
The BCAT1 CXXC Motif Provides Protection against ROS in Acute Myeloid Leukaemia Cells

 ,
,  , ,
, , 
Abstract
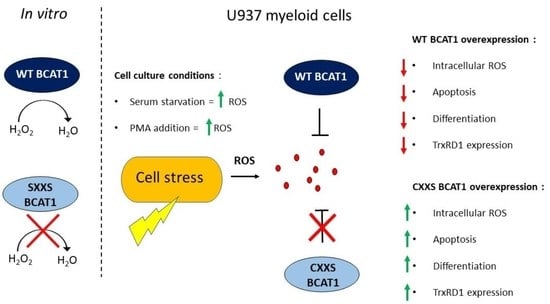
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. BCAT1 Site Directed Mutagenesis and Retroviral Transduction of U937 Cells
2.3. Overexpression and Purification of Recombinant BCAT1 Protein in E. coli
2.4. Spectrophotometric Confirmation of Cysteine Mutation by DTNB Titration
2.5. Hydrogen Peroxide Metabolism Assay
2.6. qPCR of BCAT1 and TrxRD1 in Transgenic U937 Cells
2.7. Western Blot Detection of Transgenic BCAT1 in U937 Cells
2.8. In Vitro Growth Characterisation of Transgenic U937 Cells
2.9. Intracellular ROS and Glutathione Analysis of Transgenic U937 Cells
2.10. Evaluation Transgenic U937 Cellular Differentiation
2.11. Data Analysis
3. Results
3.1. Purified WT BCAT1 Protein Metabolises Hydrogen Peroxide In Vitro
3.2. Overexpression of BCAT1 in U937 Cells Increases Growth and Survival and Is Associated with a More Reductive Redox Environment
3.3. BCAT1 CXXC Motif Provides Protection against Serum Deprivation Induced Oxidative Stress
3.4. The Effect of the BCAT1 Inhibitor, Gabapentin, on BCAT1-WT Overexpressing U937 Cells
3.5. BCAT1 CXXC Motif Modulates Intracellular ROS and Altered Myeloid Phenotypic Expression in Response to PMA Induced Differentiation
3.6. BCAT1 CXXC Motif Is Associated with Altered Expression of Nrf2 Regulated Genes in U937 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 Restricts AKG Levels in AML Stem Cells Leading to IDHmut-like DNA Hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer Progression by Reprogrammed BCAA Metabolism in Myeloid Leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Davoodi, J.; Drown, P.M.; Bledsoe, R.K.; Wallin, R.; Reinhart, G.D.; Hutson, S.M. Overexpression and Characterization of the Human Mitochondrial and Cytosolic Branched-Chain Aminotransferases. J. Biol. Chem. 1998, 273, 4982–4989. [Google Scholar] [CrossRef] [Green Version]
- Baracco, E.E.; Castoldi, F.; Durand, S.; Enot, D.P.; Tadic, J.; Kainz, K.; Madeo, F.; Chery, A.; Izzo, V.; Maiuri, M.C.; et al. α-Ketoglutarate Inhibits Autophagy. Aging 2019, 11, 3418–3431. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-Ketoglutarate Maintains the Pluripotency of Embryonic Stem Cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional Transport of Amino Acids Regulates MTOR and Autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Ananieva, E.A.; Powell, J.D.; Hutson, S.M. Leucine Metabolism in T Cell Activation: MTOR Signaling and Beyond. Adv. Nutr. Bethesda Md. 2016, 7, 798–805. [Google Scholar] [CrossRef]
- Goto, M.; Miyahara, I.; Hirotsu, K.; Conway, M.; Yennawar, N.; Islam, M.M.; Hutson, S.M. Structural Determinants for Branched-Chain Aminotransferase Isozyme-Specific Inhibition by the Anticonvulsant Drug Gabapentin. J. Biol. Chem. 2005, 280, 37246–37256. [Google Scholar] [CrossRef] [Green Version]
- Conway, M.E.; Poole, L.B.; Hutson, S.M. Roles for Cysteine Residues in the Regulatory CXXC Motif of Human Mitochondrial Branched Chain Aminotransferase Enzyme. Biochemistry 2004, 43, 7356–7364. [Google Scholar] [CrossRef]
- Conway, M.E.; Coles, S.J.; Islam, M.M.; Hutson, S.M. Regulatory Control of Human Cytosolic Branched-Chain Aminotransferase by Oxidation and S-Glutathionylation and Its Interactions with Redox Sensitive Neuronal Proteins. Biochemistry 2008, 47, 5465–5479. [Google Scholar] [CrossRef]
