
Characterization of the p.L145F and p.S135N Mutations in SOD1: Impact on the Metabolism of Fibroblasts Derived from Amyotrophic Lateral Sclerosis Patients

 ,
,  ,
,  , , , ,
, , , ,  ,
,  add
Show full author list
add
Show full author list
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
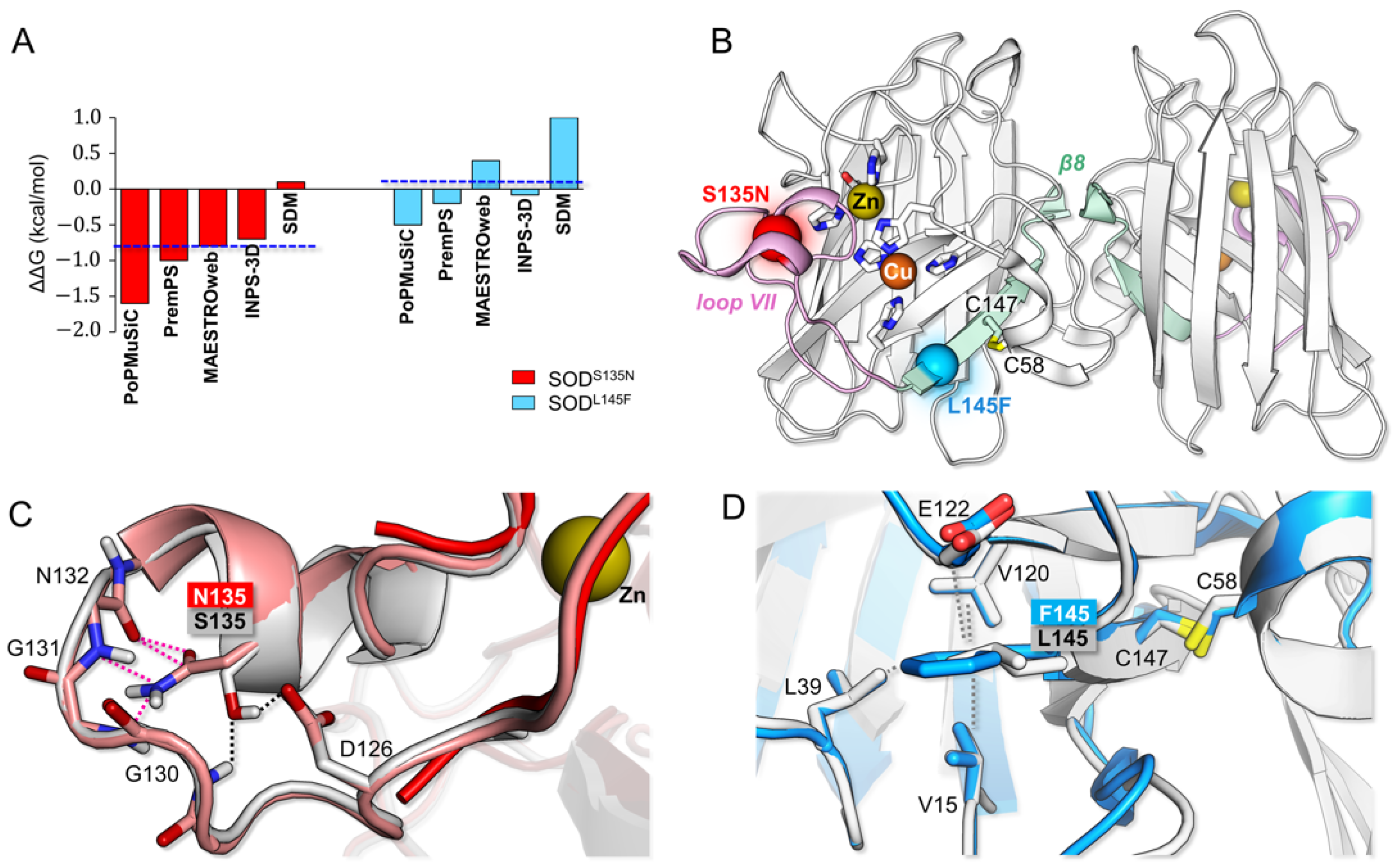
2.1. Molecular Modeling
2.2. Collection of Skin Biopsies
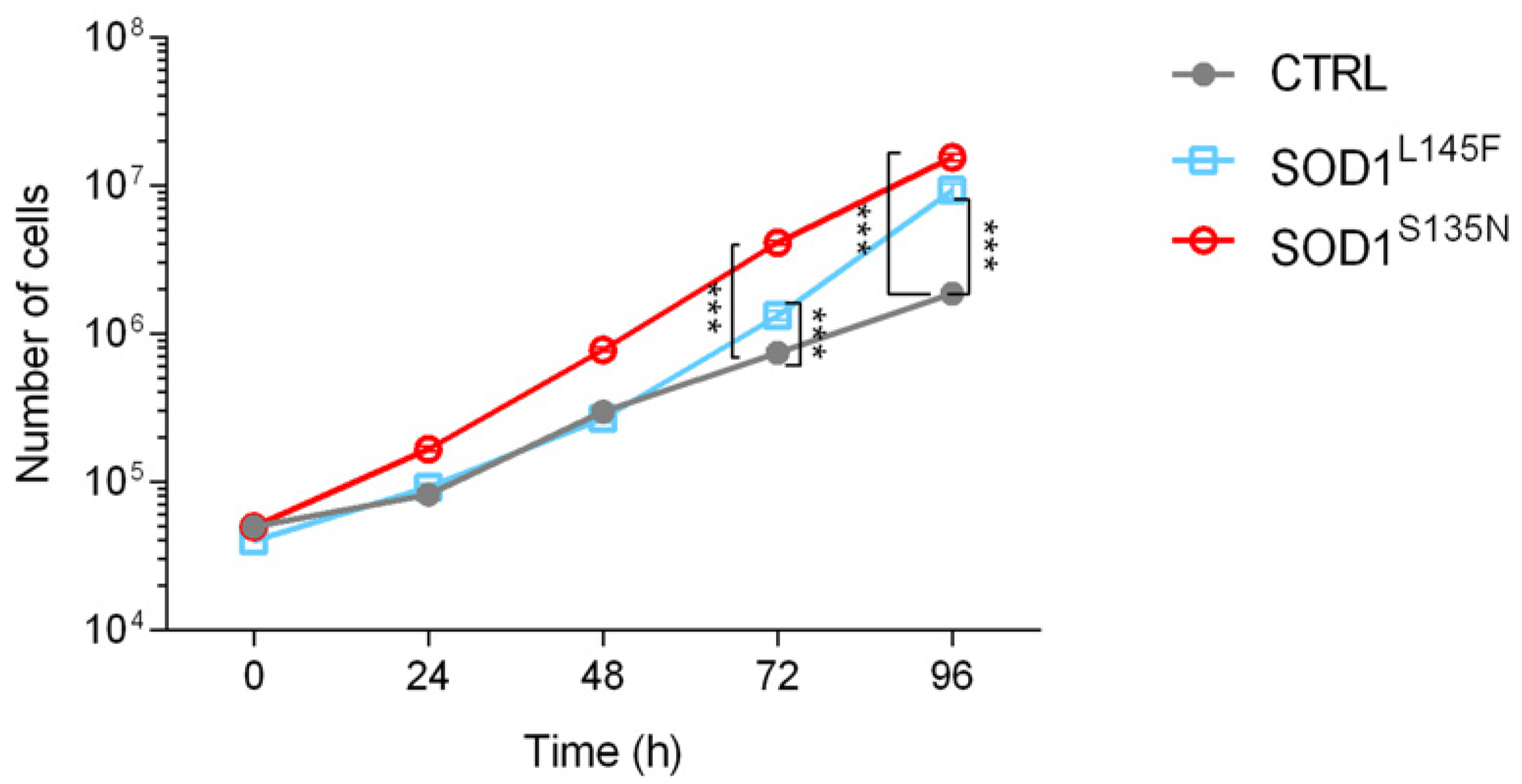
2.3. Primary Skin Fibroblast Cultures and Growth Curves
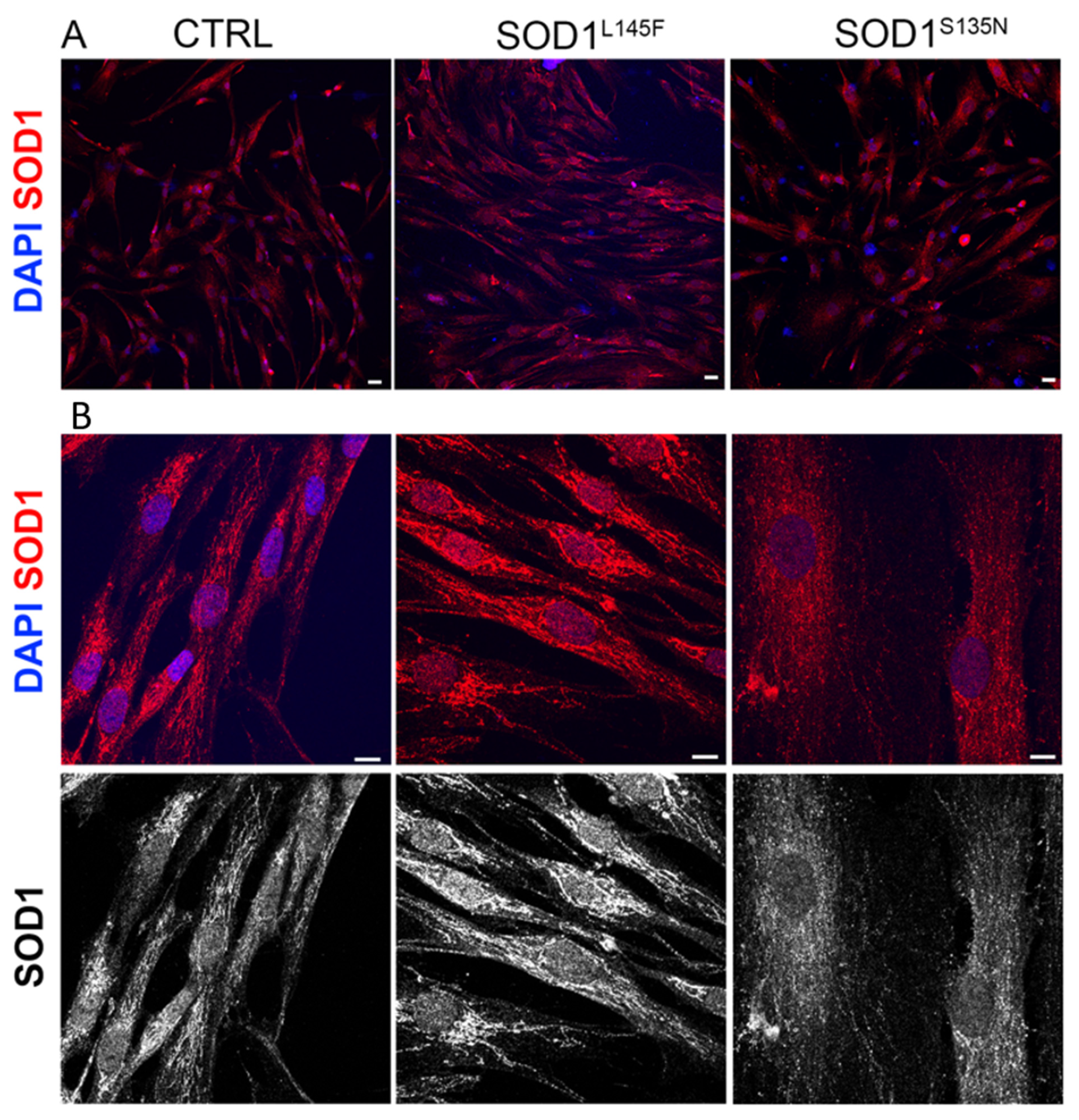
2.4. SOD1 Immunofluorescence Staining
2.5. Detection of Intracellular Cytosolic Reactive Oxygen Species (ROS)
2.6. Glutathione Detection
2.7. Oxygen Consumption Rate and Extracellular Acidification Rate Measurements
2.8. Statistical Analysis
3. Results
3.1. Clinical Evaluation of the Patients
3.1.1. SOD1L145F Patient
3.1.2. SOD1S135N Patient
3.2. Bioinformatic Analysis: p.L145F and p.S135N Differently Affect SOD1 Structural Properties
3.3. Analysis of ALS Patient-Derived Fibroblasts: SOD1L145F and SOD1S135N Cells
3.3.1. Morphology of ALS-Fibroblasts
