
Piperlongumine Inhibits Thioredoxin Reductase 1 by Targeting Selenocysteine Residues and Sensitizes Cancer Cells to Erastin

 ,
, 

Abstract
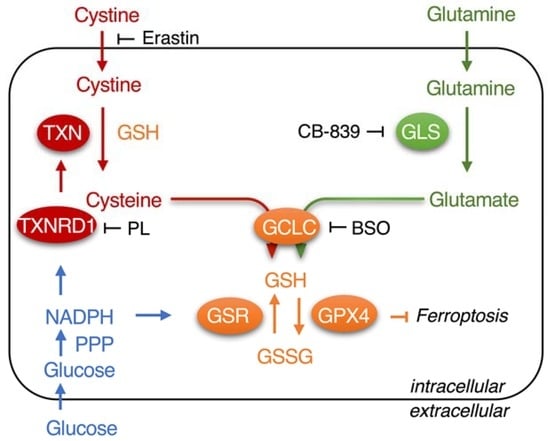
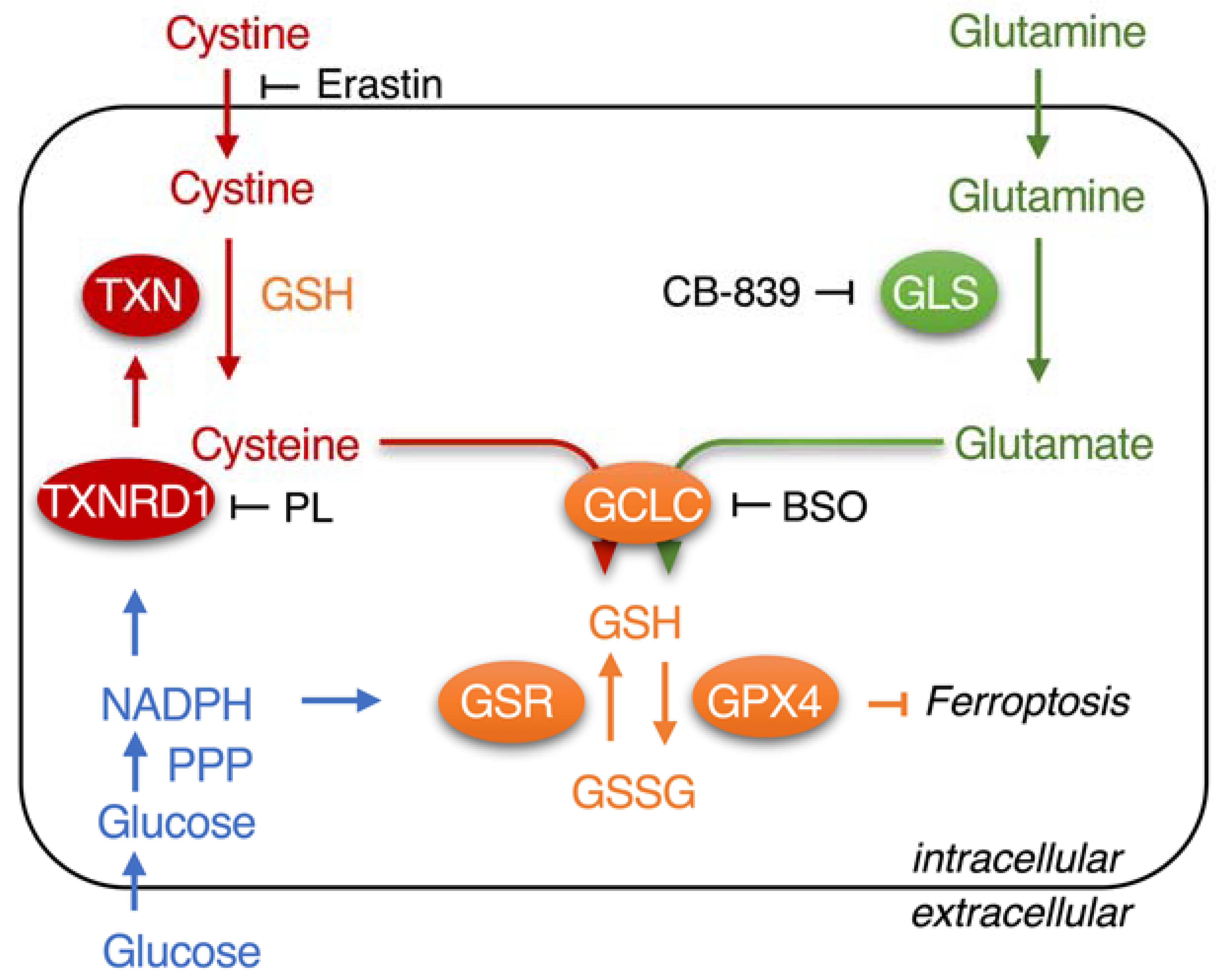
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
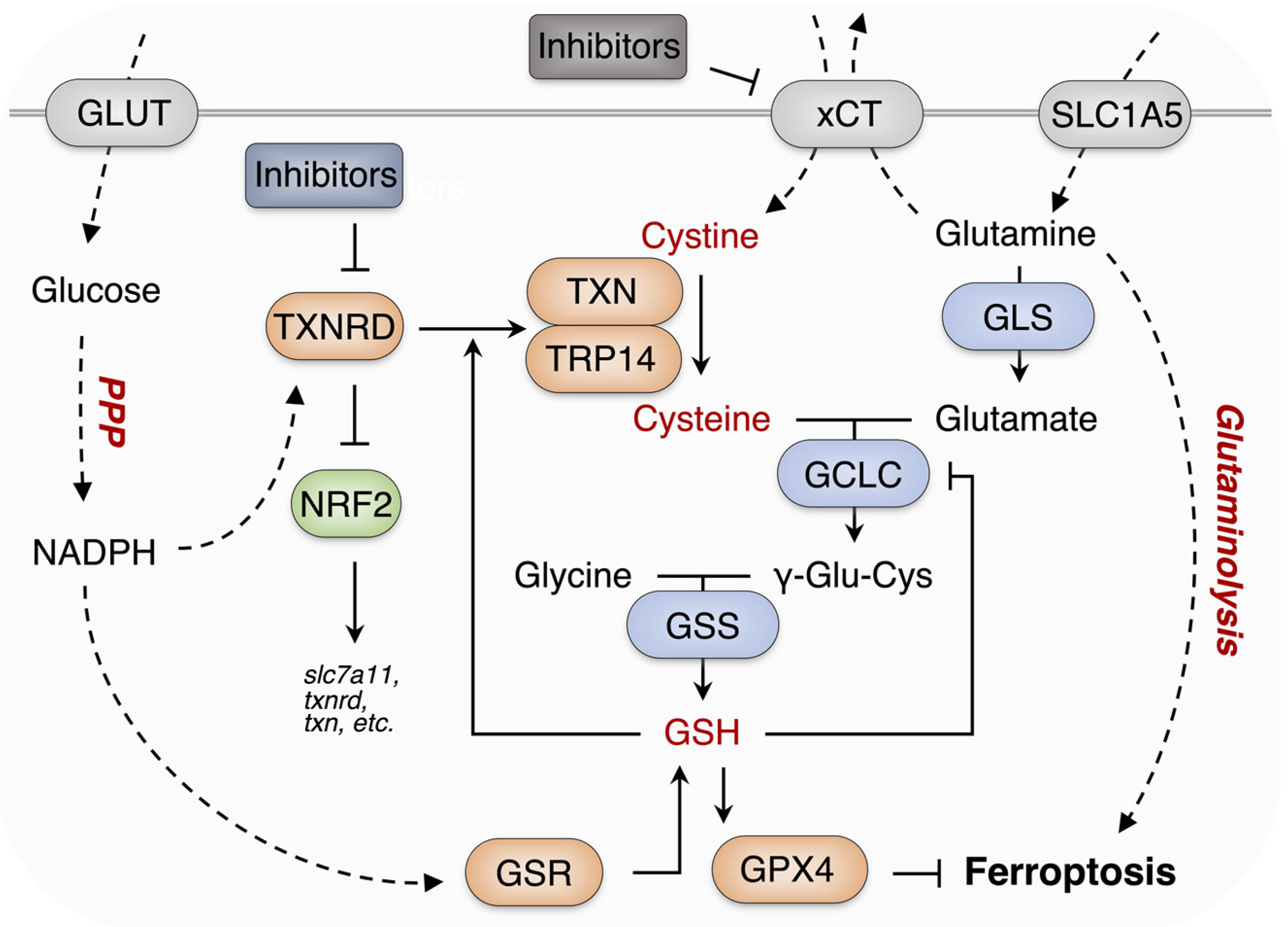{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Thioredoxin Reductase Activity Assays
2.5. NADPH Oxidase Activity Assay
2.6. Cellular TXNRD Activity Assay
2.7. Glutathione Reductase Activity Assay
2.8. Cellualr GSH Content Determination
2.9. Cellular ROS Levels Determination
2.10. Lipid Oxidation Assays
2.11. Statistics Analysis
3. Results
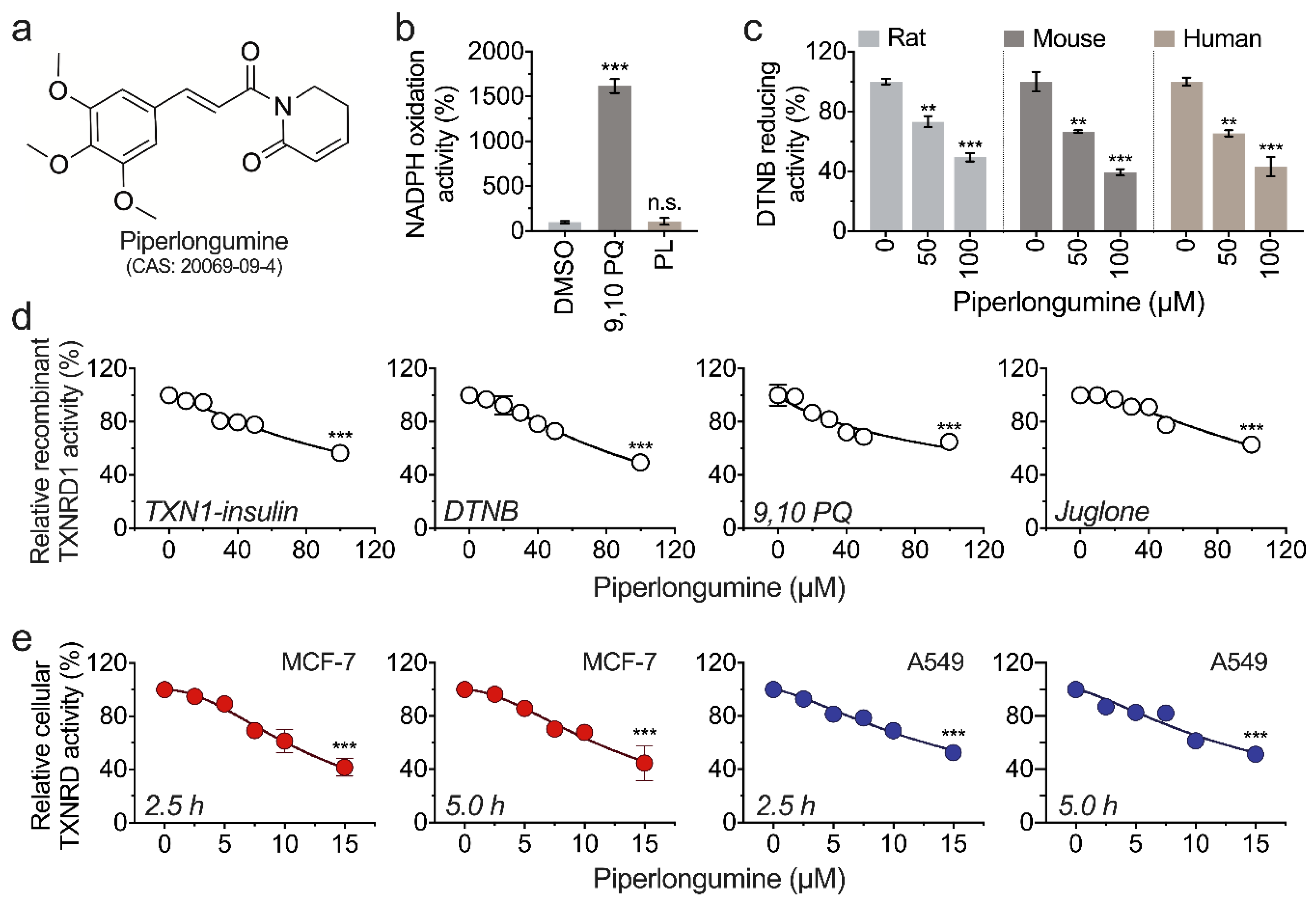
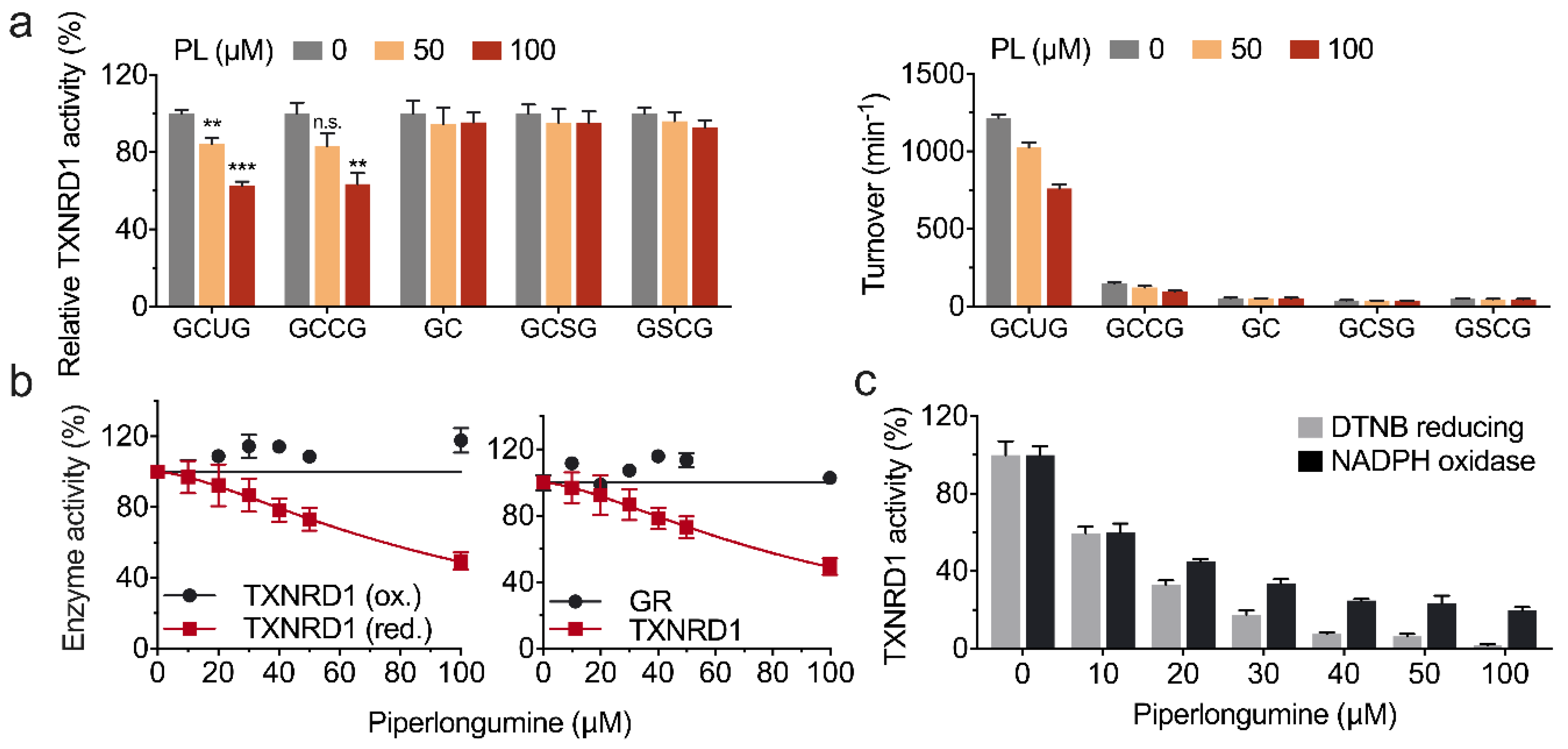
3.1. Piperlongumine Inhibits TXNRD1 in a Dose-Dependent Manner
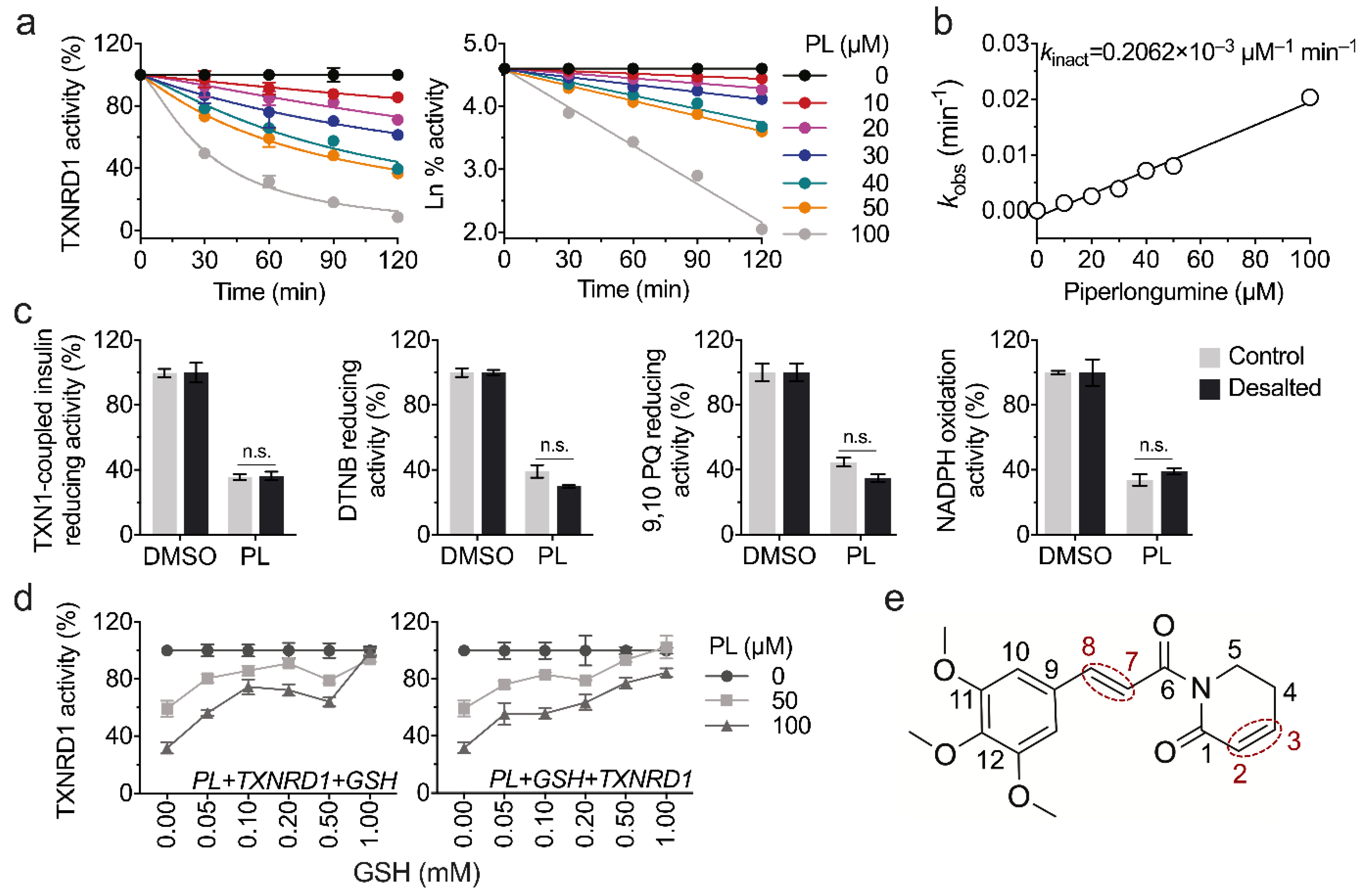
3.2. Piperlongumine Irreversibly Inhibits TXNRD1 In Vitro
3.3. Piperlongumine Targets the Sec498 of TXNRD1 and Converts the Enzyme to NADPH Oxidase
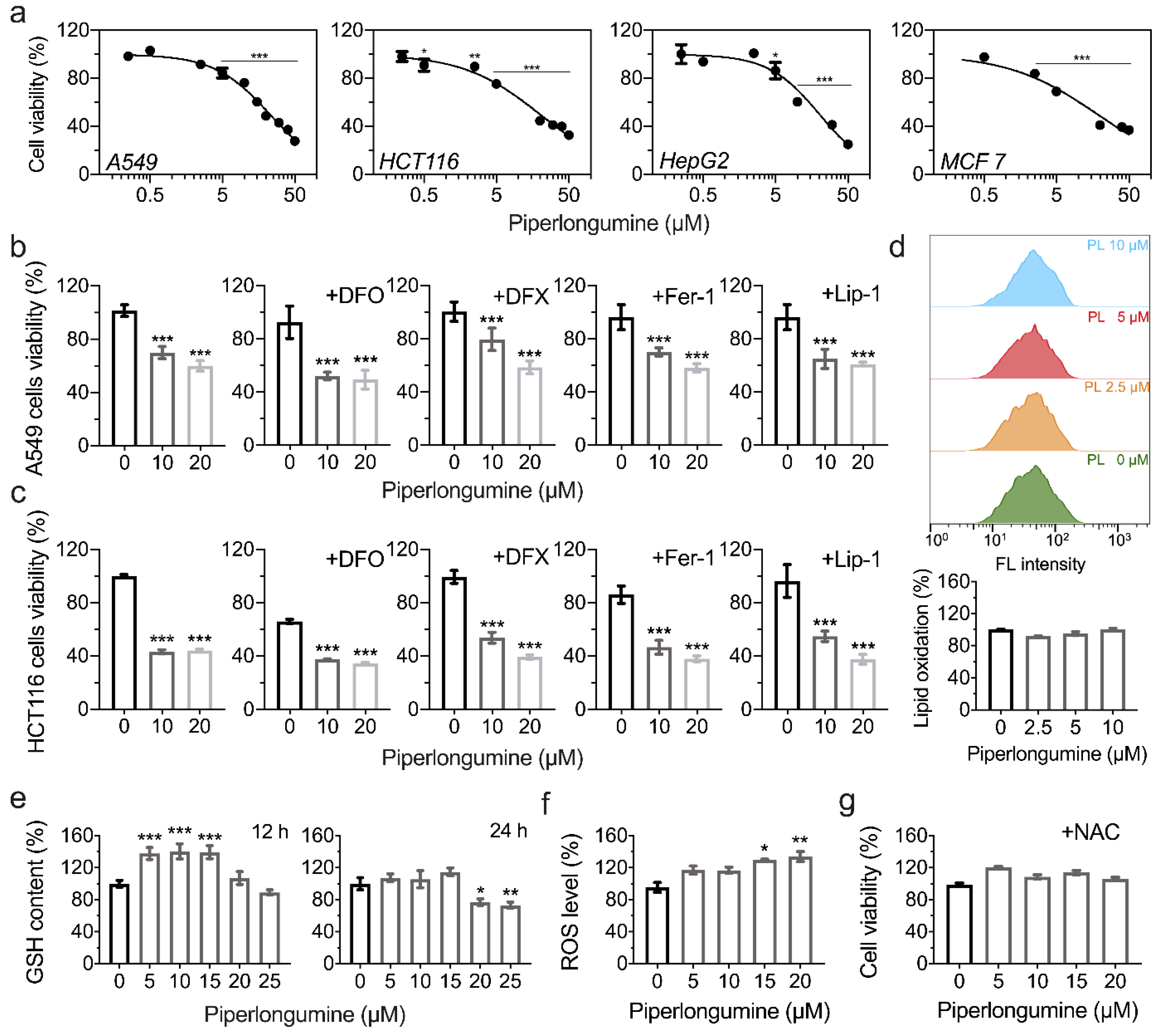
3.4. Piperlongumine Induces ROS-Dependent Cancer Cell Death but Not Ferroptosis
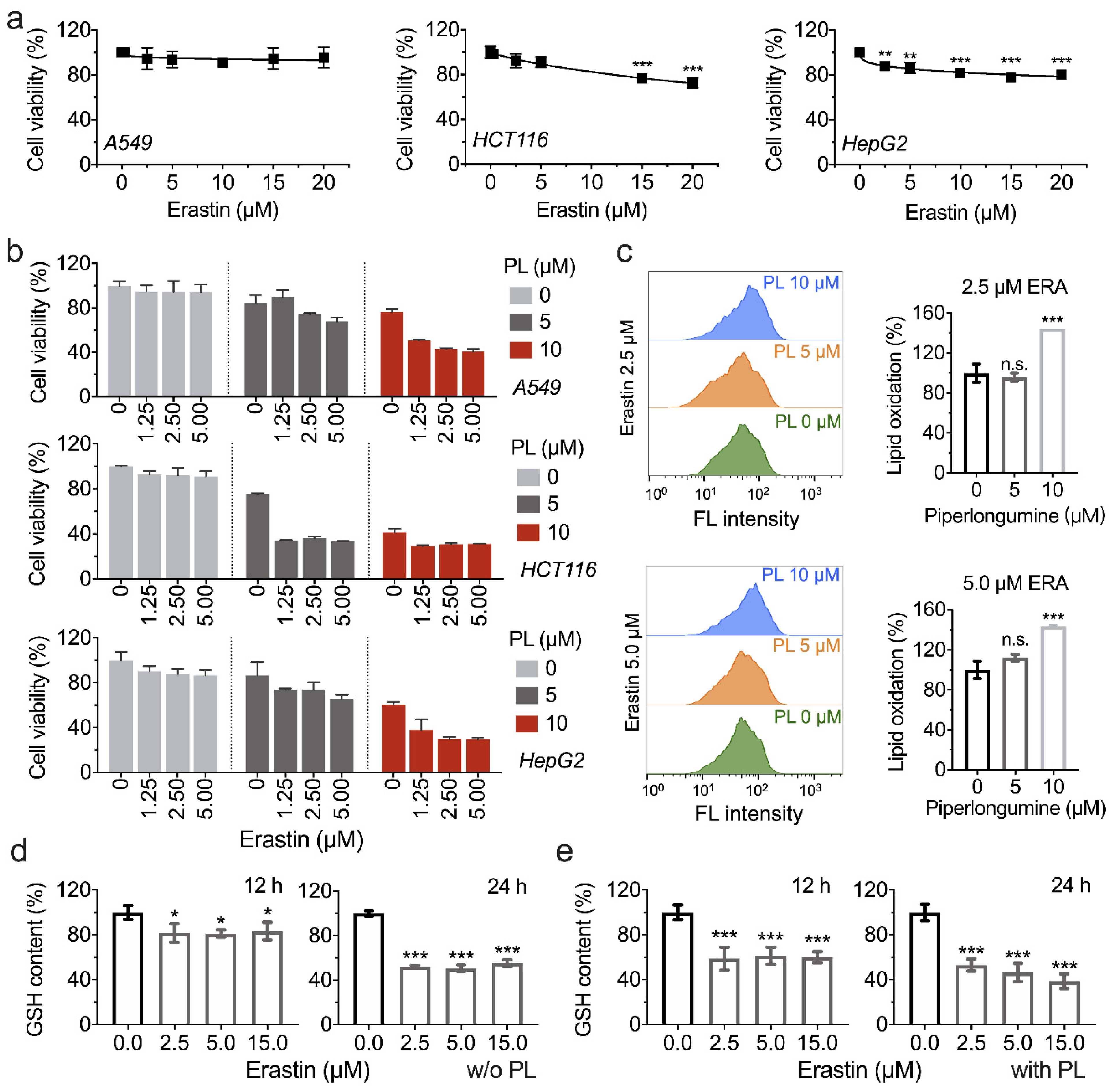
3.5. Piperlongumine Enhances Erastin-Induced Cancer Cells Death
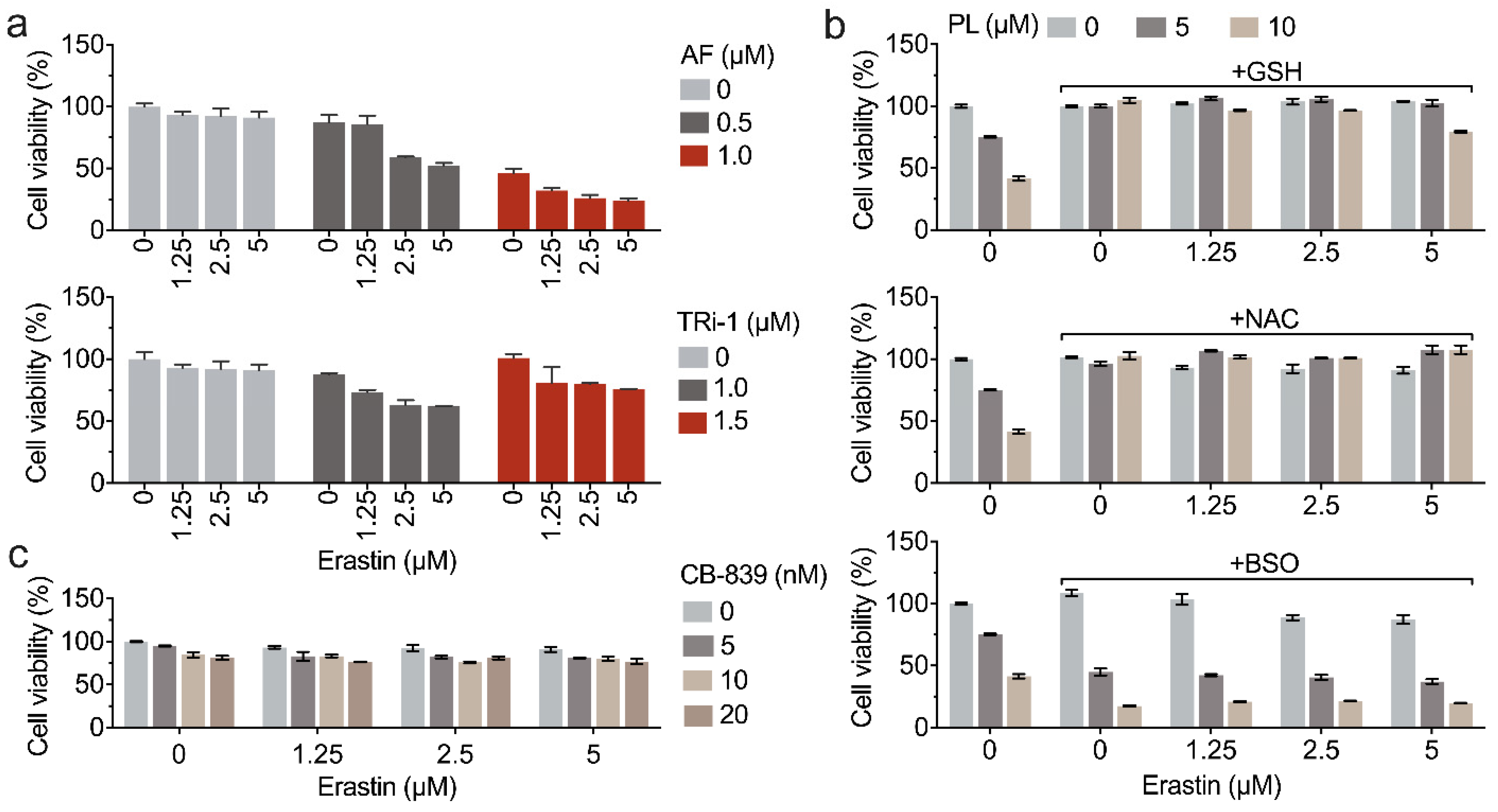
3.6. Inhibition of TXNRD1 Activity Sensitizes Cancer Cell to GSH Depletion
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gladyshev, V.N.; Arnér, E.S.; Berry, M.J.; Brigelius-Flohé, R.; Bruford, E.A.; Burk, R.F.; Carlson, B.A.; Castellano, S.; Chavatte, L.; Conrad, M.; et al. Selenoprotein gene nomenclature. J. Biol. Chem. 2016, 291, 24036–24040. [Google Scholar] [CrossRef] [Green Version]