- Coles, S.J.; Easton, P.; Sharrod, H.; Hutson, S.M.; Hancock, J.; Patel, V.B.; Conway, M.E. S-Nitrosoglutathione Inactivation of the Mitochondrial and Cytosolic BCAT Proteins: S-Nitrosation and S-Thiolation. Biochemistry 2009, 48, 645–656. [Google Scholar] [CrossRef]
- Islam, M.M.; Wallin, R.; Wynn, R.M.; Conway, M.; Fujii, H.; Mobley, J.A.; Chuang, D.T.; Hutson, S.M. A Novel Branched-Chain Amino Acid Metabolon. Protein-Protein Interactions in a Supramolecular Complex. J. Biol. Chem. 2007, 282, 11893–11903. [Google Scholar] [CrossRef] [Green Version]
- El Hindy, M.; Hezwani, M.; Corry, D.; Hull, J.; El Amraoui, F.; Harris, M.; Lee, C.; Forshaw, T.; Wilson, A.; Mansbridge, A.; et al. The Branched-Chain Aminotransferase Proteins: Novel Redox Chaperones for Protein Disulfide Isomerase—Implications in Alzheimer’s Disease. Antioxid. Redox Signal. 2014, 20, 2497–2513. [Google Scholar] [CrossRef] [Green Version]
- Coles, S.J.; Hancock, J.T.; Conway, M.E. Differential Redox Potential between the Human Cytosolic and Mitochondrial Branched-Chain Aminotransferase. Acta Biochim. Et Biophys. Sin. 2012, 44, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Sillar, J.R.; Germon, Z.P.; de Iuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.J.; Davies, S.; Darley, R.L.; Tonks, A. Reactive Oxygen Species Rewires Metabolic Activity in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 458. [Google Scholar] [CrossRef]
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J.; et al. Acute Myeloid Leukaemia. Nat. Rev. Dis. Primers 2016, 2, 1–22. [Google Scholar] [CrossRef]
- Estey, E.H. Acute Myeloid Leukemia: 2019 Update on Risk-Stratification and Management. Am. J. Hematol. 2018, 93, 1267–1291. [Google Scholar] [CrossRef] [Green Version]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.D.; Cotter, T.G. NOX-Driven ROS Formation in Cell Transformation of FLT3-ITD-Positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-Derived ROS in AML Promotes Proliferation and is Associated with Defective Oxidative Stress Signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef]
- Lam, C.F.; Yeung, H.T.; Lam, Y.M.; Ng, R.K. Reactive Oxygen Species Activate Differentiation Gene Transcription of Acute Myeloid Leukemia Cells via the JNK/c-JUN Signaling Pathway. Leuk. Res. 2018, 68, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Song, M.Y.; Lee, D.Y.; Chun, K.S.; Kim, E.H. The Role of NRF2/KEAP1 Signaling Pathway in Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Banning, A. Part of the Series: From Dietary Antioxidants to Regulators in Cellular Signaling and Gene Regulation. Sulforaphane and Selenium, Partners in Adaptive Response and Prevention of Cancer. Free Radic. Res. 2006, 40, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Karathedath, S.; Rajamani, B.M.; Aalam, S.M.M.; Abraham, A.; Varatharajan, S.; Krishnamurthy, P.; Mathews, V.; Velayudhan, S.R.; Balasubramanian, P. Role of NF-E2 Related Factor 2 (Nrf2) on Chemotherapy Resistance in Acute Myeloid Leukemia (AML) and the Effect of Pharmacological Inhibition of Nrf2. PLoS ONE 2017, 12, e0177227. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.K.; Lee, T.B.; Choi, C.H. Anti-Oxidant Adaptation in the AML Cells Supersensitive to Hydrogen Peroxide. Biochem. Biophys. Res. Commun. 2004, 319, 41–45. [Google Scholar] [CrossRef]