3.3.2. ALS Fibroblasts Showed Increased Proliferation Rate Compared to the Control
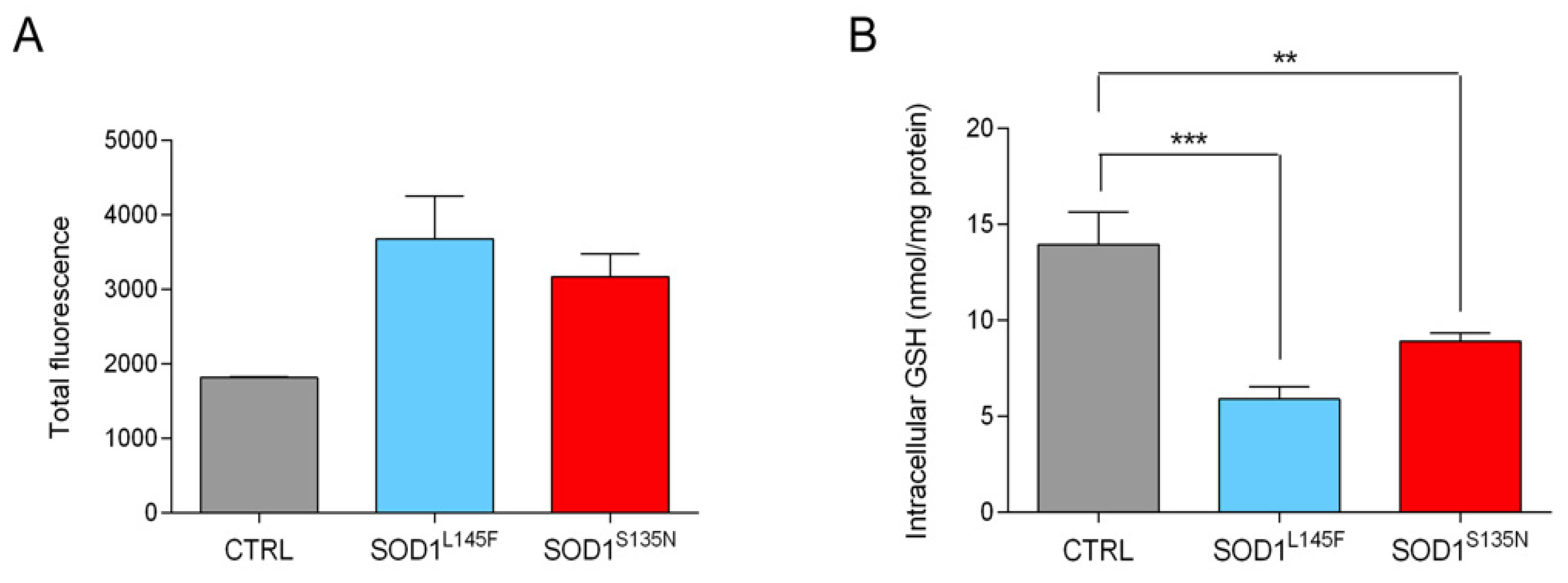
3.3.3. ALS Fibroblasts Show Reduced Anti-Oxidant Potential
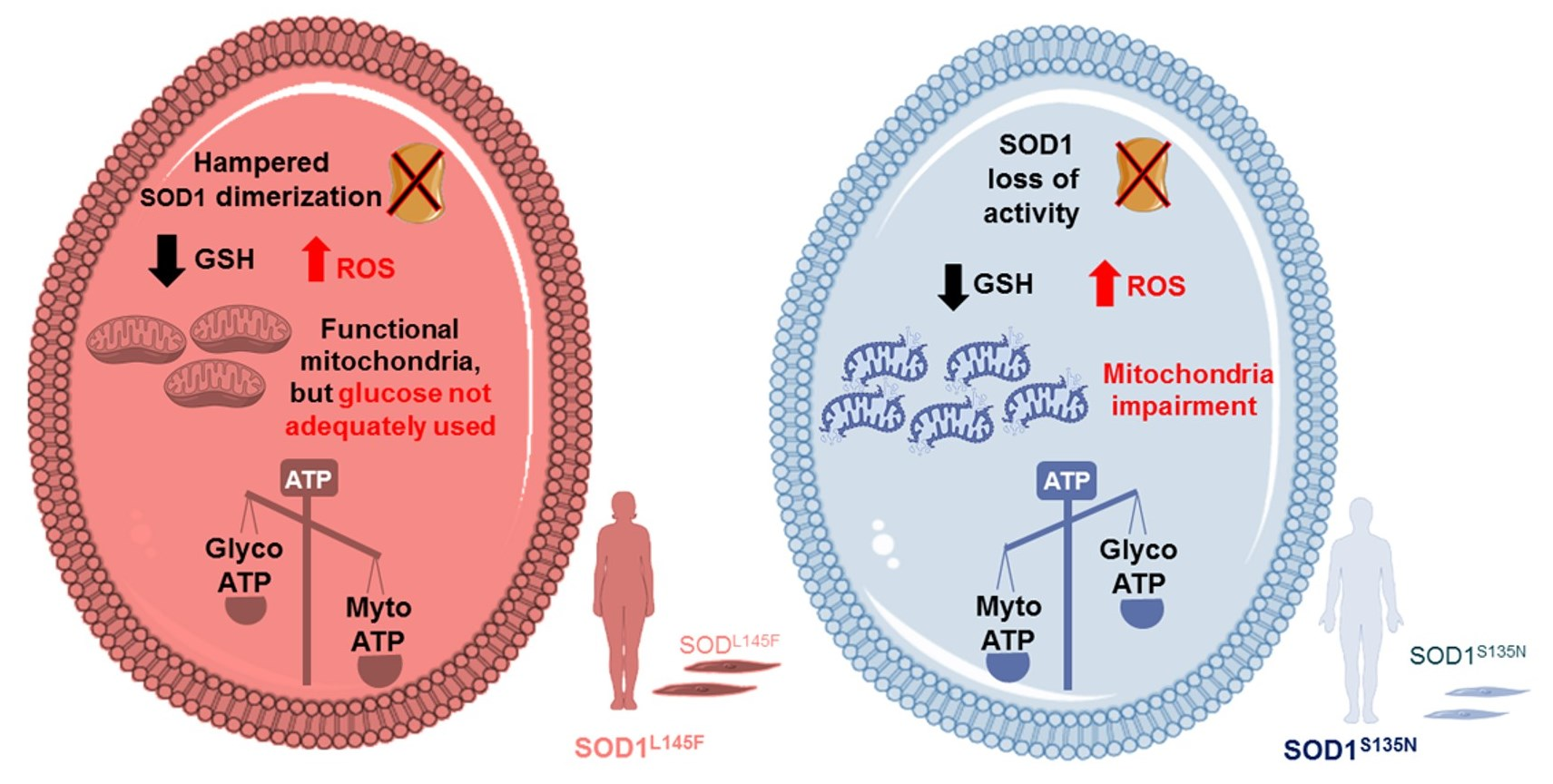
3.4. Bioenergetic Alterations in SOD1L145F and SOD1S135N Fibroblasts
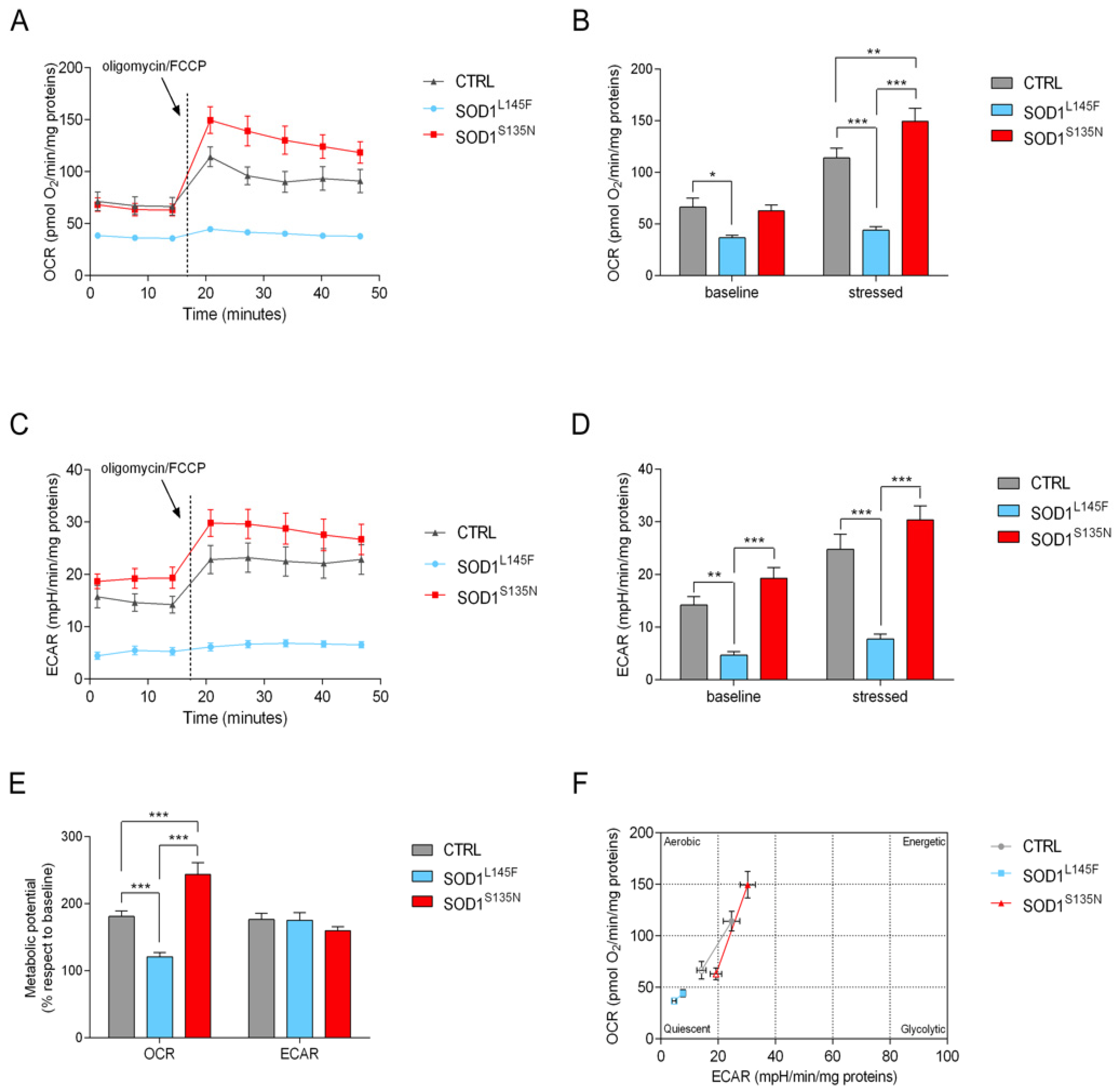
3.4.1. Cell Energy Phenotype Reveals a Different Response to Stress in Cells Expressing Mutated SOD1
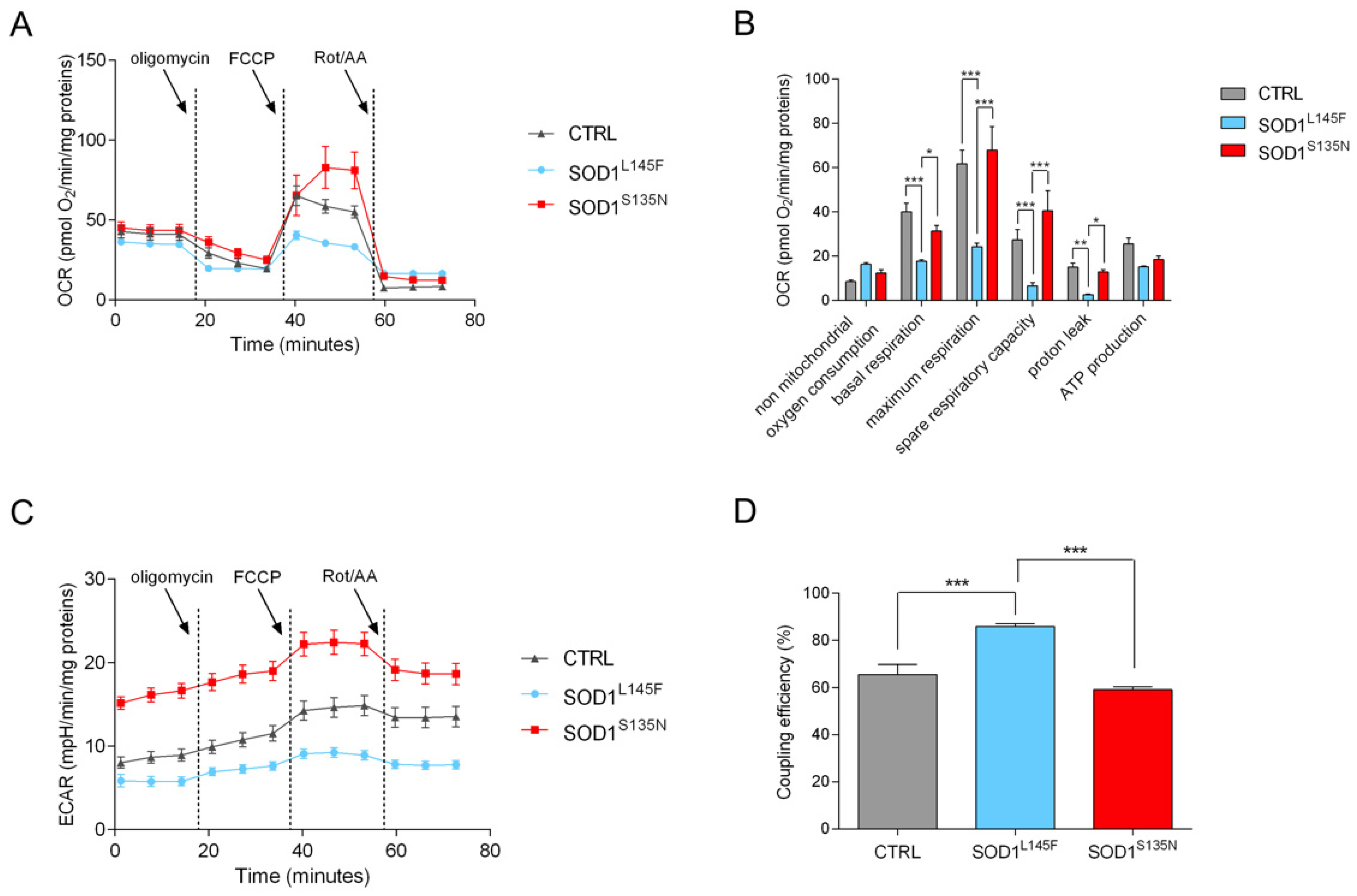
3.4.2. Mito Stress Test Shows That SOD1L145F Fibroblasts Work at Their Maximum Respiratory Rate
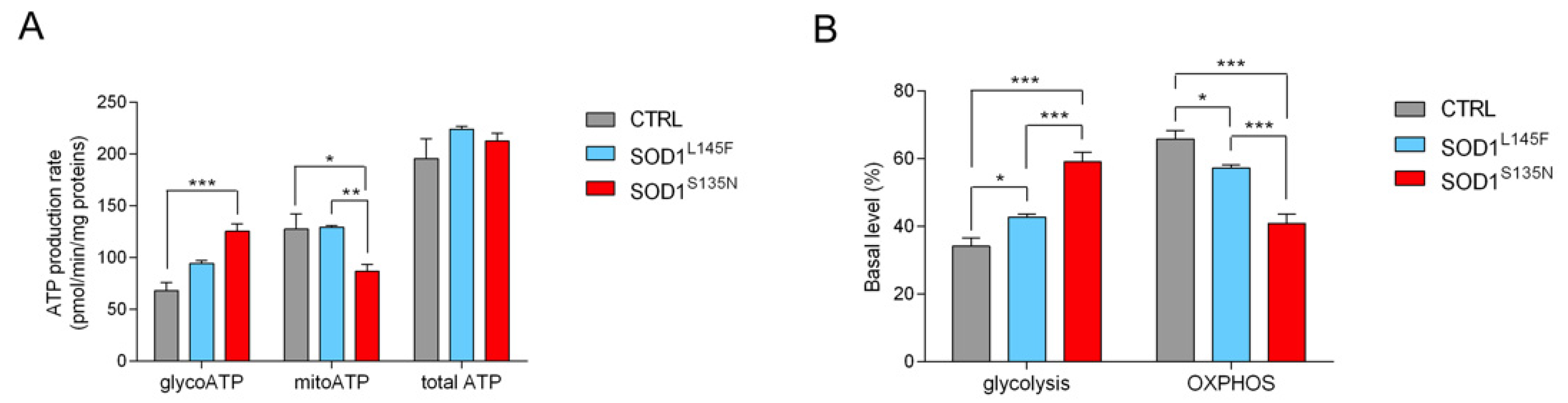
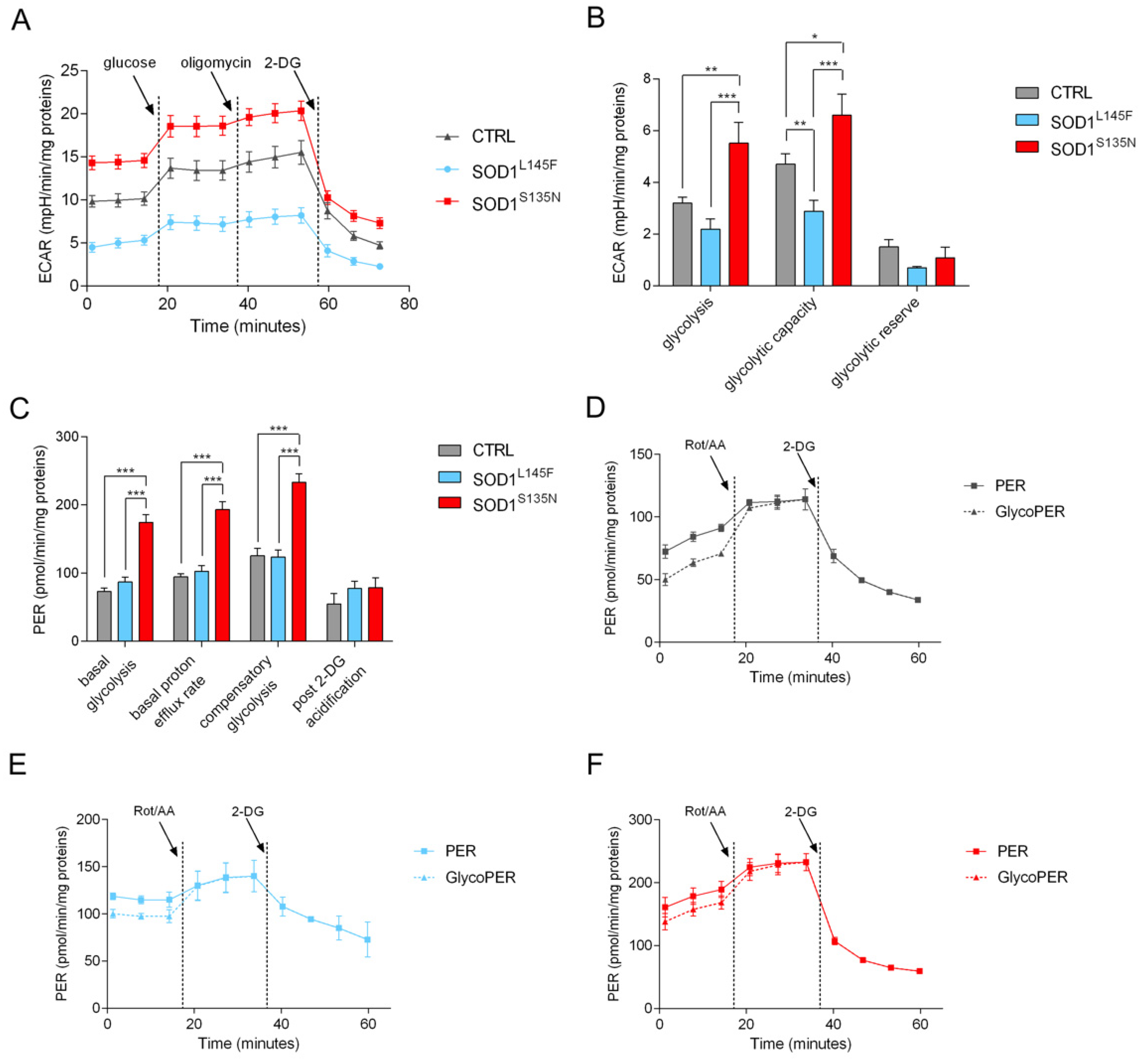
3.4.3. SODS135N Fibroblasts Rely More on Glycolysis Than on Oxidative Phosphorylation for ATP Production
3.4.4. SOD1S135N Fibroblasts Show Higher Glycolytic Rate Than the Control
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Grassano, M.; Calvo, A.; Moglia, C.; Brunetti, M.; Barberis, M.; Sbaiz, L.; Canosa, A.; Manera, U.; Vasta, R.; Corrado, L.; et al. Mutational Analysis of Known ALS Genes in an Italian Population-Based Cohort. Neurology 2021, 96, e600–e609. [Google Scholar] [CrossRef]
