
Profiling the Site of Protein CoAlation and Coenzyme A Stabilization Interactions

 ,
,  , , , , ,
, , , , , 
Abstract
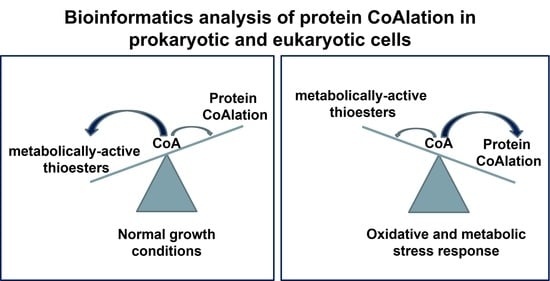
:
1. Introduction
2. Materials and Methods
2.1. Mammalian and Bacterial CoAlome (CoAlated Protein Dataset) Construction
2.2. Functional Classification of Mammalian and Bacterial CoAlated Proteins
2.3. Analysis of the Different Types of CoAlated Proteins
2.4. Analysis of CoA’s Structural Flexibility and 3D Stabilization Interactions
2.5. Construction of Mammalian and Bacterial 7-Amino-Acid-Long Peptide Datasets and 2D WebLogos
3. Results
3.1. Construction of Mammalian and Bacterial Datasets of CoAlated Proteins
3.2. Functional Characterization of Mammalian CoAlated Proteins
3.3. Molecular Function of Bacterial CoAlated Proteins
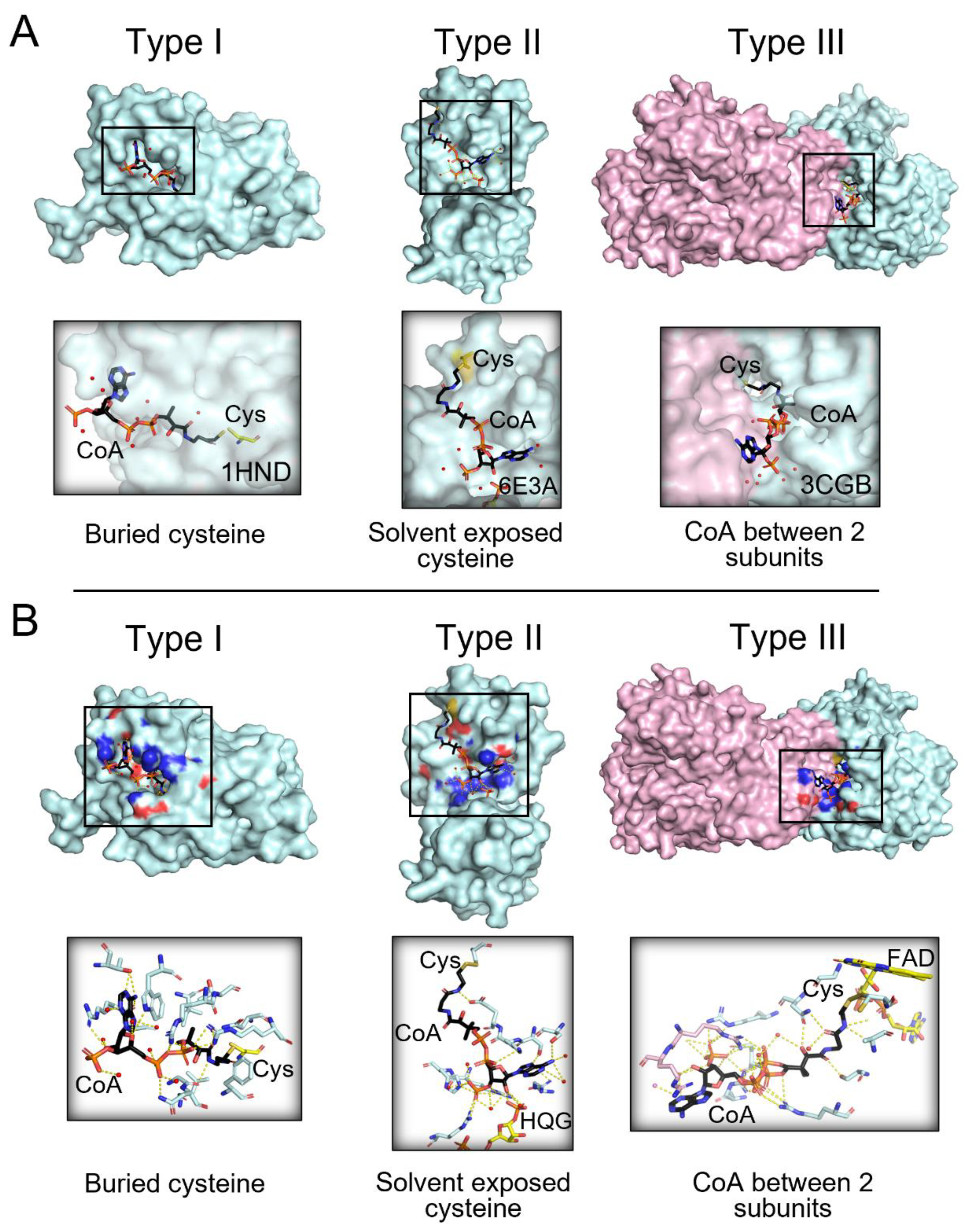
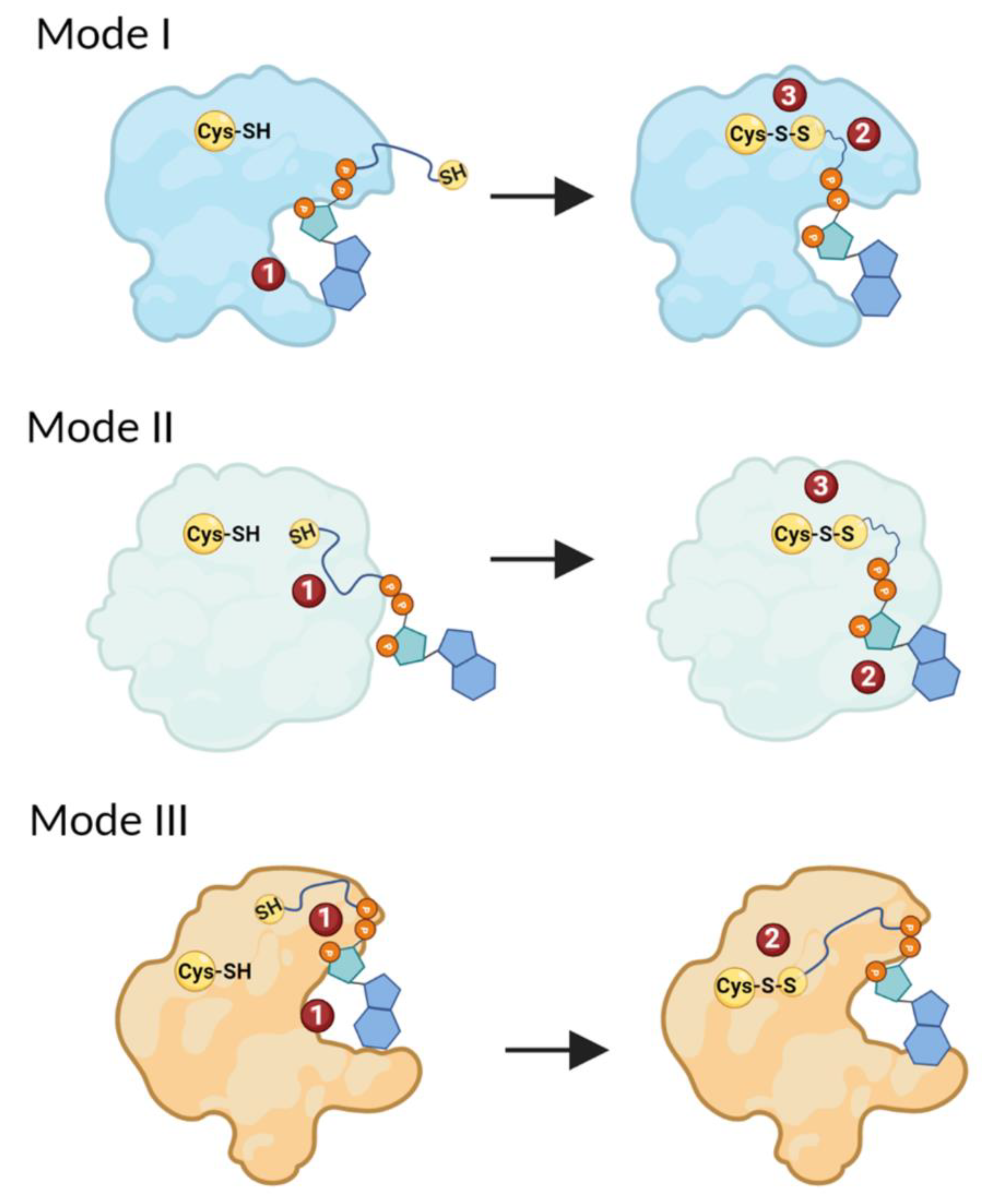
3.4. Structural Analysis Reveals Three Main Types of CoAlated Proteins
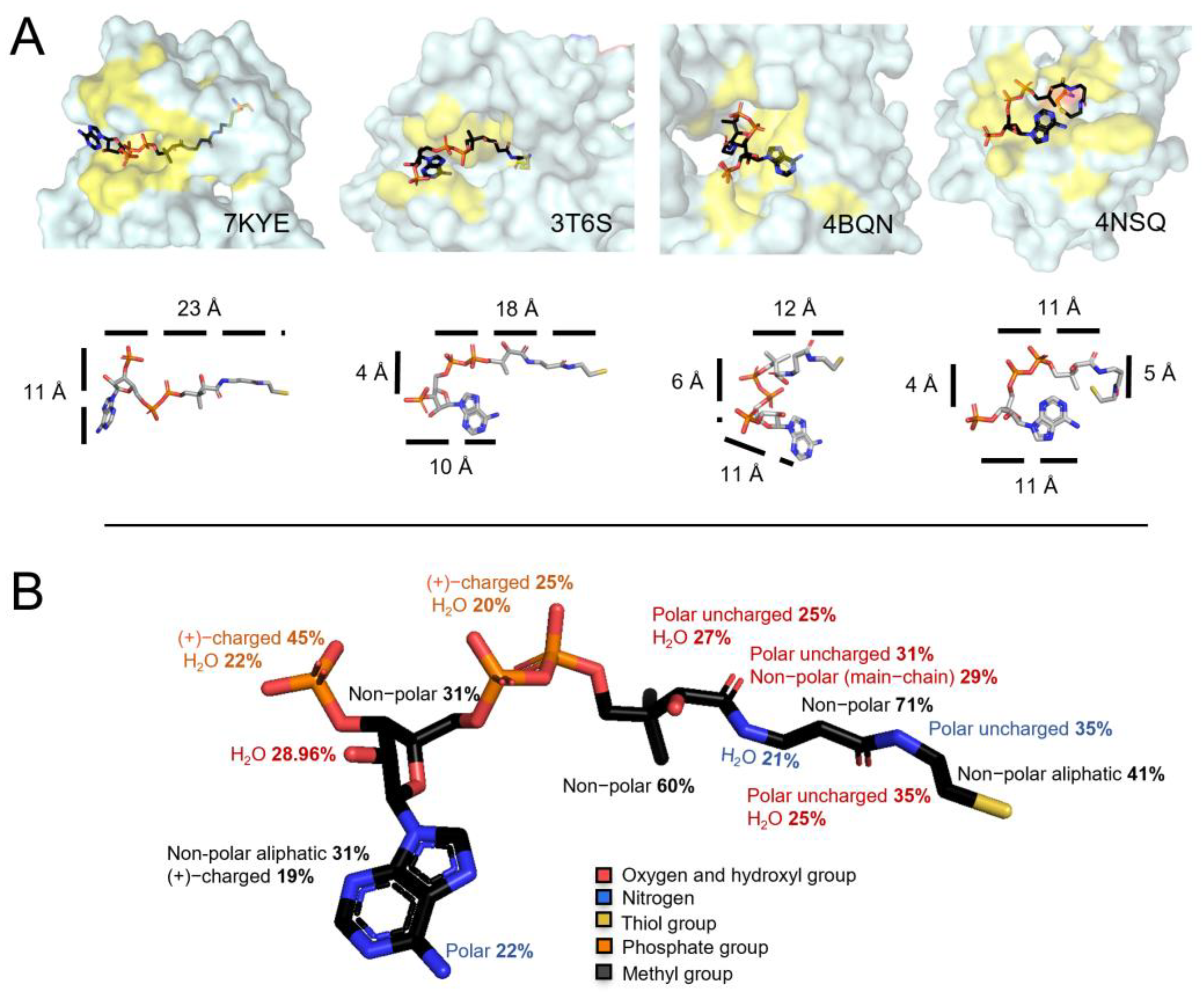
3.5. CoA Is a Bulky but Flexible Molecule When in Complex with Covalently Modified Proteins
3.6. Profiling Coenzyme A–Protein Interactions and the Amino Acids Involved
3.7. Exploring the Direct Microenvironment of the CoAlated Cysteine
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox. Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Collet, J.-F.; Messens, J. Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 2010, 13, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, B.K.; Gronau, K.; Mäder, U.; Hessling, B.; Becher, D.; Antelmann, H. S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell. Proteom. 2011, 10, M111 009506. [Google Scholar] [CrossRef] [Green Version]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1642–1653. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Peak-Chew, S.Y.; Newell, C.; Miller-Aidoo, S.; Mangal, S.; Zhyvoloup, A.; Bakovic, J.; Malanchuk, O.; Pereira, G.C.; Kotiadis, V.; et al. Protein CoAlation: A redox-regulated protein modification by coenzyme A in mammalian cells. Biochem. J. 2017, 474, 2489–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Zhyvoloup, A.; Baković, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G.; et al. Protein CoAlation and antioxidant function of coenzyme A in prokaryotic cells. Biochem. J. 2018, 475, 1909–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gout, I. Coenzyme A: A protective thiol in bacterial antioxidant defence. Biochem. Soc. Trans. 2019, 47, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Gout, I. Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Loi, V.V.; Rossius, M.; Antelmann, H. Redox regulation by reversible protein S-thiolation in bacteria. Front. Microbiol. 2015, 6, 187. [Google Scholar] [CrossRef] [Green Version]
- Tossounian, M.-A.; Truong, A.-C.K.; Buts, L.; Wahni, K.; Mourenza, Á.; Leermakers, M.; Vertommen, D.; Mateos, L.M.; Volkov, A.N.; Messens, J. Methionine sulfoxide reductase B from Corynebacterium diphtheriae catalyzes sulfoxide reduction via an intramolecular disulfide cascade. J. Biol. Chem. 2020, 295, 3664–3677. [Google Scholar] [CrossRef]
- Tossounian, M.-A.; Zhang, B.; Gout, I. The Writers, Readers, and Erasers in Redox Regulation of GAPDH. Antioxidants 2020, 9, 1288. [Google Scholar] [CrossRef]
- Rahuel-Clermont, S.; Toledano, M.B. Parsing protein sulfinic acid switches. Nat. Chem. Biol. 2018, 14, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Möller, M.N.; Alvarez, B. Biological chemistry of hydrogen sulfide and persulfides. Arch. Biochem. Biophys. 2017, 617, 9–25. [Google Scholar] [PubMed]
