
Methods for the Discovery and Identification of Small Molecules Targeting Oxidative Stress-Related Protein–Protein Interactions: An Update

 ,
,  and
and 
Abstract
: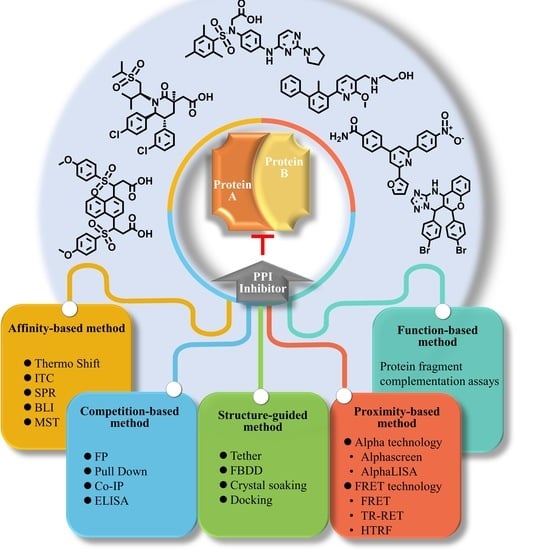
1. Introduction
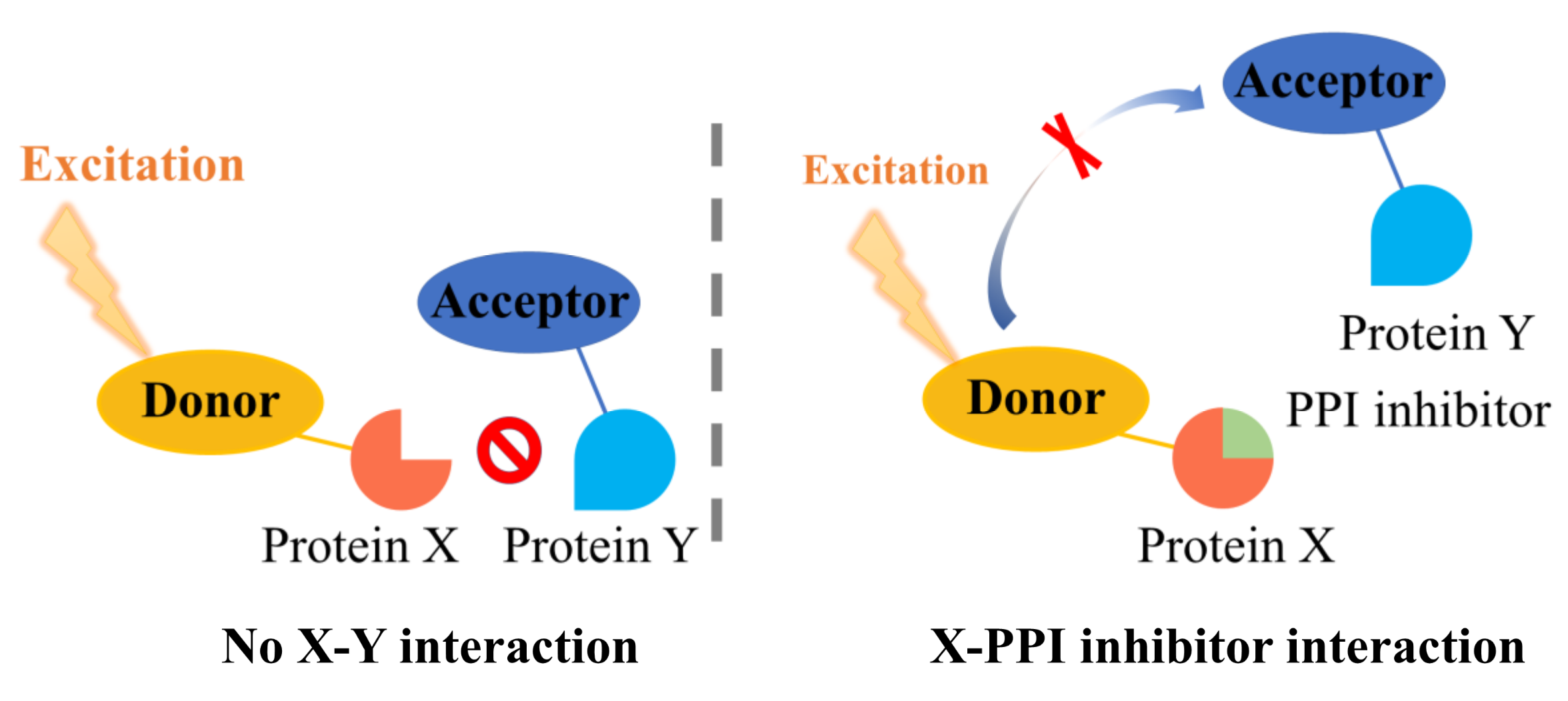
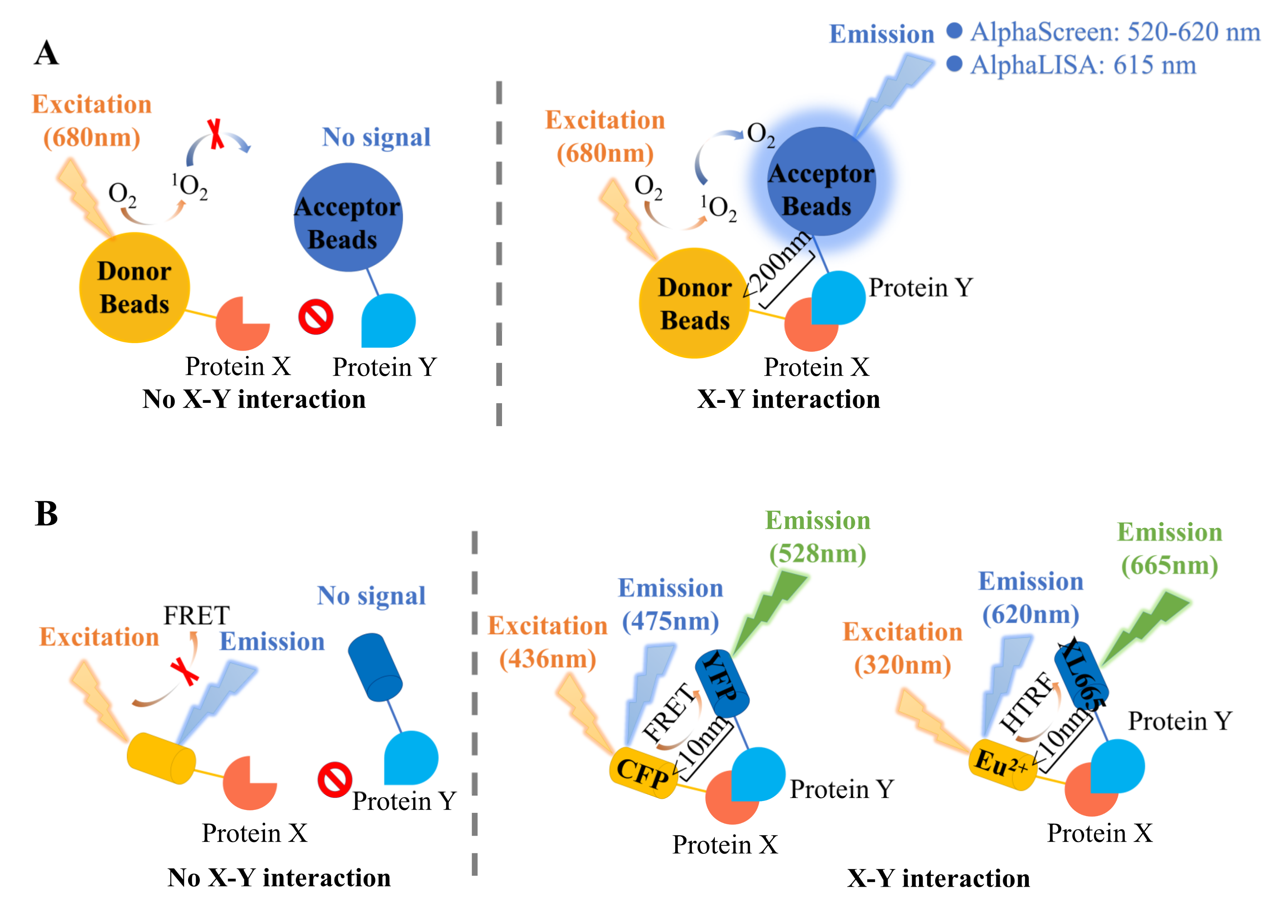
2. Proximity-Based Methods
2.1. Alpha Technology
2.2. Fluorescence Resonance Energy Transfer (FRET)
3. Affinity-Based Methods
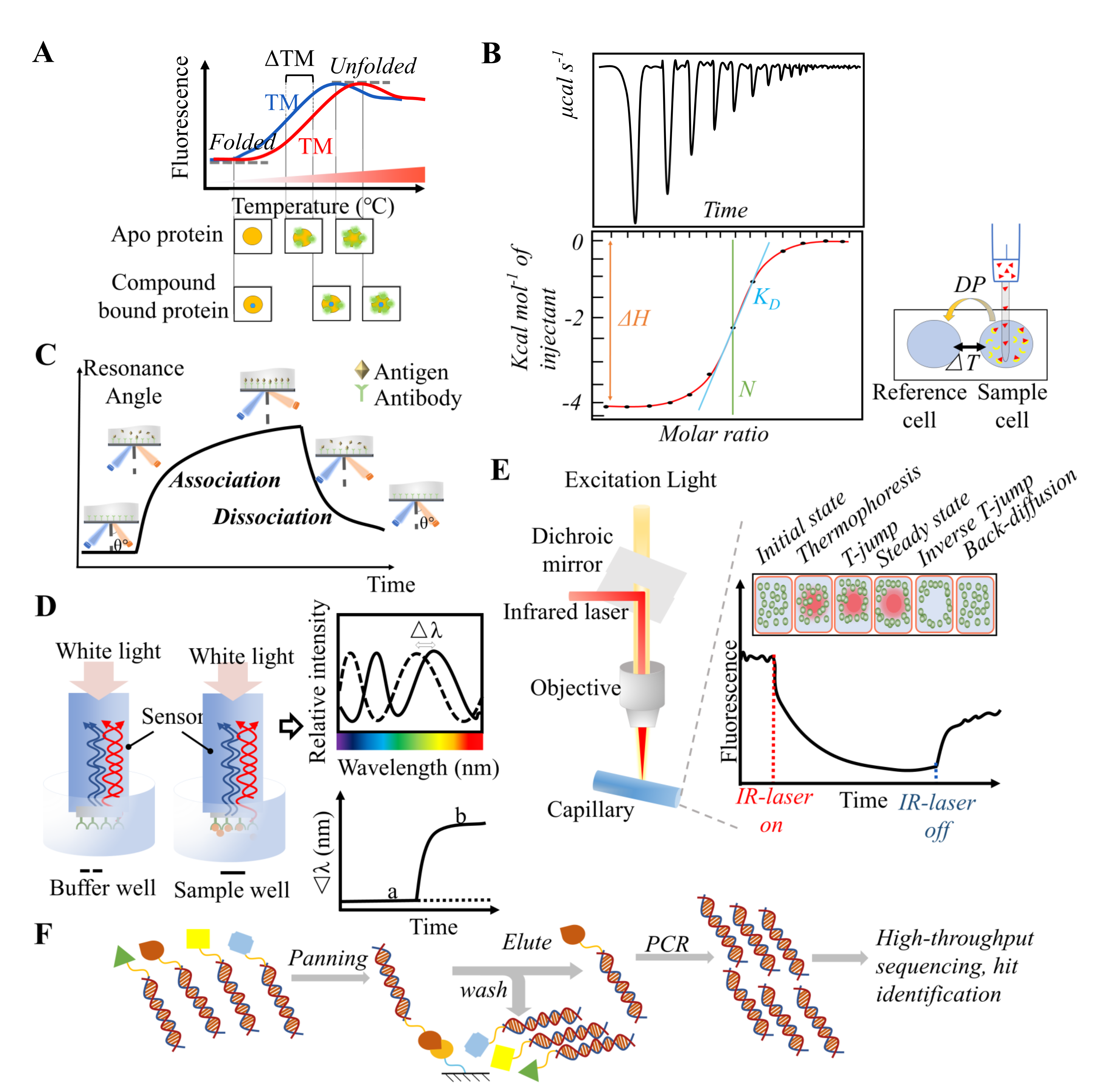
3.1. Thermal Shift Assay
3.2. Isothermal Titration Calorimetry (ITC)
3.3. Surface Plasmon Resonance (SPR)
3.4. Biolayer Interferometry (BLI)
3.5. Microscale Thermophoresis (MST)
3.6. DNA-Encoded Library
4. Competition-Based Methods
4.1. Fluorescence Polarization (FP)
4.2. Pull-Down
4.3. Co-Immunoprecipitation (Co-IP)
4.4. Enzyme Linked Immunosorbent Assay (ELISA)
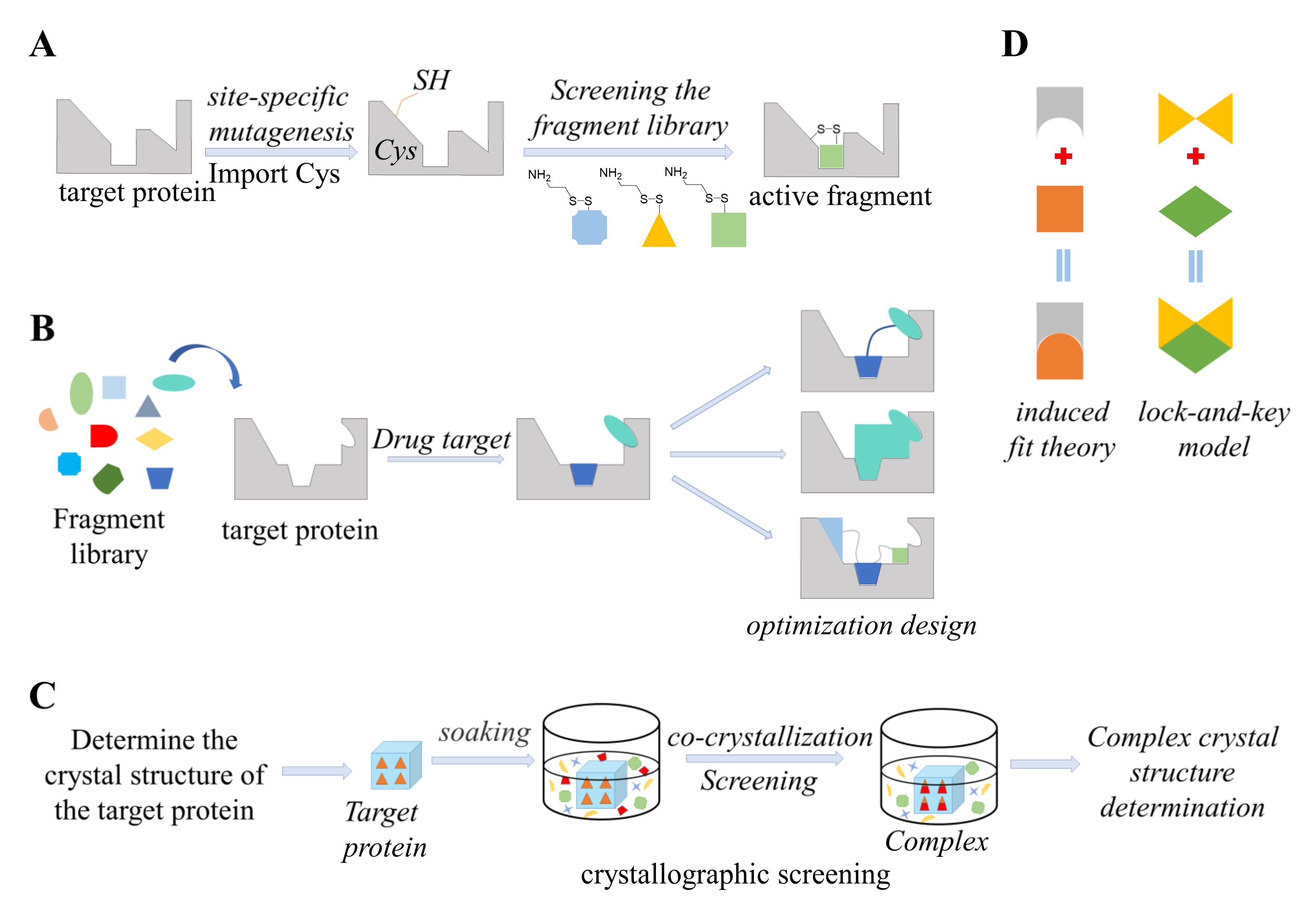
5. Structure-Guided Methods
5.1. Tethering
5.2. Fragment-Based Drug Design (FBDD)
5.3. Crystal Soaking
5.4. Computational Docking
6. Functional-Based Methods
Protein Fragment Complementation Assays (PCAs)
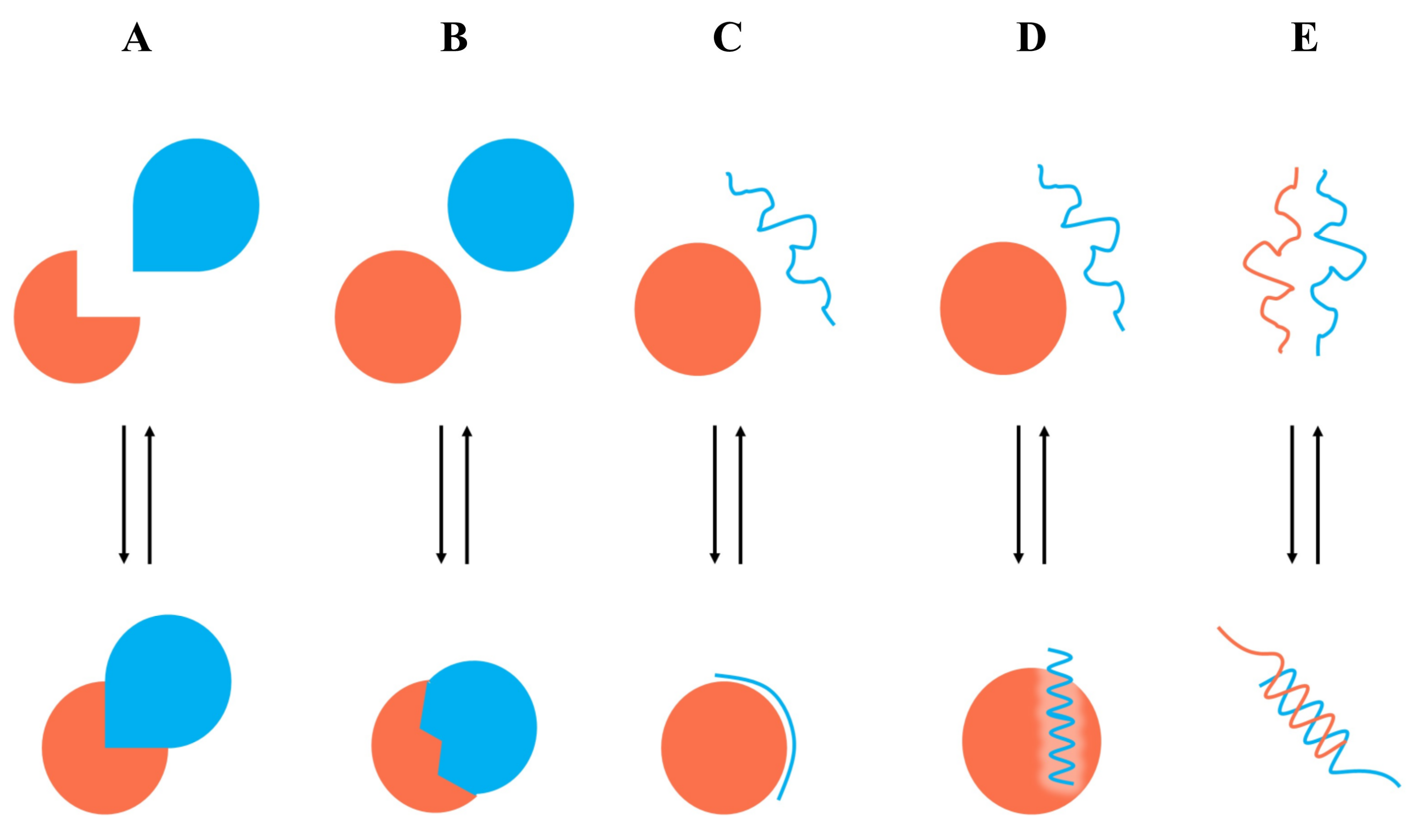
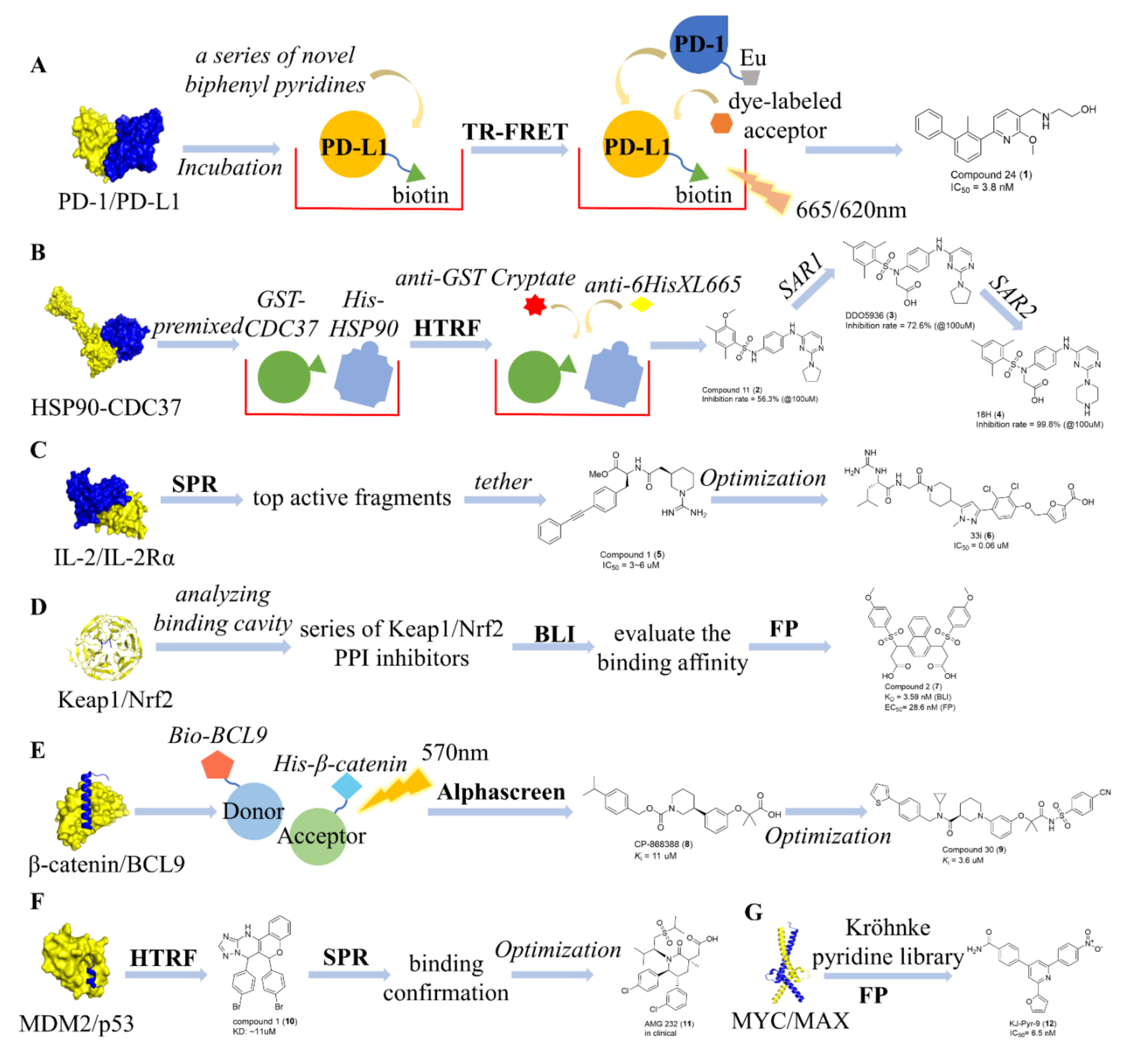
7. Case Study
7.1. Identification of Type I PPI Inhibitors
7.2. Identification of Type II PPI Inhibitors
7.3. Identification of Type III PPI Inhibitors
7.4. Identification of Type IV PPI Inhibitors
7.5. Identification of Type V PPI Inhibitors
8. Conclusions
9. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Caliceti, C.; Fogacci, F.; Giovannini, M.; Calabria, D.; Colletti, A.; Veronesi, M.; Roda, A.; Borghi, C. Effect of apple polyphenols on vascular oxidative stress and endothelium function: A translational study. Mol. Nutr. Food Res. 2017, 61, 11. [Google Scholar] [CrossRef]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J. Cell. Physiol. 2019, 234, 14951–14965. [Google Scholar] [CrossRef]
- Profumo, E.; Buttari, B.; Tinaburri, L.; D’Arcangelo, D.; Sorice, M.; Capozzi, A.; Garofalo, T.; Facchiano, A.; Businaro, R.; Kumar, P.; et al. Oxidative Stress Induces HSP90 Upregulation on the Surface of Primary Human Endothelial Cells: Role of the Antioxidant 7,8-Dihydroxy-4-methylcoumarin in Preventing HSP90 Exposure to the Immune System. Oxid. Med. Cell. Longev. 2018, 2018, 2373167. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Rajasekaran, M.; Hellstrom, W.J.G.; Naz, R.K.; Sikka, S.C. Oxidative stress and interleukins in seminal plasma during leukocytospermia. Fertil. Steril. 1995, 64, 166–171. [Google Scholar] [CrossRef]
- Nayki, C.; Gunay, M.; Kulhan, M.; Nayki, U.; Cankaya, M.; Kulhan, N.G. Serum levels of soluble interleukin-2 receptor in association with oxidative stress index in patients with different types of HPV. Ginekol. Pol. 2017, 88, 355359. [Google Scholar] [CrossRef]
- Bailly, C. Regulation of PD-L1 expression on cancer cells with ROS-modulating drugs. Life Sci. 2020, 246, 117403. [Google Scholar] [CrossRef]
- Najjar, Y.G.; Menk, A.V.; Sander, C.; Rao, U.; Karunamurthy, A.; Bhatia, R.; Zhai, S.; Kirkwood, J.M.; Delgoffe, G.M. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 2019, 4, e124989. [Google Scholar] [CrossRef]
- Beyfuss, K.; Hood, D.A. A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox. Rep. 2018, 23, 100–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, T.; D’Amico, E.; Moscatello, C.; Di Marcantonio, M.C.; Ferrone, A.; Bologna, G.; Selvaggi, F.; Lanuti, P.; Cotellese, R.; Curia, M.C.; et al. Oxidative Distress Induces Wnt/beta-Catenin Pathway Modulation in Colorectal Cancer Cells: Perspectives on APC Retained Functions. Cancers 2021, 13, 6045. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Li, W.; Cabeza-Morales, M.; Garcia-Foncillas, J. Oxidative Stress: A New Target for Pancreatic Cancer Prognosis and Treatment. J. Clin. Med. 2017, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug. Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Berg, T. Modulation of protein-protein interactions with small organic molecules. Angew. Chem. Int. Ed. Engl. 2003, 42, 2462–2481. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [Green Version]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef]