- Alsultan, A.A.; Waller, R.; Heath, P.R.; Kirby, J. The genetics of amyotrophic lateral sclerosis: Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 49–64. [Google Scholar] [CrossRef] [Green Version]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Sturtz, L.A.; Diekert, K.; Jensen, L.T.; Lill, R.; Culotta, V.C. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 2001, 276, 38084–38089. [Google Scholar] [CrossRef]
- Crapo, J.D.; Oury, T.; Rabouille, C.; Slot, J.W.; Chang, L.Y. Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc. Natl. Acad. Sci. USA 1992, 89, 10405–10409. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [Green Version]
- De vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; Mcloughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Huai, J.; Zhang, Z. Structural properties and interaction partners of familial ALS-associated SOD1 mutants. Front. Neurol. 2019, 10, 527. [Google Scholar] [CrossRef] [Green Version]
- Pacitti, D.; Privolizzi, R.; Bax, B.E. Organs to Cells and Cells to Organoids: The Evolution of in vitro Central Nervous System Modelling. Front. Cell. Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef]
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Aoki, M.; Abe, K.; Shoji, M.; Iizuka, T.; Ikeda, Y.; Hirai, S.; Kurokawa, K.; Kato, T.; Sasaki, H.; et al. A novel missense point mutation (S134N) of the Cu/Zn superoxide dismutase gene in a patient with familal motor neuron disease. Hum. Mutat. 1997, 9, 69–71. [Google Scholar] [CrossRef]
- Aoki, M.; Abe, K.; Itoyama, Y. Molecular analyses of the Cu/Zn superoxide dismutase gene in patients with familial amyotrophic lateral sclerosis (ALS) in Japan. Cell. Mol. Neurobiol. 1998, 18, 639–647. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [Green Version]
- Tripolszki, K.; Csányi, B.; Nagy, D.; Ratti, A.; Tiloca, C.; Silani, V.; Kereszty, É.; Török, N.; Vécsei, L.; Engelhardt, J.I.; et al. Genetic analysis of the SOD1 and C9ORF72 genes in Hungarian patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2017, 53, 195.e1–195.e5. [Google Scholar] [CrossRef] [Green Version]