- Li, Y.; Schellhorn, H.E. Rapid Kinetic Microassay for Catalase Activity. J. Biomol. Tech. JBT 2007, 18, 185. [Google Scholar]
- Karunanithi, S.; Liu, R.; Hou, Y.; Gonzalez, G.; Oldford, N.; Roe, A.J.; Idipilly, N.; Gupta, K.; Amara, C.S.; Putluri, S.; et al. Thioredoxin Reductase is a Major Regulator of Metabolism in Leukemia Cells. Oncogene 2021, 40, 5236. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif. 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ko, J.H.; Olona, A.; Papathanassiu, A.E.; Buang, N.; Park, K.S.; Costa, A.S.H.; Mauro, C.; Frezza, C.; Behmoaras, J. BCAT1 Affects Mitochondrial Metabolism Independently of Leucine Transamination in Activated Human Macrophages. J. Cell Sci. 2020, 133, jcs247957. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Kim, T.W.; Kim, B.S.; Lee, M.-S.; Yoo, Y. Do Serum Deprivation-Induced Reactive Oxygen Species Production is Mediated by Romo1. Apoptosis 2009, 15, 204–218. [Google Scholar] [CrossRef]
- Barbieri, S.S.; Eligini, S.; Brambilla, M.; Tremoli, E.; Colli, S. Reactive Oxygen Species Mediate Cyclooxygenase-2 Induction during Monocyte to Macrophage Differentiation: Critical Role of NADPH Oxidase. Cardiovasc. Res. 2003, 60, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Sakaguchi, N.; Hachiya, M.; Nakayama, F.; Yamakawa, M.; Akashi, M. Role of Catalase in Monocytic Differentiation of U937 Cells by TPA: Hydrogen Peroxide as a Second Messenger. Leukemia 2008, 23, 761–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the Regulation of CD36 and Stress Protein Expression in Murine Macrophages: Activation by Oxidatively Modified LDL and 4-Hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavkov-Keller, T.; Strohmeier, G.A.; Diepold, M.; Peeters, W.; Smeets, N.; Schürmann, M.; Gruber, K.; Schwab, H.; Steiner, K. Discovery and Structural Characterisation of New Fold Type IV-Transaminases Exemplify the Diversity of This Enzyme Fold. Sci. Rep. 2016, 6, 38183. [Google Scholar] [CrossRef] [Green Version]
- Poole, L.B.; Nelson, K.J. Distribution and Features of the Six Classes of Peroxiredoxins. Mol. Cells 2016, 39, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The Thioredoxin Antioxidant System. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Quan, S.; Schneider, I.; Pan, J.; von Hacht, A.; Bardwell, J.C.A. The CXXC Motif is More than a Redox Rheostat. J. Biol. Chem. 2007, 282, 28823–28833. [Google Scholar] [CrossRef] [Green Version]
- Bjørklund, G.; Zou, L.; Wang, J.; Chasapis, C.T.; Peana, M. Thioredoxin Reductase as a Pharmacological Target. Pharmacol. Res. 2021, 174, 105854. [Google Scholar] [CrossRef]
- Prasad, A.; Manoharan, R.R.; Sedlářová, M.; Pospíšil, P. Free Radical-Mediated Protein Radical Formation in Differentiating Monocytes. Int. J. Mol. Sci. 2021, 22, 9963. [Google Scholar] [CrossRef]