- Gencheva, R.; Arnér, E.S.J. Thioredoxin reductase inhibition for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2021, 62, 177–196. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Cheng, Q.; Sandalova, T.; Lindqvist, Y.; Arnér, E.S.J. Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J. Biol. Chem. 2009, 284, 3998–4008. [Google Scholar] [CrossRef] [Green Version]
- Pader, I.; Sengupta, R.; Cebula, M.; Xu, J.; Lundberg, J.O.; Holmgren, A.; Johansson, K.; Arnér, E.S.J. Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc. Natl. Acad. Sci. USA 2014, 111, 6964–6969. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, B.; Li, X.; Han, X.; Liu, R.; Fang, J. Small molecule inhibitors of mammalian thioredoxin reductase as potential anticancer agents: An update. Med. Res. Rev. 2019, 39, 5–39. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zhang, Y.; Xu, W.; Zhang, Y.; Yang, R.; Guo, J.; Guan, S.; Ma, Q.; Ma, K.; Xu, J. Chlorophyllin inhibits mammalian thioredoxin reductase 1 and triggers cancer cell death. Antioxidants 2021, 10, 1733. [Google Scholar] [CrossRef]
- Xu, J.; Fang, J. How can we improve the design of small molecules to target thioredoxin reductase for treating cancer? Expert Opin. Drug Discov. 2021, 16, 331–333. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Biswal, B.K. Piperlongumine, a potent anticancer phytotherapeutic: Perspectives on contemporary status and future possibilities as an anticancer agent. Pharmacol. Res. 2020, 156, 104772. [Google Scholar] [CrossRef]