- Nickols, N.G.; Jacobs, C.S.; Farkas, M.E.; Dervan, P.B. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem. Biol. 2007, 2, 561–571. [Google Scholar] [CrossRef]
- Fuller, J.C.; Burgoyne, N.J.; Jackson, R.M. Predicting druggable binding sites at the protein-protein interface. Drug Discov Today. 2009, 14, 155–161. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target Ther. 2020, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Ullman, E.F.; Kirakossian, H.; Singh, S.; Wu, Z.P.; Irvin, B.R.; Pease, J.S.; Switchenko, A.C.; Irvine, J.D.; Dafforn, A.; Skold, C.N.; et al. Luminescent oxygen channeling immunoassay: Measurement of particle binding kinetics by chemiluminescence. Proc. Natl. Acad. Sci. USA 1994, 91, 5426–5430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eglen, R.M.; Reisine, T.; Roby, P.; Rouleau, N.; Illy, C.; Bosse, R.; Bielefeld, M. The use of AlphaScreen technology in HTS: Current status. Curr. Chem. Genom. 2008, 1, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Peppard, J.; Glickman, F.; He, Y.; Hu, S.I.; Doughty, J.; Goldberg, R. Development of a high-throughput screening assay for inhibitors of aggrecan cleavage using luminescent oxygen channeling (AlphaScreen). J. Biomol. Screen 2003, 8, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Petersen, D.N.; Hawkins, J.; Ruangsiriluk, W.; Stevens, K.A.; Maguire, B.A.; O’Connell, T.N.; Rocke, B.N.; Boehm, M.; Ruggeri, R.B.; Rolph, T.; et al. A Small-Molecule Anti-secretagogue of PCSK9 Targets the 80S Ribosome to Inhibit PCSK9 Protein Translation. Cell. Chem. Biol. 2016, 23, 1362–1371. [Google Scholar] [CrossRef] [Green Version]
- Walport, L.J.; Low, J.K.K.; Matthews, J.M.; Mackay, J.P. The characterization of protein interactions—What, how and how much? Chem. Soc. Rev. 2021, 50, 12292–12307. [Google Scholar] [CrossRef]
- Yasgar, A.; Jadhav, A.; Simeonov, A.; Coussens, N.P. AlphaScreen-Based Assays: Ultra-High-Throughput Screening for Small-Molecule Inhibitors of Challenging Enzymes and Protein-Protein Interactions. Methods Mol. Biol. 2016, 1439, 77–98. [Google Scholar]
- Cho, E.J.; Dalby, K.N. Luminescence Energy Transfer-Based Screening and Target Engagement Approaches for Chemical Biology and Drug Discovery. SLAS Discov. 2021, 26, 984–994. [Google Scholar] [CrossRef]
- Zhou, M.; Li, Q.; Kong, W.; Wang, R. Experimental Methods Used for Identifying Small-Molecule Inhibitors of Protein-Protein Interaction. In Targeting Protein-Protein Interactions by Small Molecules; Sheng, C., Georg, G.I., Eds.; Springer: Singapore, 2018. [Google Scholar]
- Forster, T. Intermolecular Energy Migration and Fluorescence. Ann. Phys. 1948, 2, 55–75. [Google Scholar]
- Muraru, S.; Muraru, S.; Nitu, F.R.; Ionita, M. Recent Efforts and Milestones for Simulating Nucleic Acid FRET Experiments through Computational Methods. J. Chem. Inf. Model 2022, 62, 232–239. [Google Scholar] [CrossRef]
- Rogers, M.S.; Cryan, L.M.; Habeshian, K.A.; Bazinet, L.; Caldwell, T.P.; Ackroyd, P.C.; Christensen, K.A. A FRET-based high throughput screening assay to identify inhibitors of anthrax protective antigen binding to capillary morphogenesis gene 2 protein. PLoS ONE 2012, 7, e39911. [Google Scholar]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degorce, F.; Card, A.; Soh, S.; Trinquet, E.; Knapik, G.P.; Xie, B. HTRF: A technology tailored for drug discovery—A review of theoretical aspects and recent applications. Curr. Chem. Genom. 2009, 3, 22–32. [Google Scholar] [CrossRef]
- Handl, H.L.; Gillies, R.J. Lanthanide-based luminescent assays for ligand-receptor interactions. Life Sci. 2005, 77, 361–371. [Google Scholar] [CrossRef]
- Alpha, B.; Ballardini, R.; Balzani, V.; Lehn, J.-M.; Perathoner, S.; Sabbatini, N. Antenna Effect in Luminescent Lanthanide Cryptates: A Photophysical Study. Photochem. Photobiol. 1990, 52, 299–306. [Google Scholar] [CrossRef]