- Malanchuk, O.M.; Panasyuk, G.G.; Serbin, N.M.; Gout, I.T.; Filonenko, V.V. Generation and characterization of monoclonal antibodies specific to coenzyme A. Biopolym. Cell 2015, 31, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Zhyvoloup, A.; Yu, B.Y.K.; Baković, J.; Davis-Lunn, M.; Tossounian, M.-A.; Thomas, N.; Tsuchiya, Y.; Peak-Chew, S.Y.; Wigneshweraraj, S.; Filonenko, V.; et al. Analysis of disulphide bond linkage between CoA and protein cysteine thiols during sporulation and in spores of Bacillus species. FEMS Microbiol. Lett. 2020, 367, fnaa174. [Google Scholar]
- Tsuji, K.; Yoon, K.S.; Ogo, S. Glyceraldehyde-3-phosphate dehydrogenase from Citrobacter sp. S-77 is post-translationally modified by CoA (protein CoAlation) under oxidative stress. FEBS Open Bio 2019, 9, 53–73. [Google Scholar] [CrossRef] [Green Version]
- Barinova, K.V.; Serebryakova, M.V.; Muronetz, V.I.; Schmalhausen, E.V. S-glutathionylation of glyceraldehyde-3-phosphate dehydrogenase induces formation of C150-C154 intrasubunit disulfide bond in the active site of the enzyme. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3167–3177. [Google Scholar] [CrossRef]
- Hillion, M.; Imber, M.; Pedre, B.; Bernhardt, J.; Saleh, M.; Van Loi, V.; Maaß, S.; Becher, D.; Rosado, L.A.; Adrian, L.; et al. The glyceraldehyde-3-phosphate dehydrogenase GapDH of Corynebacterium diphtheriae is redox-controlled by protein S-mycothiolation under oxidative stress. Sci. Rep. 2017, 7, 5020. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Baković, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T.; et al. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318. [Google Scholar] [CrossRef]
