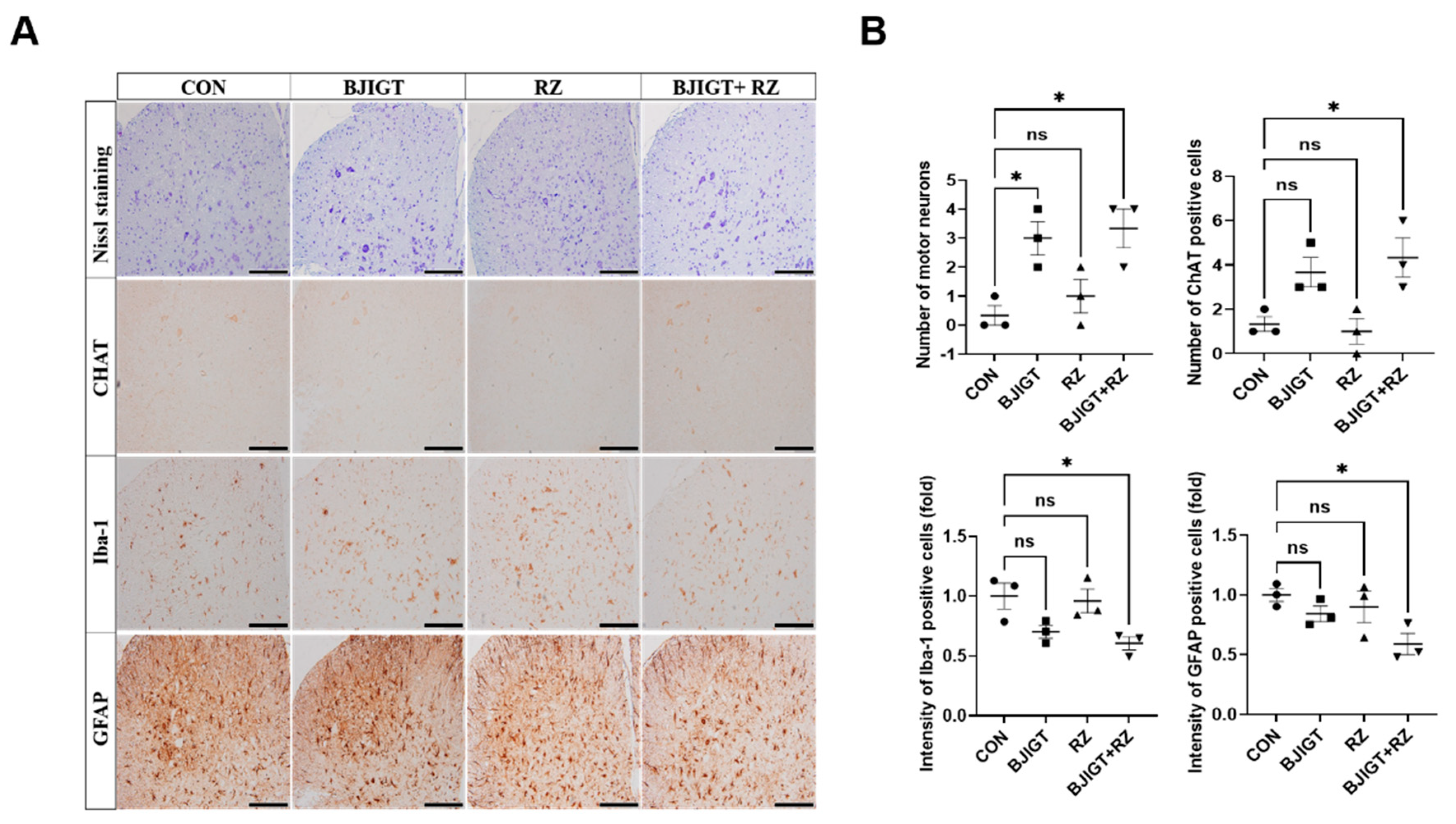
3.5. Combined BJIGT/RZ Treatment Protects Motor Neurons and Reduces Neuroinflammation in the SP of the hSOD1G93A ALS Mouse Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P. The Glutamate Hypothesis in ALS: Pathophysiology and Drug Development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Bonafede, R.; Mariotti, R. ALS Pathogenesis and Therapeutic Approaches: The Role of Mesenchymal Stem Cells and Extracellular Vesicles. Front. Cell. Neurosci. 2017, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2017, 10, 611. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Lamanauskas, N.; Nistri, A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur. J. Neurosci. 2008, 27, 2501–2514. [Google Scholar] [CrossRef]
- Doble, A. The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, S233–S241. [Google Scholar] [CrossRef]
- Gurney, M.E.; Fleck, T.J.; Himes, C.S.; Hall, E.D. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology 1998, 50, 62–66. [Google Scholar] [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Fang, T.; Al Khleifat, A.; Meurgey, J.-H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Lou, Y.; Kong, M.; Luo, Q.; Liu, Z.; Wu, J. A Systematic Review of Phytochemistry, Pharmacology and Pharmacokinetics on Astragali Radix: Implications for Astragali Radix as a Personalized Medicine. Int. J. Mol. Sci. 2019, 20, 1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Lv, C.; Lu, J. Natural occurring polysaccharides from Panax ginseng C. A. Meyer: A review of isolation, structures, and bioactivities. Int. J. Biol. Macromol. 2019, 133, 324–336. [Google Scholar] [CrossRef]
- Lim, H.-S.; Kim, Y.J.; Sohn, E.; Yoon, J.; Kim, B.-Y.; Jeong, S.-J. Bojungikgi-Tang, a Traditional Herbal Formula, Exerts Neuroprotective Effects and Ameliorates Memory Impairments in Alzheimer’s Disease-Like Experimental Models. Nutrients 2018, 10, 1952. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Lee, S.H.; Yang, E.J. Bojungikgi-tang Improves Muscle and Spinal Cord Function in an Amyotrophic Lateral Sclerosis Model. Mol. Neurobiol. 2019, 56, 2394–2407. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, X.; Deng, T.; Liu, X. Summary of professor Deng Tietao’s experience in treating amyotrophic lateral sclerosis. J. Guangzhou Univ. Tradit. Chin. Med. 2010, 27, 310–312. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, N. Jiaji electroacupuncture combined with traditional Chinese medicine to improve amyotrophic lateral sclerosis: A case. J. Clin. Acupunct. Moxibustion 2014, 30, 28–29. [Google Scholar]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Park, H.R.; Yang, E.J. Combined Treatment with Herbal Medicine and Drug Ameliorates Inflammation and Metabolic Abnormalities in the Liver of an Amyotrophic Lateral Sclerosis Mouse Model. Antioxidants 2022, 11, 173. [Google Scholar] [CrossRef]
- Crippa, V.; Boncoraglio, A.; Galbiati, M.; Aggarwal, T.; Rusmini, P.; Giorgetti, E.; Cristofani, R.; Carra, S.; Pennuto, M.; Poletti, A. Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front. Cell. Neurosci. 2013, 7, 234. [Google Scholar] [CrossRef] [Green Version]