- Gu, S.M.; Yun, J.; Son, D.J.; Kim, H.Y.; Nam, K.T.; Kim, H.D.; Choi, M.G.; Choi, J.S.; Kim, Y.M.; Han, S.-B.; et al. Piperlongumine attenuates experimental autoimmune encephalomyelitis through inhibition of NF-kappaB activity. Free Radic. Biol. Med. 2017, 103, 133–145. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Eckols, T.K.; Kolosov, M.; Kasembeli, M.M.; Adam, A.; Torres, D.; Zhang, X.; Dobrolecki, L.E.; Wei, W.; Lewis, M.T.; et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene 2015, 34, 1341–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Chen, W.; Lv, X.; Weng, Q.; Chen, M.; Cui, R.; Liang, G.; Ji, J. Piperlongumine, a novel TrxR1 inhibitor, induces apoptosis in hepatocellular carcinoma cells by ROS-mediated ER stress. Front. Pharmacol. 2019, 10, 1180. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Xia, Y.; Ji, J.; Chen, W.; Zhang, J.; Chen, X.; Rajamanickam, V.; Chen, G.; Wang, Z.; Chen, L.; et al. Piperlongumine as a direct TrxR1 inhibitor with suppressive activity against gastric cancer. Cancer Lett. 2016, 375, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Banjac Canak, A.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System x(c)− and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef] [Green Version]
- Anestål, K.; Prast-Nielsen, S.; Cenas, N.; Arnér, E.S.J. Cell death by SecTRAPs: Thioredoxin reductase as a prooxidant killer of cells. PLoS ONE 2008, 3, e1846. [Google Scholar] [CrossRef]
- Cheng, Q.; Antholine, W.E.; Myers, J.M.; Kalyanaraman, B.; Arnér, E.S.J.; Myers, C.R. The selenium-independent inherent pro-oxidant NADPH oxidase activity of mammalian thioredoxin reductase and its selenium-dependent direct peroxidase activities. J. Biol. Chem. 2010, 285, 21708–21723. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Xu, W.; Zhou, H.; Zhang, Y.; Zhang, J.; Li, X.; Li, B.; Ma, K.; Xu, J. Efficient purification of selenoprotein thioredoxin reductase 1 by using chelating reagents to protect the affinity resins and rescue the enzyme activities. Process Biochem. 2021, 101, 256–265. [Google Scholar] [CrossRef]