- Baković, J.; Yu, B.Y.K.; Silva, D.; Chew, S.P.; Kim, S.; Ahn, S.-H.; Palmer, L.; Aloum, L.; Stanzani, G.; Malanchuk, O.; et al. A key metabolic integrator, coenzyme A, modulates the activity of peroxiredoxin 5 via covalent modification. Mol. Cell. Biochem. 2019, 461, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.Y.K.; Tossounian, M.-A.; Hristov, S.D.; Lawrence, R.; Arora, P.; Tsuchiya, Y.; Peak-Chew, S.Y.; Filonenko, V.; Oxenford, S.; Angell, R.; et al. Regulation of metastasis suppressor NME1 by a key metabolic cofactor coenzyme A. Redox Biol. 2021, 44, 101978. [Google Scholar] [CrossRef]
- Baković, J.; Yu, B.; Silva, D.; Baczynska, M.; Peak-Chew, S.; Switzer, A.; Burchell, L.; Wigneshweraraj, S.; Vandanashree, M.; Gopal, B.; et al. Redox Regulation of the Quorum-sensing Transcription Factor AgrA by Coenzyme A. Antioxidants 2021, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Tossounian, M.A.; Costello Heaven, N.; Wallworth, S.; Peak-Chew, S.; Bradshaw, A.; Cooper, J.M.; de Silva, R.; Srai, S.K.; Malanchuk, O.; et al. Extensive Anti-CoA Immunostaining in Alzheimer’s Disease and Covalent Modification of Tau by a Key Cellular Metabolite Coenzyme A. Front. Cell. Neurosci. 2021, 15, 739425. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, M.Z.; Meyer, A.G.; Sydykova, D.K.; Spielman, S.J.; Wilke, C.O. Maximum allowed solvent accessibilites of residues in proteins. PLoS ONE 2013, 8, e80635. [Google Scholar]
- Savojardo, C.; Manfredi, M.; Martelli, P.L.; Casadio, R. Solvent Accessibility of Residues Undergoing Pathogenic Variations in Humans: From Protein Structures to Protein Sequences. Front. Mol. Biosci. 2020, 7, 626363. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Ishikita, H.; Saito, K. Proton transfer reactions and hydrogen-bond networks in protein environments. J. R. Soc. Interface 2014, 11, 20130518. [Google Scholar] [CrossRef] [PubMed]