- Ngo, S.; Steyn, F. The interplay between metabolic homeostasis and neurodegeneration: Insights into the neurometabolic nature of amyotrophic lateral sclerosis. Cell Regen. 2015, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Tsitkanou, S.; Della Gatta, P.A.; Russell, A.P. Skeletal Muscle Satellite Cells, Mitochondria, and MicroRNAs: Their Involvement in the Pathogenesis of ALS. Front. Physiol. 2016, 7, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective Mitochondrial Dynamics Is an Early Event in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Iłżecka, J.; Stelmasiak, Z.; Dobosz, B. Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine 2002, 20, 239–243. [Google Scholar] [CrossRef]
- Hogg, M.; Halang, L.; Woods, I.; Coughlan, K.S.; Prehn, J.H.M. Riluzole does not improve lifespan or motor function in three ALS mouse models. Amyotroph. Lateral Scler. Frontotemp. Degener. 2018, 19, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Komine, O.; Yamanaka, K. Neuroinflammation in motor neuron disease. Nagoya J. Med. Sci. 2015, 77, 537–549. [Google Scholar] [PubMed]
- Xiong, L.; McCoy, M.; Komuro, H.; West, X.Z.; Yakubenko, V.; Gao, D.; Dudiki, T.; Milo, A.; Chen, J.; Podrez, E.A.; et al. Inflammation-dependent oxidative stress metabolites as a hallmark of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2021, 178, 125–133. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, C.; Yi, J.; Wu, S.; Luo, G.; Xu, X.; Lin, P.-H.; Sun, J.; Zhou, J. Suppressed autophagy flux in skeletal muscle of an amyotrophic lateral sclerosis mouse model during disease progression. Physiol. Rep. 2015, 3, e12271. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Yang, E.J. Molecular factors related to skeletal muscle atrophy in a mouse model of amyotrophic lateral sclerosis. J. Biomed. Transl. Res. 2020, 21, 143–151. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.; Estrela, J. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Ioannides, Z.A.; Ngo, S.T.; Henderson, R.D.; McCombe, P.A.; Steyn, F.J. Altered Metabolic Homeostasis in Amyotrophic Lateral Sclerosis: Mechanisms of Energy Imbalance and Contribution to Disease Progression. Neurodegener. Dis. 2016, 16, 382–397. [Google Scholar] [CrossRef]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.; Wu, J.; Estall, J.; Irving, B.; et al. A PGC-1α Isoform Induced by Resistance Training Regulates Skeletal Muscle Hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Thau, N.; Knippenberg, S.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA Expression of PGC-1α and PGC-1α-Regulated Factors in the SOD1G93AALS Mouse Model and in Human Sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [Green Version]
- Cannavino, J.; Brocca, L.; Sandri, M.; Bottinelli, R.; Pellegrino, M.A. PGC1-α over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J. Physiol. 2014, 592, 4575–4589. [Google Scholar] [CrossRef]
- Bae, J.H.; Seo, D.Y.; Lee, S.H.; Shin, C.; Jamrasi, P.; Han, J.; Song, W. Effects of exercise on AKT/PGC1-alpha/FOXO3a pathway and muscle atrophy in cisplatin-administered rat skeletal muscle. Korean J. Physiol. Pharmacol. 2021, 25, 585–592. [Google Scholar] [CrossRef]