- D’Anzi, A.; Altieri, F.; Perciballi, E.; Ferrari, D.; Bernardini, L.; Goldoni, M.; Mazzini, L.; De Marchi, F.; Di Pierro, A.; D’Alfonso, S.; et al. Generation of an induced pluripotent stem cell line, CSSi011-A (6534), from an Amyotrophic lateral sclerosis patient with heterozygous L145F mutation in SOD1 gene. Stem Cell Res. 2020, 47, 101924. [Google Scholar] [CrossRef]
- Masè, G.; Ros, S.; Gemma, A.; Bonfigli, L.; Carraro, N.; Cazzato, G.; Rolfo, M.; Zanconati, F.; Sepcic, J.; Jurjevic, A.; et al. ALS with variable phenotypes in a six-generation family caused by leu144phe mutation in the SOD1 gene. J. Neurol. Sci. 2001, 191, 11–18. [Google Scholar] [CrossRef]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Muyderman, H.; Hutson, P.G.; Matusica, D.; Rogers, M.-L.; Rush, R.A. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochem. Res. 2009, 34, 1847–1856. [Google Scholar] [CrossRef]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [CrossRef]
- Kirk, K.; Gennings, C.; Hupf, J.C.; Tadesse, S.; D’Aurelio, M.; Kawamata, H.; Valsecchi, F.; Mitsumoto, H.; Manfredi, G. Bioenergetic markers in skin fibroblasts of sporadic amyotrophic lateral sclerosis and progressive lateral sclerosis patients. Ann. Neurol. 2014, 76, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Gilis, D.; Rooman, M. PoPMuSiC, an algorithm for predicting protein mutant stability changes: Application to prion proteins. Protein Eng. 2000, 13, 849–856. [Google Scholar] [CrossRef] [Green Version]
- Kepp, K.P. Computing stability effects of mutations in human superoxide dismutase 1. J. Phys. Chem. B 2014, 118, 1799–1812. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [Green Version]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A web server for structure-based protein stability prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, H.; Zhang, N.; Zhu, Z.; Wang, S.; Li, M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput. Biol. 2020, 16, e1008543. [Google Scholar] [CrossRef]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-4; Maestro, Schrödinger, LLC: New York, NY, USA, 2021.