- Bacart, J.; Corbel, C.; Jockers, R.; Bach, S.; Couturier, C. The BRET technology and its application to screening assays. Biotechnol. J. 2008, 3, 311–324. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen 2001, 6, 429–440. [Google Scholar] [CrossRef]
- Huynh, K.; Partch, C.L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 2015, 79, 2891–28914. [Google Scholar] [CrossRef]
- Lo, M.C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Layton, C.J.; Hellinga, H.W. Quantitation of protein-protein interactions by thermal stability shift analysis. Protein Sci. 2011, 20, 1439–1450. [Google Scholar] [CrossRef] [Green Version]
- Crowther, G.J.; He, P.; Rodenbough, P.P.; Thomas, A.P.; Kovzun, K.V.; Leibly, D.J.; Bhandari, J.; Castaneda, L.J.; Hol, W.G.; Gelb, M.H.; et al. Use of thermal melt curves to assess the quality of enzyme preparations. Anal. Biochem. 2010, 399, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voter, A.F.; Manthei, K.A.; Keck, J.L. A High-Throughput Screening Strategy to Identify Protein-Protein Interaction Inhibitors That Block the Fanconi Anemia DNA Repair Pathway. J. Biomol. Screen 2016, 21, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzi, S.; Restouin, A.; Opi, S.; Arold, S.T.; Parrot, I.; Guerlesquin, F.; Morelli, X.; Collette, Y. Protein protein interaction inhibition (2P2I) combining high throughput and virtual screening: Application to the HIV-1 Nef protein. Proc. Natl. Acad. Sci. USA 2007, 104, 19256–19261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkel, A.F.; Engel, C.K.; Margerie, D.; Kannt, A.; Szillat, H.; Glombik, H.; Kallus, C.; Ruf, S.; Gussregen, S.; Riedel, J.; et al. Characterization of RA839, a Noncovalent Small Molecule Binder to Keap1 and Selective Activator of Nrf2 Signaling. J. Biol. Chem. 2015, 290, 28446–28455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, M.; Bloch, I.; Shechter, N.; Romanenko, O.; Shir, O.M. Efficient Isothermal Titration Calorimetry Technique Identifies Direct Interaction of Small Molecule Inhibitors with the Target Protein. Comb. Chem. High Throughput Screen 2016, 19, 4–13. [Google Scholar] [CrossRef]
- Lin, K.; Wu, G. Isothermal Titration Calorimetry Assays to Measure Binding Affinities In Vitro. Methods Mol. Biol. 2019, 1893, 257–272. [Google Scholar]
- Falconer, R.J.; Penkova, A.; Jelesarov, I.; Collins, B.M. Survey of the year 2008: Applications of isothermal titration calorimetry. J. Mol. Recognit. 2010, 23, 395–413. [Google Scholar] [CrossRef]
- Krainer, G.; Keller, S. Single-experiment displacement assay for quantifying high-affinity binding by isothermal titration calorimetry. Methods 2015, 76, 116–123. [Google Scholar] [CrossRef]
- Karlsson, R.; Michaelsson, A.; Mattsson, L. Kinetic analysis of monoclonal antibody-antigen interactions with a new biosensor based analytical system. J. Immunol. Methods 1991, 145, 229–240. [Google Scholar] [CrossRef]
- Jönsson, U.; Malmqvist, M. Real time biospecific interaction analysis. The integration of surface plasmon resonance. Detection, general biospecific interface chemistry and microfluidics into one analytical system. Adv. Biosens. 1992, 2, 291–336. [Google Scholar]
- McDonnell, J.M. Surface plasmon resonance: Towards an understanding of the mechanisms of biological molecular recognition. Curr. Opin. Chem. Biol. 2001, 5, 572–577. [Google Scholar] [CrossRef]
- Englebienne, P.; Hoonacker, A.V.; Verhas, M. Surface plasmon resonance: Principles, methods and applications in biomedical sciences. Spectroscopy 2003, 17, 255–273. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface plasmon resonance: A versatile technique for biosensor applications. Sensors 2015, 15, 10481–10510. [Google Scholar] [CrossRef] [Green Version]
- Abdulhalim, I.; Zourob, M.; Lakhtakia, A. Surface Plasmon Resonance for Biosensing: A Mini-Review. Electromagnetics 2008, 28, 214–242. [Google Scholar] [CrossRef]
- Azzouz, A.; Hejji, L.; Kim, K.H.; Kukkar, D.; Souhail, B.; Bhardwaj, N.; Brown, R.J.C.; Zhang, W. Advances in surface plasmon resonance-based biosensor technologies for cancer biomarker detection. Biosens. Bioelectron. 2022, 197, 113767. [Google Scholar] [CrossRef]