- Xu, J.; Croitoru, V.; Rutishauser, D.; Cheng, Q.; Arnér, E.S. Wobble decoding by the Escherichia coli selenocysteine insertion machinery. Nucleic Acids Res. 2013, 41, 9800–9811. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Li, W.; Zhu, G.; Chi, H.; Yin, Y.; Li, Y.; Zong, Y.; Guo, Z.; Wang, L.; Xu, W.; et al. PEGylated DOX-coated nano graphene oxide as pH-responsive multifunctional nanocarrier for targeted drug delivery. J. Drug Target. 2021, 29, 884–891. [Google Scholar] [CrossRef]
- Ma, K.; Li, W.; Zhu, G.; Sun, S.; Chi, H.; Yin, Y.; Diao, H.; Xing, X.; Guo, Z.; Wang, L.; et al. Functionalized PDA/DEX-PEI@HA nanoparticles combined with sleeping-beauty transposons for multistage targeted delivery of CRISPR/Cas9 gene. Biomed. Pharmacother. 2021, 142, 112061. [Google Scholar] [CrossRef]
- Xu, J.; Cheng, Q.; Arnér, E.S. Details in the catalytic mechanism of mammalian thioredoxin reductase 1 revealed using point mutations and juglone-coupled enzyme activities. Free Radic. Biol. Med. 2016, 94, 110–120. [Google Scholar] [CrossRef]
- Sun, S.; Xu, W.; Zhang, Y.; Yang, Y.; Ma, Q.; Xu, J. Menadione inhibits thioredoxin reductase 1 via arylation at the Sec498 residue and enhances both NADPH oxidation and superoxide production in Sec(498) to Cys(498) substitution. Free Radic. Biol. Med. 2021, 172, 482–489. [Google Scholar] [CrossRef]
- Arnér, E.S.; Holmgren, A. Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. 2005, 24, 7-4. [Google Scholar] [CrossRef]