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 2014, 82, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2020.
- Gardiner, B.; Dougherty, J.A.; Ponnalagu, D.; Singh, H.; Angelos, M.; Chen, C.-A.; Khan, M. Measurement of Oxidative Stress Markers In Vitro Using Commercially Available Kits; Berliner, L.J., Parinandi, N.L., Eds.; Springer: Cham, Switzerland, 2020; pp. 39–60. ISBN 978-3-030-47317-4. [Google Scholar]
- Bovio, F.; Melchioretto, P.; Forcella, M.; Fusi, P.; Urani, C. Cadmium promotes glycolysis upregulation and glutamine dependency in human neuronal cells. Neurochem. Int. 2021, 149, 105144. [Google Scholar] [CrossRef]
- Ludolph, A.; Drory, V.; Hardiman, O.; Nakano, I.; Ravits, J.; Robberecht, W.; Shefner, J. A revision of the El Escorial criteria—2015. Amyotroph Lateral Scler Front. Degener 2015, 16, 291–292. [Google Scholar] [CrossRef]
- Kumar, V.; Rahman, S.; Choudhry, H.; Zamzami, M.A.; Sarwar Jamal, M.; Islam, A.; Ahmad, F.; Hassan, M.I. Computing disease-linked SOD1 mutations: Deciphering protein stability and patient-phenotype relations. Sci. Rep. 2017, 7, 4678. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.S.A.; Antonyuk, S.V.; Hasnain, S.S. The biophysics of superoxide dismutase-1 and amyotrophic lateral sclerosis. Q. Rev. Biophys. 2019, 52, e12. [Google Scholar] [CrossRef]
- Hayward, L.J.; Rodriguez, J.A.; Kim, J.W.; Tiwari, A.; Goto, J.J.; Cabelli, D.E.; Valentine, J.S.; Brown, R.H.J. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 15923–15931. [Google Scholar] [CrossRef] [Green Version]
- Elam, J.S.; Taylor, A.B.; Strange, R.; Antonyuk, S.; Doucette, P.A.; Rodriguez, J.A.; Hasnain, S.S.; Hayward, L.J.; Valentine, J.S.; Yeates, T.O.; et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 2003, 10, 461–467. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; D’Amelio, N.; Gaggelli, E.; Libralesso, E.; Matecko, I.; Turano, P.; Valentine, J.S. Fully metallated S134N Cu,Zn-superoxide dismutase displays abnormal mobility and intermolecular contacts in solution. J. Biol. Chem. 2005, 280, 35815–35821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, E.F.; Cervantes, L. An in silico study of the effect of SOD1 electrostatic loop dynamics on amyloid-like filament formation. Eur. Biophys. J. 2016, 45, 853–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, E.; Rajasekaran, R. Computational Investigation on Electrostatic Loop Mutants Instigating Destabilization and Aggregation on Human SOD1 Protein Causing Amyotrophic Lateral Sclerosis. Protein J. 2019, 38, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.J.; Byström, R.; Boknäs, N.; Andersen, P.M.; Oliveberg, M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc. Natl. Acad. Sci. USA 2005, 102, 9754–9759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.J.; Normark, J.; Holmgren, A.; Oliveberg, M. Folding of human superoxide dismutase: Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc. Natl. Acad. Sci. USA 2004, 101, 15893–15898. [Google Scholar] [CrossRef] [Green Version]
- Doucette, P.A.; Whitson, L.J.; Cao, X.; Schirf, V.; Demeler, B.; Valentine, J.S.; Hansen, J.C.; Hart, P.J. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J. Biol. Chem. 2004, 279, 54558–54566. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Reports 2017, 8, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Carrera-Juliá, S.; Moreno, M.L.; Barrios, C.; de la Rubia Ortí, J.E.; Drehmer, E. Antioxidant Alternatives in the Treatment of Amyotrophic Lateral Sclerosis: A Comprehensive Review. Front. Physiol. 2020, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Richardson, K.; Allen, S.P.; Mortiboys, H.; Grierson, A.J.; Wharton, S.B.; Ince, P.G.; Shaw, P.J.; Heath, P.R. The effect of SOD1 mutation on cellular bioenergetic profile and viability in response to oxidative stress and influence of mutation-type. PLoS ONE 2013, 8, e68256. [Google Scholar] [CrossRef]
- Suzuki, M.; Mikami, H.; Watanabe, T.; Yamano, T.; Yamazaki, T.; Nomura, M.; Yasui, K.; Ishikawa, H.; Ono, S. Increased expression of TDP-43 in the skin of amyotrophic lateral sclerosis. Acta Neurol. Scand. 2010, 122, 367–372. [Google Scholar] [CrossRef]
- Sabatelli, M.; Zollino, M.; Conte, A.; Del Grande, A.; Marangi, G.; Lucchini, M.; Mirabella, M.; Romano, A.; Piacentini, R.; Bisogni, G.; et al. Primary fibroblasts cultures reveal TDP-43 abnormalities in amyotrophic lateral sclerosis patients with and without SOD1 mutations. Neurobiol. Aging 2015, 36, 2005.e5–2005.e13. [Google Scholar] [CrossRef]
- Oketa, Y.; Higashida, K.; Fukasawa, H.; Tsukie, T.; Ono, S. Abundant FUS-immunoreactive pathology in the skin of sporadic amyotrophic lateral sclerosis. Acta Neurol. Scand. 2013, 128, 257–264. [Google Scholar] [CrossRef]
- Ishikawa, H.; Yasui, K.; Oketa, Y.; Suzuki, M.; Ono, S. Increased expression of valosin-containing protein in the skin of patients with amyotrophic lateral sclerosis. J. Clin. Neurosci. 2012, 19, 522–526. [Google Scholar] [CrossRef]