- Onofrio, A.; Parisi, G.; Punzi, G.; Todisco, S.; Di Noia, M.A.; Bossis, F.; Turi, A.; De Grassi, A.; Pierri, C.L. Distance-dependent hydrophobic-hydrophobic contacts in protein folding simulations. Phys. Chem. Chem. Phys. 2014, 16, 18907–18917. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Engel, C.; Wierenga, R. The diverse world of coenzyme A binding proteins. Curr. Opin. Struct. Biol. 1996, 6, 790–797. [Google Scholar] [CrossRef]
- Spies, H.S.C.; Steenkamp, D.J. Thiols of intracellular pathogens. Identification of ovothiol A in Leishmania donovani and structural analysis of a novel thiol from Mycobacterium bovis. Eur. J. Biochem. 1994, 224, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Imber, M.; Huyen, N.T.T.; Pietrzyk-Brzezinska, A.J.; Van Loi, V.; Hillion, M.; Bernhardt, J.; Thärichen, L.; Kolšek, K.; Saleh, M.; Hamilton, C.J.; et al. Protein S-Bacillithiolation Functions in Thiol Protection and Redox Regulation of the Glyceraldehyde-3-Phosphate Dehydrogenase Gap in Staphylococcus aureus Under Hypochlorite Stress. Antioxid. Redox Signal. 2018, 28, 410–430. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Shi, Z.-Z.; Zhou, X.; Chen, L.; Zhao, X.-M. Prediction of S-glutathionylation sites based on protein sequences. PLoS ONE 2013, 8, e55512. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Lu, C.-T.; Lee, T.-Y. dbGSH: A database of S-glutathionylation. Bioinformatics 2014, 30, 2386–2388. [Google Scholar] [CrossRef]
- Anashkina, A.A.; Poluektov, Y.M.; Dmitriev, V.A.; Kuznetsov, E.N.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. A novel approach for predicting protein S-glutathionylation. BMC Bioinform. 2020, 21, 282. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J. Structure in Protein Chemistry, 2nd ed.; Garland Science: New York, NY, USA, 2007. [Google Scholar]