- Xia, L.; Jiang, Y.; Zhang, X.H.; Wang, X.R.; Wei, R.; Qin, K.; Lu, Y. SUMOylation Disassembles the Tetrameric Pyruvate Kinase M2 to Block Myeloid Differentiation of Leukemia Cells. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sundström, C.; Nilsson, K. Establishment and Characterization of a Human Histiocytic Lymphoma Cell Line (U-937). Int. J. Cancer 1976, 17, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Liu, J.; Gao, X.; Li, H. Inhibition of Proliferation in U937 Cells Treated by Blue Light Irradiation and Combined Blue Light Irradiation/Drug. Int. J. Mol. Sci. 2018, 19, 1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, H.A.; Yourish, H.B.; Bunaciu, R.P.; Varner, J.D.; Yen, A. Induced Myelomonocytic Differentiation in Leukemia Cells is Accompanied by Noncanonical Transcription Factor Expression. FEBS Open Bio 2015, 5, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunaka, M.; Shinki, H.; Koyama, T. Cell-Based Evaluation of Changes in Coagulation Activity Induced by Antineoplastic Drugs for the Treatment of Acute Myeloid Leukemia. PLoS ONE 2017, 12, e0175765. [Google Scholar] [CrossRef] [PubMed]
- Chanput, W.; Peters, V.; Wichers, H. THP-1 and U937 Cells. Impact Food Bioact. Health Vitr. Ex Vivo Models 2015, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Baek, Y.S.; Haas, S.; Hackstein, H.; Bein, G.; Hernandez-Santana, M.; Lehrach, H.; Sauer, S.; Seitz, H. Identification of Novel Transcriptional Regulators Involved in Macrophage Differentiation and Activation in U937 Cells. BMC Immunol. 2009, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Sedlářová, M.; Balukova, A.; Ovsii, A.; Rác, M.; Křupka, M.; Kasai, S.; Pospíšil, P. Reactive Oxygen Species Imaging in U937 Cells. Front. Physiol. 2020, 11, 1309. [Google Scholar] [CrossRef]
- Digre, A.; Lindskog, C. The Human Protein Atlas-Spatial Localization of the Human Proteome in Health and Disease. Protein Sci. A Publ. Protein Soc. 2021, 30, 218–233. [Google Scholar] [CrossRef]
- Stubbins, R.J.; Karsan, A. Differentiation Therapy for Myeloid Malignancies: Beyond Cytotoxicity. Blood Cancer J. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Kim, S.H.; Oh, J.; Choi, J.Y.; Jang, J.Y.; Kang, M.W.; Lee, C.E. Identification of Human Thioredoxin as a Novel IFN-Gamma-Induced Factor: Mechanism of Induction and Its Role in Cytokine Production. BMC Immunol. 2008, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- Grankvist, N.; Lagerborg, K.A.; Jain, M.; Nilsson, R. Gabapentin Can Suppress Cell Proliferation Independent of the Cytosolic Branched-Chain Amino Acid Transferase 1 (BCAT1). Biochemistry 2018, 57, 6762–6766. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Pearn, L.; Tonks, A.J.; James, P.E.; Burnett, A.K.; Darley, R.L.; Tonks, A. Ras-Induced Reactive Oxygen Species Promote Growth Factor-Independent Proliferation in Human CD34+ Hematopoietic Progenitor Cells. Blood 2010, 115, 1238–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Yotnda, P. Production and Detection of Reactive Oxygen Species (ROS) in Cancers. J. Vis. Exp. JoVE 2011, 57, e3357. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox Environment of the Cell as Viewed through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Wadley, A.J.; Holliday, A.; Morgan, R.G.; Heesom, K.J.; Aldred, S.; Peters, D.M.; Bueno, A.A.; Coles, S.J. Preliminary Evidence of Reductive Stress in Human Cytotoxic T Cells Following Exercise. J. Appl. Physiol. 2018, 125, 586–595. [Google Scholar] [CrossRef]
- Collet, J.F.; Messens, J. Structure, Function, and Mechanism of Thioredoxin Proteins. Antioxid. Redox Signal. 2010, 13, 1205–1216. [Google Scholar] [CrossRef]
- Božok, V.; Yu, L.Y.; Palgi, J.; Arumäe, U. Antioxidative CXXC Peptide Motif From Mesencephalic Astrocyte-Derived Neurotrophic Factor Antagonizes Programmed Cell Death. Front. Cell Dev. Biol. 2018, 6, 106. [Google Scholar] [CrossRef]