- Homola, J. Present and future of surface plasmon resonance biosensors. Anal. Bioanal. Chem. 2003, 377, 528–539. [Google Scholar] [CrossRef]
- Dalal, K.; Ban, F.; Li, H.; Morin, H.; Roshan-Moniri, M.; Tam, K.J.; Shepherd, A.; Sharma, A.; Peacock, J.; Carlson, M.L.; et al. Selectively targeting the dimerization interface of human androgen receptor with small-molecules to treat castration-resistant prostate cancer. Cancer Lett. 2018, 437, 35–43. [Google Scholar] [CrossRef]
- Liu, S.; Su, Y.; Lin, M.Z.; Ronald, J.A. Brightening up Biology: Advances in Luciferase Systems for in Vivo Imaging. ACS Chem. Biol. 2021, 16, 2707–2718. [Google Scholar] [CrossRef]
- Concepcion, J.; Witte, K.; Wartchow, C.; Choo, S.; Yao, D.; Persson, H.; Wei, J.; Li, P.; Heidecker, B.; Ma, W.; et al. Label-free detection of biomolecular interactions using BioLayer interferometry for kinetic characterization. Comb. Chem. High Throughput Screen 2009, 12, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Lee, J.E. Measuring protein-protein and protein-nucleic Acid interactions by biolayer interferometry. Curr. Protoc. Protein Sci. 2015, 79, 19251–192526. [Google Scholar] [CrossRef] [PubMed]
- Wartchow, C.A.; Podlaski, F.; Li, S.; Rowan, K.; Zhang, X.; Mark, D.; Huang, K.S. Biosensor-based small molecule fragment screening with biolayer interferometry. J. Comput. Aided Mol. Des. 2011, 25, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Gogate, P.N.; Ethirajan, M.; Kurenova, E.V.; Magis, A.T.; Pandey, R.K.; Cance, W.G. Design, synthesis, and biological evaluation of novel FAK scaffold inhibitors targeting the FAK-VEGFR3 protein-protein interaction. Eur. J. Med. Chem. 2014, 80, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 100. [Google Scholar] [CrossRef] [Green Version]
- Linke, P.; Amaning, K.; Maschberger, M.; Vallee, F.; Steier, V.; Baaske, P.; Duhr, S.; Breitsprecher, D.; Rak, A. An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J. Biomol. Screen 2016, 21, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Cole, P.A. N-Terminal Protein Labeling with N-Hydroxysuccinimide Esters and Microscale Thermophoresis Measurements of Protein-Protein Interactions Using Labeled Protein. Curr. Protoc. 2021, 1, e14. [Google Scholar] [CrossRef]
- Mao, Y.; Yu, L.; Yang, R.; Qu, L.B.; Harrington, P.B. A novel method for the study of molecular interaction by using microscale thermophoresis. Talanta 2015, 132, 894–901. [Google Scholar] [CrossRef]
- Seidel, S.A.; Dijkman, P.M.; Lea, W.A.; van den Bogaart, G.; Jerabek-Willemsen, M.; Lazic, A.; Joseph, J.S.; Srinivasan, P.; Baaske, P.; Simeonov, A.; et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 2013, 59, 301–315. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockers, M.; Paul, N.W.; Efferth, T. Bisphenolic compounds alter gene expression in MCF-7 cells through interaction with estrogen receptor alpha. Toxicol. Appl. Pharmacol. 2020, 399, 115030. [Google Scholar] [CrossRef]
- Brenner, S.; Lerner, R.A. Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. USA 1992, 89, 5381–5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gironda-Martinez, A.; Donckele, E.J.; Samain, F.; Neri, D. DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. Transl. Sci. 2021, 4, 1265–1279. [Google Scholar] [CrossRef]
- Blakskjaer, P.; Heitner, T.; Hansen, N.J. Fidelity by design: Yoctoreactor and binder trap enrichment for small-molecule DNA-encoded libraries and drug discovery. Curr. Opin. Chem. Biol. 2015, 26, 62–71. [Google Scholar] [CrossRef]
- Perrin, F. Polarisation de la lumière de fluorescence. Vie moyenne des molécules dans l’etat excité. J. Phys. Radium 1926, 7, 390–401. [Google Scholar] [CrossRef]
- Weiel, J.; Hershey, J.W. The binding of fluorescein-labeled protein synthesis initiation factor 2 to Escherichia coli 30 S ribosomal subunits determined by fluorescence polarization. J. Biol. Chem. 1982, 257, 1215–1220. [Google Scholar] [CrossRef]