- Roos, G.; Foloppe, N.; Messens, J. Understanding the pK(a) of redox cysteines: The key role of hydrogen bonding. Antioxid. Redox Signal. 2013, 18, 94–127. [Google Scholar] [CrossRef] [PubMed]
- Shekhter, T.; Metanis, N.; Dawson, P.E.; Keinan, E. A residue outside the active site CXXC motif regulates the catalytic efficiency of Glutaredoxin 3. Mol. BioSyst. 2010, 6, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foloppe, N.; Nilsson, L. Stabilizing of the catalytic thiolate in a mammalian glutaredoxin: Structure, dynamics and electrostatics of reduced pig glutaredoxin and its mutants. J. Mol. Biol. 2007, 372, 798–816. [Google Scholar] [CrossRef]
- Gane, P.J.; Freedman, R.B.; Warwicker, J. A Molecular-Model for the Redox Potential Difference between Thioredoxin and Dsba, based on Electrostatics Calculations. J. Mol. Biol. 1995, 249, 376–387. [Google Scholar] [CrossRef]
- Hol, W.G.J.; Van Duijnen, P.T.; Berendsen, H.J.C. The alpha-helix dipole and the properties of proteins. Nature 1978, 273, 443–446. [Google Scholar] [CrossRef]
- Kortemme, T.; Creighton, T.E. Ionization of Cysteine Residues at the Termini of Model Alpha-Helical Peptides. Relevance to Unusual Thiol Pk(a) Values in Proteins of the Thioredoxin Family. J. Mol. Biol. 1995, 253, 799–812. [Google Scholar] [CrossRef]
- Kortemme, T.; Darby, N.J.; Creighton, T.E. Electrostatic interactions in the active site of the N-terminal thioredoxin-like domain of protein disulfide isomerase. Biochemistry 1996, 35, 14503–14511. [Google Scholar] [CrossRef]
- Madzelan, P.; Labunska, T.; Wilson, M.A. Influence of peptide dipoles and hydrogen bonds on reactive cysteine pKa values in fission yeast DJ-1. FEBS J. 2012, 279, 4111–4120. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 445–473. [Google Scholar] [CrossRef]