- Olsson, I.; Bergh, G.; Ehinger, M.; Gullberg, U. Cell Differentiation in Acute Myeloid Leukemia. Eur. J. Haematol. 1996, 57, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pedre, B.; Barayeu, U.; Ezeriņa, D.; Dick, T.P. The Mechanism of Action of N-Acetylcysteine (NAC): The Emerging Role of H2S and Sulfane Sulfur Species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Rhee, J.G.; Suntharalingam, M.; Walsh, S.A.; Spitz, D.R.; Lee, Y.J. Role of Glutaredoxin in Metabolic Oxidative Stress. Glutaredoxin as a Sensor of Oxidative Stress Mediated by H2O2. J. Biol. Chem. 2002, 277, 46566–46575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.F.; Bodnar, Y.; Klein, C.; Salas, C.O.; Arnér, E.S.J.; Gellert, M.; Lillig, C.H. Molecular Basis for the Interactions of Human Thioredoxins with Their Respective Reductases. Oxidative Med. Cell. Longev. 2021, 2021, 6621292. [Google Scholar] [CrossRef] [PubMed]
- Bagger, F.O.; Sasivarevic, D.; Sohi, S.H.; Laursen, L.G.; Pundhir, S.; Sønderby, C.K.; Winther, O.; Rapin, N.; Porse, B.T. BloodSpot: A Database of Gene Expression Profiles and Transcriptional Programs for Healthy and Malignant Haematopoiesis. Nucleic Acids Res. 2016, 44, 917–924. [Google Scholar] [CrossRef]
- Locy, M.L.; Rogers, L.K.; Prigge, J.R.; Schmidt, E.E.; Elias, E.S.; Tipple, T.E. Thioredoxin Reductase Inhibition Elicits Nrf2-Mediated Responses in Clara Cells: Implications for Oxidant-Induced Lung Injury. Antioxid. Redox Signal. 2012, 17, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, Redox Defense and Much More. Redox Biol. 2021, 43, 101975. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vector | WT BCAT1 | CXXS BACT1 | |
|---|---|---|---|
| IC50 based on viability (%) | 38.6 ± 0.30 mM | 33.92 ± 0.22 mM | 36.88 ± 0.36 mM |
| R2 | 0.98 | 0.991 | 0.975 |
| IC50 based on density (cell/mL) | 33.77 ± 1.19 mM | 28.67 ± 1.07 mM | 32.41 ± 2.61 mM |
| R2 | 0.919 | 0.947 | 0.875 |
| X0 based on viability (%) | 29.01 ± 0.60 mM | 24.38 ± 0.72 mM | 28.65 ± 0.79 mM |
| R2 | 0.983 | 0.97 | 0.966 |
| X0 based on density (cell/mL) | 23.99 ± 1.38 mM | 18.35 ± 1.29 mM | 15.89 ± 3.91 mM |
| R2 | 0.908 | 0.934 | 0.867 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hillier, J.; Allcott, G.J.; Guest, L.A.; Heaselgrave, W.; Tonks, A.; Conway, M.E.; Cherry, A.L.; Coles, S.J. The BCAT1 CXXC Motif Provides Protection against ROS in Acute Myeloid Leukaemia Cells. Antioxidants 2022, 11, 683. https://doi.org/10.3390/antiox11040683
Hillier J, Allcott GJ, Guest LA, Heaselgrave W, Tonks A, Conway ME, Cherry AL, Coles SJ. The BCAT1 CXXC Motif Provides Protection against ROS in Acute Myeloid Leukaemia Cells. Antioxidants. 2022; 11(4):683. https://doi.org/10.3390/antiox11040683
Chicago/Turabian StyleHillier, James, Gemma J. Allcott, Laura A. Guest, Wayne Heaselgrave, Alex Tonks, Myra E. Conway, Amy L. Cherry, and Steven J. Coles. 2022. "The BCAT1 CXXC Motif Provides Protection against ROS in Acute Myeloid Leukaemia Cells" Antioxidants 11, no. 4: 683. https://doi.org/10.3390/antiox11040683
APA StyleHillier, J., Allcott, G. J., Guest, L. A., Heaselgrave, W., Tonks, A., Conway, M. E., Cherry, A. L., & Coles, S. J. (2022). The BCAT1 CXXC Motif Provides Protection against ROS in Acute Myeloid Leukaemia Cells. Antioxidants, 11(4), 683. https://doi.org/10.3390/antiox11040683