- Kim, Y.T.; Tabor, S.; Churchich, J.E.; Richardson, C.C. Interactions of gene 2.5 protein and DNA polymerase of bacteriophage T7. J. Biol. Chem. 1992, 267, 15032–15040. [Google Scholar] [CrossRef]
- Wang, Z.; Bhattacharya, A.; Ivanov, D.N. Identification of Small-Molecule Inhibitors of the HuR/RNA Interaction Using a Fluorescence Polarization Screening Assay Followed by NMR Validation. PLoS ONE 2015, 10, e0138780. [Google Scholar]
- Du, Y. Fluorescence polarization assay to quantify protein-protein interactions in an HTS format. Methods Mol. Biol. 2015, 1278, 529–544. [Google Scholar]
- Rusinova, E.; Tretyachenko-Ladokhina, V.; Vele, O.E.; Senear, D.F.; Alexander Ross, J.B. Alexa and Oregon Green dyes as fluorescence anisotropy probes for measuring protein–protein and protein–nucleic acid interactions. Anal. Biochem. 2002, 308, 18–25. [Google Scholar] [CrossRef]
- Turek-Etienne, T.C.; Lei, M.; Terracciano, J.S.; Langsdorf, E.F.; Bryant, R.W.; Hart, R.F.; Horan, A.C. Use of red-shifted dyes in a fluorescence polarization AKT kinase assay for detection of biological activity in natural product extracts. J. Biomol. Screen 2004, 9, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.D.; Yasgar, A.; Peryea, T.; Braisted, J.C.; Jadhav, A.; Simeonov, A.; Coussens, N.P. Fluorescence polarization assays in high-throughput screening and drug discovery: A review. Methods Appl. Fluoresc. 2016, 4, 022001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolovska-Coleska, Z.; Wang, R.; Fang, X.; Pan, H.; Tomita, Y.; Li, P.; Roller, P.P.; Krajewski, K.; Saito, N.G.; Stuckey, J.A.; et al. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 2004, 332, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Owicki, J.C. Fluorescence polarization and anisotropy in high throughput screening: Perspectives and primer. J. Biomol. Screen 2000, 5, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Turek-Etienne, T.C.; Small, E.C.; Soh, S.C.; Xin, T.A.; Gaitonde, P.V.; Barrabee, E.B.; Hart, R.F.; Bryant, R.W. Evaluation of fluorescent compound interference in 4 fluorescence polarization assays: 2 kinases, 1 protease, and 1 phosphatase. J. Biomol. Screen 2003, 8, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G.; Pallas, D.C.; DeCaprio, J.A.; Kaye, F.J.; Livingston, D.M. Identification of cellular proteins that can interact specifically with the T/ElA-binding region of the retinoblastoma gene product. Cell 1991, 64, 521–532. [Google Scholar] [CrossRef]
- Einarson, M.B.; Pugacheva, E.N.; Orlinick, J.R. GST Pull-down. CSH Protoc. 2007, 2007, pdb.prot4757. [Google Scholar] [CrossRef]
- Shao, H.; Xu, X.; Jing, N.; Tweardy, D.J. Unique structural determinants for Stat3 recruitment and activation by the granulocyte colony-stimulating factor receptor at phosphotyrosine ligands 704 and 744. J. Immunol. 2006, 176, 2933–2941. [Google Scholar] [CrossRef] [Green Version]
- Cendrowicz, E.; van Kessel, S.P.; van Bezouwen, L.S.; Kumar, N.; Boekema, E.J.; Scheffers, D.J. Bacillus subtilis SepF binds to the C-terminus of FtsZ. PLoS ONE 2012, 7, e43293. [Google Scholar]
- Mochizuki, Y.; Kohno, F.; Nishigaki, K.; Nemoto, N. A pull-down method with a biotinylated bait protein prepared by cell-free translation using a puromycin linker. Anal. Biochem. 2013, 434, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Coimmunoprecipitation assay. Methods Mol. Biol. 2007, 362, 401–406. [Google Scholar] [PubMed]
- Dwane, S.; Kiely, P.A. Tools used to study how protein complexes are assembled in signaling cascades. Bioeng. Bugs 2011, 2, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Markham, K.; Bai, Y.; Schmitt-Ulms, G. Co-immunoprecipitations revisited: An update on experimental concepts and their implementation for sensitive interactome investigations of endogenous proteins. Anal. Bioanal. Chem. 2007, 389, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Berggard, T.; Linse, S.; James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics 2007, 7, 2833–2842. [Google Scholar] [CrossRef]