- Fukazawa, H.; Tsukie, T.; Higashida, K.; Fujikura, M.; Ono, S. An immunohistochemical study of increased tumor necrosis factor-α in the skin of patients with amyotrophic lateral sclerosis. J. Clin. Neurosci. 2013, 20, 1371–1376. [Google Scholar] [CrossRef]
- Ono, S.; Hu, J.; Shimizu, N.; Imai, T.; Nakagawa, H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 187, 27–34. [Google Scholar] [CrossRef]
- Fang, L.; Huber-Abel, F.; Teuchert, M.; Hendrich, C.; Dorst, J.; Schattauer, D.; Zettlmeissel, H.; Wlaschek, M.; Scharffetter-Kochanek, K.; Tumani, H.; et al. Linking neuron and skin: Matrix metalloproteinases in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 2009, 285, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Bicchi, I.; Morena, F.; Argentati, C.; Nodari, L.R.; Emiliani, C.; Gelati, M.; Vescovi, A.L.; Martino, S. Storage of mutant human sod1 in non-neural cells from the type-1 amyotrophic lateral sclerosis ratg93a model correlated with the lysosomes’ dysfunction. Biomedicines 2021, 9, 1080. [Google Scholar] [CrossRef]
- Shenberger, J.S.; Zhang, L.; Hughlock, M.K.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R. Roles of mitogen-activated protein kinase signal-integrating kinases 1 and 2 in oxidant-mediated eIF4E phosphorylation. Int. J. Biochem. Cell Biol. 2007, 39, 1828–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’h, P.; Andres, C.R.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Can. J. Neurol. Sci. 2017, 44, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Calcagno, E.; Tartari, S.; Rizzardini, M.; Invernizzi, R.W.; Cantoni, L. Glutamate and glutathione interplay in a motor neuronal model of amyotrophic lateral sclerosis reveals altered energy metabolism. Neurobiol. Dis. 2011, 43, 346–355. [Google Scholar] [CrossRef]
- Kim, K. Glutathione in the nervous system as a potential therapeutic target to control the development and progression of amyotrophic lateral sclerosis. Antioxidants 2021, 10, 1011. [Google Scholar] [CrossRef]
- Martinez-Banaclocha, M. N-Acetyl-Cysteine: Modulating the Cysteine Redox Proteome in Neurodegenerative Diseases. Antioxidants 2022, 11, 416. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador, R.; Estrela, J.M.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L. Oxidative stress, neuroinflammation and mitochondria in the pathophysiology of amyotrophic lateral sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Perera, N.D.; Turner, B.J. AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res. 2016, 41, 544–553. [Google Scholar] [CrossRef]
- Buck, E.; Bayer, H.; Lindenberg, K.S.; Hanselmann, J.; Pasquarelli, N.; Ludolph, A.C.; Weydt, P.; Witting, A. Comparison of sirtuin 3 levels in ALS and huntington’s disease—differential effects in human tissue samples vs. Transgenic mouse models. Front. Mol. Neurosci. 2017, 10, 156. [Google Scholar] [CrossRef]
- Sonntag, K.C.; Ryu, W.I.; Amirault, K.M.; Healy, R.A.; Siegel, A.J.; McPhie, D.L.; Forester, B.; Cohen, B.M. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017, 7, 14038. [Google Scholar] [CrossRef] [Green Version]
- Aldana, B.I.; Zhang, Y.; Jensen, P.; Chandrasekaran, A.; Christensen, S.K.; Nielsen, T.T.; Nielsen, J.E.; Hyttel, P.; Larsen, M.R.; Waagepetersen, H.S.; et al. Glutamate-glutamine homeostasis is perturbed in neurons and astrocytes derived from patient iPSC models of frontotemporal dementia. Mol. Brain 2020, 13, 125. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perciballi, E.; Bovio, F.; Rosati, J.; Arrigoni, F.; D’Anzi, A.; Lattante, S.; Gelati, M.; De Marchi, F.; Lombardi, I.; Ruotolo, G.; et al. Characterization of the p.L145F and p.S135N Mutations in SOD1: Impact on the Metabolism of Fibroblasts Derived from Amyotrophic Lateral Sclerosis Patients. Antioxidants 2022, 11, 815. https://doi.org/10.3390/antiox11050815
Perciballi E, Bovio F, Rosati J, Arrigoni F, D’Anzi A, Lattante S, Gelati M, De Marchi F, Lombardi I, Ruotolo G, et al. Characterization of the p.L145F and p.S135N Mutations in SOD1: Impact on the Metabolism of Fibroblasts Derived from Amyotrophic Lateral Sclerosis Patients. Antioxidants. 2022; 11(5):815. https://doi.org/10.3390/antiox11050815
Chicago/Turabian StylePerciballi, Elisa, Federica Bovio, Jessica Rosati, Federica Arrigoni, Angela D’Anzi, Serena Lattante, Maurizio Gelati, Fabiola De Marchi, Ivan Lombardi, Giorgia Ruotolo, and et al. 2022. "Characterization of the p.L145F and p.S135N Mutations in SOD1: Impact on the Metabolism of Fibroblasts Derived from Amyotrophic Lateral Sclerosis Patients" Antioxidants 11, no. 5: 815. https://doi.org/10.3390/antiox11050815
APA StylePerciballi, E., Bovio, F., Rosati, J., Arrigoni, F., D’Anzi, A., Lattante, S., Gelati, M., De Marchi, F., Lombardi, I., Ruotolo, G., Forcella, M., Mazzini, L., D’Alfonso, S., Corrado, L., Sabatelli, M., Conte, A., De Gioia, L., Martino, S., Vescovi, A. L., ... Ferrari, D. (2022). Characterization of the p.L145F and p.S135N Mutations in SOD1: Impact on the Metabolism of Fibroblasts Derived from Amyotrophic Lateral Sclerosis Patients. Antioxidants, 11(5), 815. https://doi.org/10.3390/antiox11050815