- Wang, H.; Sun, S.; Ren, Y.; Yang, R.; Guo, J.; Zong, Y.; Zhang, Q.; Zhao, J.; Zhang, W.; Xu, W.; et al. Selenite ameliorates cadmium-induced cytotoxicity through downregulation of ROS levels and upregulation of selenoprotein thioredoxin reductase 1 in SH-SY5Y cells. Biol. Trace Elem. Res. 2022, 1–10. [Google Scholar] [CrossRef]
- Stafford, W.C.; Peng, X.; Olofsson, M.H.; Zhang, X.; Luci, D.K.; Lu, L.; Cheng, Q.; Trésaugues, L.; Dexheimer, T.S.; Coussens, N.P.; et al. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci. Transl. Med. 2018, 10, eaaf7444. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Arnér, E.S. Pyrroloquinoline quinone modulates the kinetic parameters of the mammalian selenoprotein thioredoxin reductase 1 and is an inhibitor of glutathione reductase. Biochem. Pharmacol. 2012, 83, 815–820. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 2017, 6, e28083. [Google Scholar] [CrossRef]
- LeBoeuf, S.E.; Wu, W.L.; Karakousi, T.R.; Karadal, B.; Jackson, S.R.; Davidson, S.M.; Wong, K.K.; Koralov, S.B.; Sayin, V.I.; Papagiannakopoulos, T. Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell Metab. 2020, 31, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, Z.; Wu, K.; Huang, S.M.; Wu, W.L.; LeBoeuf, S.E.; Pillai, R.G.; Rabinowitz, J.D.; Papagiannakopoulos, T. Activation of the NRF2 antioxidant program sensitizes tumors to G6PD inhibition. Sci. Adv. 2021, 7, eabk1023. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Duan, D.; Song, Z.-L.; Zhang, J.; Fang, J. Sanguinarine as a new chemical entity of thioredoxin reductase inhibitor to elicit oxidative stress and promote tumor cell apoptosis. Free Radic. Biol. Med. 2020, 152, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Bian, M.; Wang, X.; Sun, Y.; Liu, W. Synthesis and biological evaluation of gold(III) Schiff base complexes for the treatment of hepatocellular carcinoma through attenuating TrxR activity. Eur. J. Med. Chem. 2020, 193, 112234. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, D.; Song, Z.; Liu, T.; Hou, Y.; Fang, J. Small molecules regulating reactive oxygen species homeostasis for cancer therapy. Med. Res. Rev. 2021, 41, 342–394. [Google Scholar] [CrossRef]
- Adams, D.J.; Dai, M.; Pellegrino, G.; Wagner, B.K.; Stern, A.M.; Shamji, A.F.; Schreiber, S.L. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc. Natl. Acad. Sci. USA 2012, 109, 15115–15120. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jiang, H.; Corbet, C.; de Mey, S.; Law, K.; Gevaert, T.; Feron, O.; De Ridder, M. Piperlongumine increases sensitivity of colorectal cancer cells to radiation: Involvement of ROS production via dual inhibition of glutathione and thioredoxin systems. Cancer Lett. 2019, 450, 42–52. [Google Scholar] [CrossRef]
- Duan, D.; Zhang, B.; Yao, J.; Liu, Y.; Sun, J.; Ge, C.; Peng, S.; Fang, J. Gambogic acid induces apoptosis in hepatocellular carcinoma SMMC-7721 cells by targeting cytosolic thioredoxin reductase. Free Radic. Biol. Med. 2014, 69, 15–25. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Duan, D.; Yao, J.; Gao, K.; Fang, J. Inhibition of thioredoxin reductase by alantolactone prompts oxidative stress-mediated apoptosis of HeLa cells. Biochem. Pharmacol. 2016, 102, 34–44. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, Y.; Xu, W.; Yang, R.; Yang, Y.; Guo, J.; Ma, Q.; Ma, K.; Zhang, J.; Xu, J. Plumbagin reduction by thioredoxin reductase 1 possesses synergy effects with GLUT1 inhibitor on KEAP1-mutant NSCLC cells. Biomed. Pharmacother. 