- Armstrong, R.N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997, 10, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; von Rosenvinge, E.C.; Johnson, W.W.; Tomarev, S.I.; Piatigorsky, J.; Armstrong, R.N.; Gilliland, G.L. Three-dimensional structure, catalytic properties, and evolution of a sigma class glutathione transferase from squid, a progenitor of the lens S-crystallins of cephalopods. Biochemistry 1995, 34, 5317–5328. [Google Scholar] [CrossRef]
- Kolm, R.H.; Sroga, G.E.; Mannervik, B. Participation of the phenolic hydroxyl group of Tyr-8 in the catalytic mechanism of human glutathione transferase P1-1. Biochem. J. 1992, 285 Pt 2, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhang, P.; Ji, X.; Johnson, W.W.; Gilliland, G.L.; Armstrong, R.N. Contribution of tyrosine 6 to the catalytic mechanism of isoenzyme 3-3 of glutathione S-transferase. J. Biol. Chem. 1992, 267, 4296–4299. [Google Scholar] [CrossRef]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.T.; Rossmann, M.G. Comparison of super-secondary structures in proteins. J. Mol. Biol. 1973, 76, 241–256. [Google Scholar] [CrossRef]
- Zhang, S.; Nelson, O.D.; Price, I.R.; Zhu, C.; Lu, X.; Fernandez, I.R.; Weiss, R.S.; Lin, H. Long-chain fatty acyl coenzyme A inhibits NME1/2 and regulates cancer metastasis. Proc. Natl. Acad. Sci. USA 2022, 119, e2117013119. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
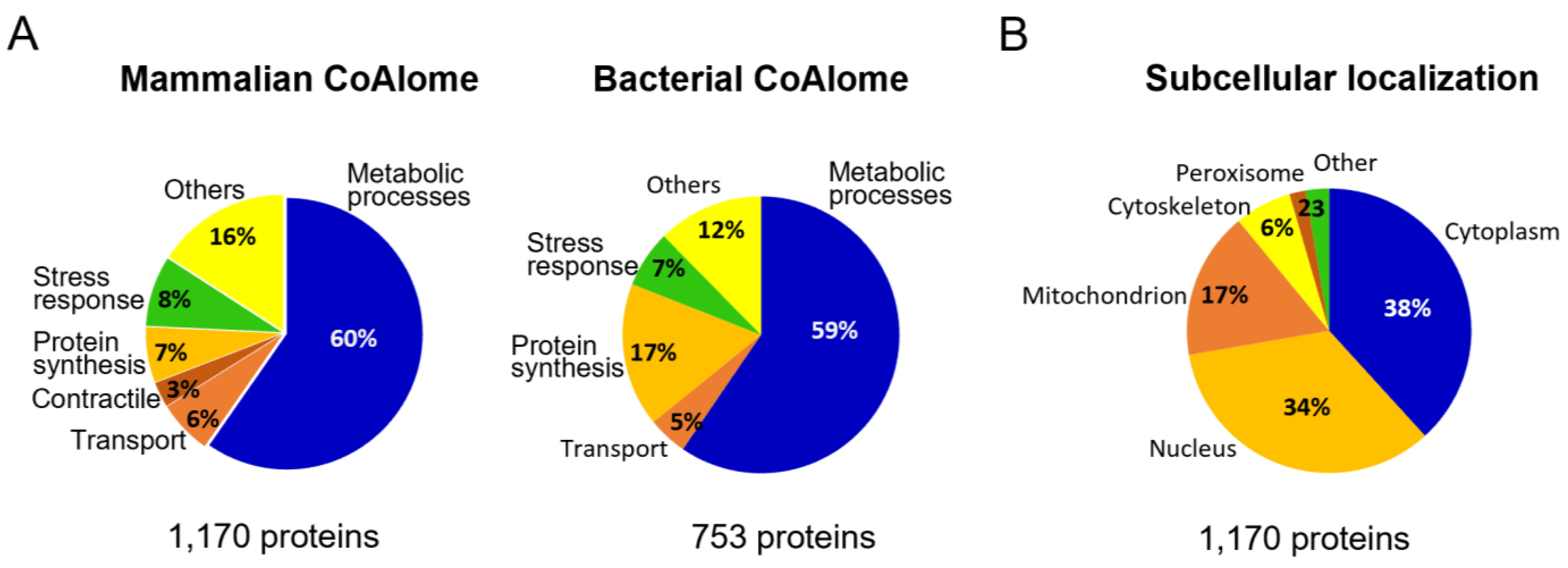{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mammalian CoAlome | |||
| Dataset | Organism, Cell or Tissue Type | CoAlated Proteins (Peptides) | Total Proteins (Peptides) |
| 1 | R. norvegicus perfused heart [11] | 26 (64) | 1170 (1728) |