- Chiong, M.; Cartes-Saavedra, B.; Norambuena-Soto, I.; Mondaca-Ruff, D.; Morales, P.E.; Garcãa-Miguel, M.; Mellado, R.; Cartes-Saavedra, B.; García-Miguel, M. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front. Cell Dev. Biol. 2014, 2, 72. [Google Scholar] [CrossRef] [Green Version]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [Green Version]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2016, 2016, 5930621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-García, M.J.; Burden, S.J. Increasing MuSK Activity Delays Denervation and Improves Motor Function in ALS Mice. Cell Rep. 2012, 2, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, S.R.; Gnanasambandan, K. Structure and activation of MuSK, a receptor tyrosine kinase central to neuromuscular junction formation. Biochim. Biophys. Acta 2013, 1834, 2166–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattenlöhner, S.; Schneider, C.; Thamer, C.; Klein, R.; Roggendorf, W.; Gohlke, F.; Niethammer, C.; Czub, S.; Vincent, A.; Müller-Hermelink, H.; et al. Expression of foetal type acetylcholine receptor is restricted to type 1 muscle fibres in human neuromuscular disorders. Brain 2002, 125, 1309–1319. [Google Scholar] [CrossRef]
- Kostrominova, T.Y.; MacPherson, P.C.D.; Carlson, B.M.; Goldman, D. Regulation of myogenin protein expression in denervated muscles from young and old rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R179–R188. [Google Scholar] [CrossRef] [PubMed]
- Ceccanti, M.; Pozzilli, V.; Cambieri, C.; Libonati, L.; Onesti, E.; Frasca, V.; Fiorini, I.; Petrucci, A.; Garibaldi, M.; Palma, E.; et al. Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis. Cells 2020, 9, 1174. [Google Scholar] [CrossRef]
- Ito, D.; Hashizume, A.; Hijikata, Y.; Yamada, S.; Iguchi, Y.; Iida, M.; Kishimoto, Y.; Moriyoshi, H.; Hirakawa, A.; Katsuno, M. Elevated serum creatine kinase in the early stage of sporadic amyotrophic lateral sclerosis. J. Neurol. 2019, 266, 2952–2961. [Google Scholar] [CrossRef]
- Klivenyi, P.; Ferrante, R.J.; Matthews, R.T.; Bogdanov, M.B.; Klein, A.M.; Andreassen, O.A.; Mueller, G.; Wermer, M.; Kaddurah-Daouk, R.; Beal, M.F. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat. Med. 1999, 5, 347–350. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, M.; Yang, E.J. Combined Treatment with Bojungikgi-Tang and Riluzole Regulates Muscle Metabolism and Dysfunction in the hSOD1G93A Mouse Model. Antioxidants 2022, 11, 579. https://doi.org/10.3390/antiox11030579
Cai M, Yang EJ. Combined Treatment with Bojungikgi-Tang and Riluzole Regulates Muscle Metabolism and Dysfunction in the hSOD1G93A Mouse Model. Antioxidants. 2022; 11(3):579. https://doi.org/10.3390/antiox11030579
Chicago/Turabian StyleCai, Mudan, and Eun Jin Yang. 2022. "Combined Treatment with Bojungikgi-Tang and Riluzole Regulates Muscle Metabolism and Dysfunction in the hSOD1G93A Mouse Model" Antioxidants 11, no. 3: 579. https://doi.org/10.3390/antiox11030579
APA StyleCai, M., & Yang, E. J. (2022). Combined Treatment with Bojungikgi-Tang and Riluzole Regulates Muscle Metabolism and Dysfunction in the hSOD1G93A Mouse Model. Antioxidants, 11(3), 579. https://doi.org/10.3390/antiox11030579