2022, 146, 112546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, S.; Xu, W.; Yang, R.; Yang, Y.; Guo, J.; Ma, K.; Xu, J. Thioredoxin reductase 1 inhibitor shikonin promotes cell necroptosis via SecTRAPs generation and oxygen-coupled redox cycling. Free Radic. Biol. Med. 2021, 180, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Dagnell, M.; Schmidt, E.E.; Arnér, E.S. The A to Z of modulated cell patterning by mammalian thioredoxin reductases. Free Radic. Biol. Med. 2018, 115, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arnér, E.S.J. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019, 294, 12330–12338. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Santhosh, S.M.; Coppo, L.; Ogata, F.T.; Lu, J.; Holmgren, A. The combination of ascorbate and menadione causes cancer cell death by oxidative stress and replicative stress. Free Radic. Biol. Med. 2019, 134, 350–358. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.P.; Seibt, T.; et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 2018, 172, 409–422.e12. [Google Scholar] [CrossRef] [Green Version]
- Torrente, L.; Prieto-Farigua, N.; Falzone, A.; Elkins, C.M.; Boothman, D.A.; Haura, E.B.; DeNicola, G.M. Inhibition of TXNRD or SOD1 overcomes NRF2-mediated resistance to β-lapachone. Redox Biol. 2020, 30, 101440. [Google Scholar] [CrossRef]
- Kang, Y.P.; Torrente, L.; Falzone, A.; Elkins, C.M.; Liu, M.; Asara, J.M.; Dibble, C.C.; DeNicola, G.M. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. eLife 2019, 8, e45572. [Google Scholar] [CrossRef]