- Lin, J.S.; Lai, E.M. Protein-Protein Interactions: Co-Immunoprecipitation. Methods Mol. Biol. 2017, 1615, 211–219. [Google Scholar]
- Biesiadecki, B.J.; Jin, J.P. A high-throughput solid-phase microplate protein-binding assay to investigate interactions between myofilament proteins. J. Biomed. Biotechnol. 2011, 2011, 421701. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; High, K.; Antonello, J.; Washabaugh, M.W.; Zhao, Q. Enhanced sensitivity and precision in an enzyme-linked immunosorbent assay with fluorogenic substrates compared with commonly used chromogenic substrates. Anal. Biochem. 2005, 345, 227–236. [Google Scholar] [CrossRef]
- Wang, Z.Z.; Shi, X.X.; Huang, G.Y.; Hao, G.F.; Yang, G.F. Fragment-based drug design facilitates selective kinase inhibitor discovery. Trends Pharmacol. Sci. 2021, 42, 551–565. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Deacon, A.M.; Duncton, M.A.J.; Pellicena, P.; Georgiadis, M.M.; Yeh, A.P.; Arvai, A.S.; Moiani, D.; Tainer, J.A.; Das, D. Fragment- and structure-based drug discovery for developing therapeutic agents targeting the DNA Damage Response. Prog. Biophys. Mol. Biol. 2021, 163, 130–142. [Google Scholar] [CrossRef]
- Nienaber, V.L.; Richardson, P.L.; Klighofer, V.; Bouska, J.J.; Giranda, V.L.; Greer, J. Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nat. Biotechnol. 2000, 18, 1105–1108. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guo, R.; Zong, Q.; Ling, G. Application of molecular docking in elaborating molecular mechanisms and interactions of supramolecular cyclodextrin. Carbohydr. Polym. 2022, 276, 118644. [Google Scholar] [CrossRef] [PubMed]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L.A. Machine-learning methods for ligand-protein molecular docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, S.; Wang, M.; Zhang, W.; Zhang, K.; Chen, D.; Li, Z.; Jiang, S. Novel Biphenyl Pyridines as Potent Small-Molecule Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2021, 64, 7390–7403. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Q.; You, Q. Targeting the HSP90-CDC37-kinase chaperone cycle: A promising therapeutic strategy for cancer. Med. Res. Rev. 2021, 42, 156–182. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.X.; Li, L.; Jiang, J.S.; Zheng, Z.; Shang, J.L.; Wang, C.X.; Chen, W.L.; Bao, Q.C.; Xu, X.L.; et al. Small-molecule inhibitor targeting the Hsp90-Cdc37 protein-protein interaction in colorectal cancer. Sci. Adv. 2019, 5, eaax2277. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jiang, J.S.; Zhang, L.X.; Zhang, Q.Y.; Zhou, J.R.; Li, L.; Xu, X.L.; You, Q.D. Discovery and Optimization of Small Molecules Targeting the Protein-Protein Interaction of Heat Shock Protein 90 (Hsp90) and Cell Division Cycle 37 as Orally Active Inhibitors for the Treatment of Colorectal Cancer. J. Med. Chem. 2020, 63, 1281–1297. [Google Scholar] [CrossRef]
- Rubin, L.A.; Nelson, D.L. The soluble interleukin-2 receptor: Biology, function, and clinical application. Ann. Intern Med. 1990, 113, 619–627. [Google Scholar] [CrossRef]
- Nelson, B.H.; Willeford, D.M. Biology of the interleukin-2 receptor. Adv. Immunol. 1998, 70, 1–81. [Google Scholar]
- Waldmann, T.A.; Dubois, S.; Tagaya, Y. Contrasting Roles of IL-2 and IL-15 in the Life and Death of Lymphocytes. Immunity 2001, 14, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Nelson, B.H. Interleukin-2 signaling and the maintenance of self-tolerance. Curr. Dir. Autoimmun. 2002, 5, 92–112. [Google Scholar] [PubMed]
- Church, A.C. Clinical advances in therapies targeting the interleukin-2 receptor. QJM 2003, 96, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmann, T.A. The IL-2/IL-2 receptor system: A target for rational immune intervention. Immunol. Today 1993, 14, 264–270. [Google Scholar] [CrossRef]
- Berard, J.L.; Velez, R.L.; Freeman, R.B.; Tsunoda, S.M. A review of interleukin-2 receptor antagonists in solid organ transplantation. Pharmacotherapy 1999, 19, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; O’Shea, J. The use of antibodies against the IL-2 receptor in transplantation. Curr. Opin. Immunol. 1998, 10, 507–512. [Google Scholar] [CrossRef]