| R. norvegicus liver mitochondria [11] | 18 (30) | ||
| HEK293/Pank1β cells | 1126 (1634) | ||
| Bacterial CoAlome | |||
| Dataset | Organism, Cell or Tissue Type | CoAlated Proteins (Peptides) | Total Proteins (Peptides) |
| 2 | B. megaterium [24] | 355 (439) | 923 (1134) |
| S. aureus [12] | 362 (448) | ||
| B. subtilis | 206 (247) | ||
| Total CoAlated Proteins (Peptides) = 2093 (2862) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tossounian, M.-A.; Baczynska, M.; Dalton, W.; Newell, C.; Ma, Y.; Das, S.; Semelak, J.A.; Estrin, D.A.; Filonenko, V.; Trujillo, M.; et al. Profiling the Site of Protein CoAlation and Coenzyme A Stabilization Interactions. Antioxidants 2022, 11, 1362. https://doi.org/10.3390/antiox11071362
Tossounian M-A, Baczynska M, Dalton W, Newell C, Ma Y, Das S, Semelak JA, Estrin DA, Filonenko V, Trujillo M, et al. Profiling the Site of Protein CoAlation and Coenzyme A Stabilization Interactions. Antioxidants. 2022; 11(7):1362. https://doi.org/10.3390/antiox11071362
Chicago/Turabian StyleTossounian, Maria-Armineh, Maria Baczynska, William Dalton, Charlie Newell, Yilin Ma, Sayoni Das, Jonathan Alexis Semelak, Dario Ariel Estrin, Valeriy Filonenko, Madia Trujillo, and et al. 2022. "Profiling the Site of Protein CoAlation and Coenzyme A Stabilization Interactions" Antioxidants 11, no. 7: 1362. https://doi.org/10.3390/antiox11071362
APA StyleTossounian, M. -A., Baczynska, M., Dalton, W., Newell, C., Ma, Y., Das, S., Semelak, J. A., Estrin, D. A., Filonenko, V., Trujillo, M., Peak-Chew, S. Y., Skehel, M., Fraternali, F., Orengo, C., & Gout, I. (2022). Profiling the Site of Protein CoAlation and Coenzyme A Stabilization Interactions. Antioxidants, 11(7), 1362. https://doi.org/10.3390/antiox11071362