- Koppula, P.; Olszewski, K.; Zhang, Y.; Kondiparthi, L.; Liu, X.; Lei, G.; Das, M.; Fang, B.; Poyurovsky, M.V.; Gan, B. KEAP1 deficiency drives glucose dependency and sensitizes lung cancer cells and tumors to GLUT inhibition. iScience 2021, 24, 102649. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.L.; Ruberto, R.A.; Ryan, M.J.; Eaton, J.K.; Schreiber, S.L.; Viswanathan, V.S. Modulation of ferroptosis sensitivity by TXNRD1 in pancreatic cancer cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Arnér, E.S.J. Effects of mammalian thioredoxin reductase inhibitors. Handb. Exp. Pharmacol. 2021, 264, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Sun, S.; Xu, W.; Zhang, Y.; Yang, R.; Ma, K.; Zhang, J.; Xu, J. Piperlongumine Inhibits Thioredoxin Reductase 1 by Targeting Selenocysteine Residues and Sensitizes Cancer Cells to Erastin. Antioxidants 2022, 11, 710. https://doi.org/10.3390/antiox11040710
Yang Y, Sun S, Xu W, Zhang Y, Yang R, Ma K, Zhang J, Xu J. Piperlongumine Inhibits Thioredoxin Reductase 1 by Targeting Selenocysteine Residues and Sensitizes Cancer Cells to Erastin. Antioxidants. 2022; 11(4):710. https://doi.org/10.3390/antiox11040710
Chicago/Turabian StyleYang, Yijia, Shibo Sun, Weiping Xu, Yue Zhang, Rui Yang, Kun Ma, Jie Zhang, and Jianqiang Xu. 2022. "Piperlongumine Inhibits Thioredoxin Reductase 1 by Targeting Selenocysteine Residues and Sensitizes Cancer Cells to Erastin" Antioxidants 11, no. 4: 710. https://doi.org/10.3390/antiox11040710
APA StyleYang, Y., Sun, S., Xu, W., Zhang, Y., Yang, R., Ma, K., Zhang, J., & Xu, J. (2022). Piperlongumine Inhibits Thioredoxin Reductase 1 by Targeting Selenocysteine Residues and Sensitizes Cancer Cells to Erastin. Antioxidants, 11(4), 710. https://doi.org/10.3390/antiox11040710