- Rickert, M.; Wang, X.; Boulanger, M.J.; Goriatcheva, N.; Garcia, K.C. The structure of interleukin-2 complexed with its alpha receptor. Science 2005, 308, 1477–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimundo, B.C.; Oslob, J.D.; Braisted, A.C.; Hyde, J.; McDowell, R.S.; Randal, M.; Waal, N.D.; Wilkinson, J.; Yu, C.H.; Arkin, M.R. Integrating fragment assembly and biophysical methods in the chemical advancement of small-molecule antagonists of IL-2: An approach for inhibiting protein-protein interactions. J. Med. Chem. 2004, 47, 3111–3130. [Google Scholar] [CrossRef]
- Bartolini, D.; Torquato, P.; Piroddi, M.; Galli, F. Targeting glutathione S-transferase P and its interactome with selenium compounds in cancer therapy. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 130–143. [Google Scholar] [CrossRef]
- Ali, A.; Shah, A.S.; Ahmad, A. Gain-of-function of mutant p53: Mutant p53 enhances cancer progression by inhibiting KLF17 expression in invasive breast carcinoma cells. Cancer Lett. 2014, 354, 87–96. [Google Scholar] [CrossRef]
- Bartolini, D.; Giustarini, D.; Pietrella, D.; Rossi, R.; Galli, F. Glutathione S-transferase P influences the Nrf2-dependent response of cellular thiols to seleno-compounds. Cell. Biol. Toxicol. 2020, 36, 379–386. [Google Scholar] [CrossRef]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Wisniewski, J.A.; Ji, H. AlphaScreen selectivity assay for beta-catenin/B-cell lymphoma 9 inhibitors. Anal. Biochem. 2015, 469, 43–53. [Google Scholar] [CrossRef]
- Salik, B.; Yi, H.; Hassan, N.; Santiappillai, N.; Vick, B.; Connerty, P.; Duly, A.; Trahair, T.; Woo, A.J.; Beck, D.; et al. Targeting RSPO3-LGR4 Signaling for Leukemia Stem Cell Eradication in Acute Myeloid Leukemia. Cancer Cell. 2020, 38, 263–278.e6. [Google Scholar] [CrossRef]
- Soleas, J.P.; D’Arcangelo, E.; Huang, L.; Karoubi, G.; Nostro, M.C.; McGuigan, A.P.; Waddell, T.K. Assembly of lung progenitors into developmentally-inspired geometry drives differentiation via cellular tension. Biomaterials 2020, 254, 120128. [Google Scholar] [CrossRef]
- Choi, B.R.; Cave, C.; Na, C.H.; Sockanathan, S. GDE2-Dependent Activation of Canonical Wnt Signaling in Neurons Regulates Oligodendrocyte Maturation. Cell Rep. 2020, 31, 107540. [Google Scholar] [CrossRef]
- Zhang, M.; Weng, W.; Zhang, Q.; Wu, Y.; Ni, S.; Tan, C.; Xu, M.; Sun, H.; Liu, C.; Wei, P.; et al. The lncRNA NEAT1 activates Wnt/beta-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J. Hematol. Oncol. 2018, 11, 113. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Tang, S. WNT/beta-catenin signaling in the development of liver cancers. Biomed. Pharmacother. 2020, 132, 110851. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, M.; Luo, W.; Zhang, Y.; Ji, H. Discovery of 2-(3-(3-Carbamoylpiperidin-1-yl)phenoxy)acetic Acid Derivatives as Novel Small-Molecule Inhibitors of the beta-Catenin/B-Cell Lymphoma 9 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 5886–5904. [Google Scholar] [CrossRef]
- Allen, J.G.; Bourbeau, M.P.; Wohlhieter, G.E.; Bartberger, M.D.; Michelsen, K.; Hungate, R.; Gadwood, R.C.; Gaston, R.D.; Evans, B.; Mann, L.W.; et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein-protein interaction. J. Med. Chem. 2009, 52, 7044–7053. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.P.; DeGraffenreid, M.; Fox, B.; Allen, J.G.; Rew, Y.; Schneider, S.; Saiki, A.Y.; Yu, D.; Oliner, J.D.; Salyers, K.; et al. Improvement of the synthesis and pharmacokinetic properties of chromenotriazolopyrimidine MDM2-p53 protein-protein inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2752–2755. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Sun, D.; Gonzalez-Lopez De Turiso, F.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B.M.; et al. Structure-based design of novel inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2012, 55, 4936–4954. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Gisselberg, J.E.; Herrera, Z.; Orchard, L.M.; Llinas, M.; Yeh, E. Specific Inhibition of the Bifunctional Farnesyl/Geranylgeranyl Diphosphate Synthase in Malaria Parasites via a New Small-Molecule Binding Site. Cell Chem. Biol. 2018, 25, 185–193.e5. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Ji, H.; Wu, G.; Zhan, X.; Nolan, A.; Koh, C.; De Marzo, A.; Doan, H.M.; Fan, J.; Cheadle, C.; Fallahi, M.; et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 2011, 6, e26057. [Google Scholar] [CrossRef]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.K.; Burley, S.K. X-Ray Structures of Myc-Max and Mad-Max Recognizing DNA. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Hart, J.R.; Garner, A.L.; Yu, J.; Ito, Y.; Sun, M.; Ueno, L.; Rhee, J.K.; Baksh, M.M.; Stefan, E.; Hartl, M.; et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proc. Natl. Acad. Sci. USA 2014, 111, 12556–12561. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Alpha Technology | FRET Technology | |||
|---|---|---|---|---|---|
| Alpha- Screen | Alpha- LISA | FRET | TR-RET | HTRF | |
| Excitation wavelength (nm) | 680 | 436 | 320 | ||
| Emission wavelength (nm) | 520–620 | 615 | 528 | 665 | |
| Distance (nm) | <200 | <10 | |||
| Advantage |
|
| |||
| Disadvantage |
|
| |||
| Applicable PPI type | I, II | I, II | |||
| HTS suitability | Yes | Yes | |||
| Assay | Advantages | Disadvantages | Applicable PPI Type | HTS Suitability |
|---|---|---|---|---|
| Thermal Shift |
| Not suitable for some proteins that have no ideal thermal denaturation curve and stability information cannot be extracted | All types | No |
| ITC |
|
| All types | No |
| SPR |
|
| All types | No |
| BLI |
|
| All types | No |
| MST |
| No accurate information on stoichiometry can be obtained. | All types | No |
| Assay | Advantage | Disadvantage | Applicable PPI Type | HTS Suitability |
|---|---|---|---|---|
| FP | Simple operation High throughput Real-time and homogeneity Friendly to experimenters | Interference from light scattering, quenching, and auto-fluorescence. | All types | Yes |
| Pull Down | Direct protein–protein interactions can be verified Conjugated beads with strong affinity and high elution purity | Cannot fully reflect the true state of intracellular protein interaction, and the fusion expressed GST tag may change the original folding of the target protein structure. | All types | No |
| Co-IP | Reflect the real interaction of target PPI in intact cells | Low affinity and transient protein–protein interactions may not be detected. May not reflect direct interaction, a third party may act as a bridge in between. | All types | No |
| ELISA | Highly sensitivity | Time-consuming Weak interactions are hard to detect | All types | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Zhang, Q.; Guo, Y.; Zhang, H.; Guo, X.; You, Q.; Wang, L. Methods for the Discovery and Identification of Small Molecules Targeting Oxidative Stress-Related Protein–Protein Interactions: An Update. Antioxidants 2022, 11, 619. https://doi.org/10.3390/antiox11040619
Wu X, Zhang Q, Guo Y, Zhang H, Guo X, You Q, Wang L. Methods for the Discovery and Identification of Small Molecules Targeting Oxidative Stress-Related Protein–Protein Interactions: An Update. Antioxidants. 2022; 11(4):619. https://doi.org/10.3390/antiox11040619
Chicago/Turabian StyleWu, Xuexuan, Qiuyue Zhang, Yuqi Guo, Hengheng Zhang, Xiaoke Guo, Qidong You, and Lei Wang. 2022. "Methods for the Discovery and Identification of Small Molecules Targeting Oxidative Stress-Related Protein–Protein Interactions: An Update" Antioxidants 11, no. 4: 619. https://doi.org/10.3390/antiox11040619
APA StyleWu, X., Zhang, Q., Guo, Y., Zhang, H., Guo, X., You, Q., & Wang, L. (2022). Methods for the Discovery and Identification of Small Molecules Targeting Oxidative Stress-Related Protein–Protein Interactions: An Update. Antioxidants, 11(4), 619. https://doi.org/10.3390/antiox11040619