
Hormesis and Oxidative Distress: Pathophysiology of Reactive Oxygen Species and the Open Question of Antioxidant Modulation and Supplementation


 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
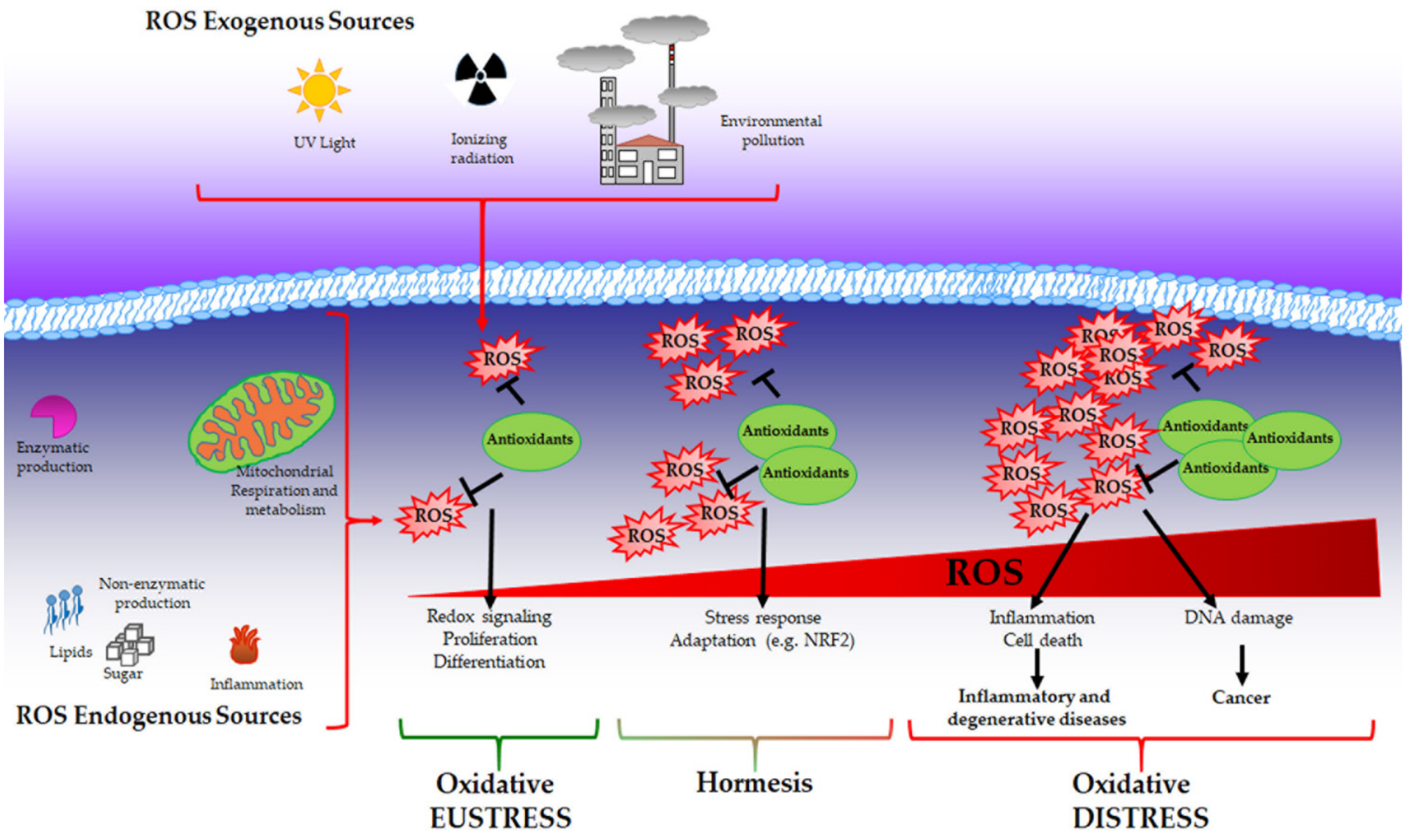
1. Reactive Oxygen Species
2. Protection against ROS Generation
3. Modulation of Redox Equilibrium
3.1. Redox Signaling
3.2. The Nuclear Factor Erythroid 2 p45-Related Factor 2 (NRF2)
3.3. MicroRNAs
4. Oxidative (di)Stress and Disease
4.1. Oxidative Stress and Aging
4.2. Oxidative Stress in Inflammation and Inflammatory Diseases
4.3. Oxidative Stress and Metabolic Diseases
4.4. Oxidative Stress and Degenerative Diseases
4.5. Oxidative Stress and Cancer
GSH and GSH-Related Pathways in Cancer Progression
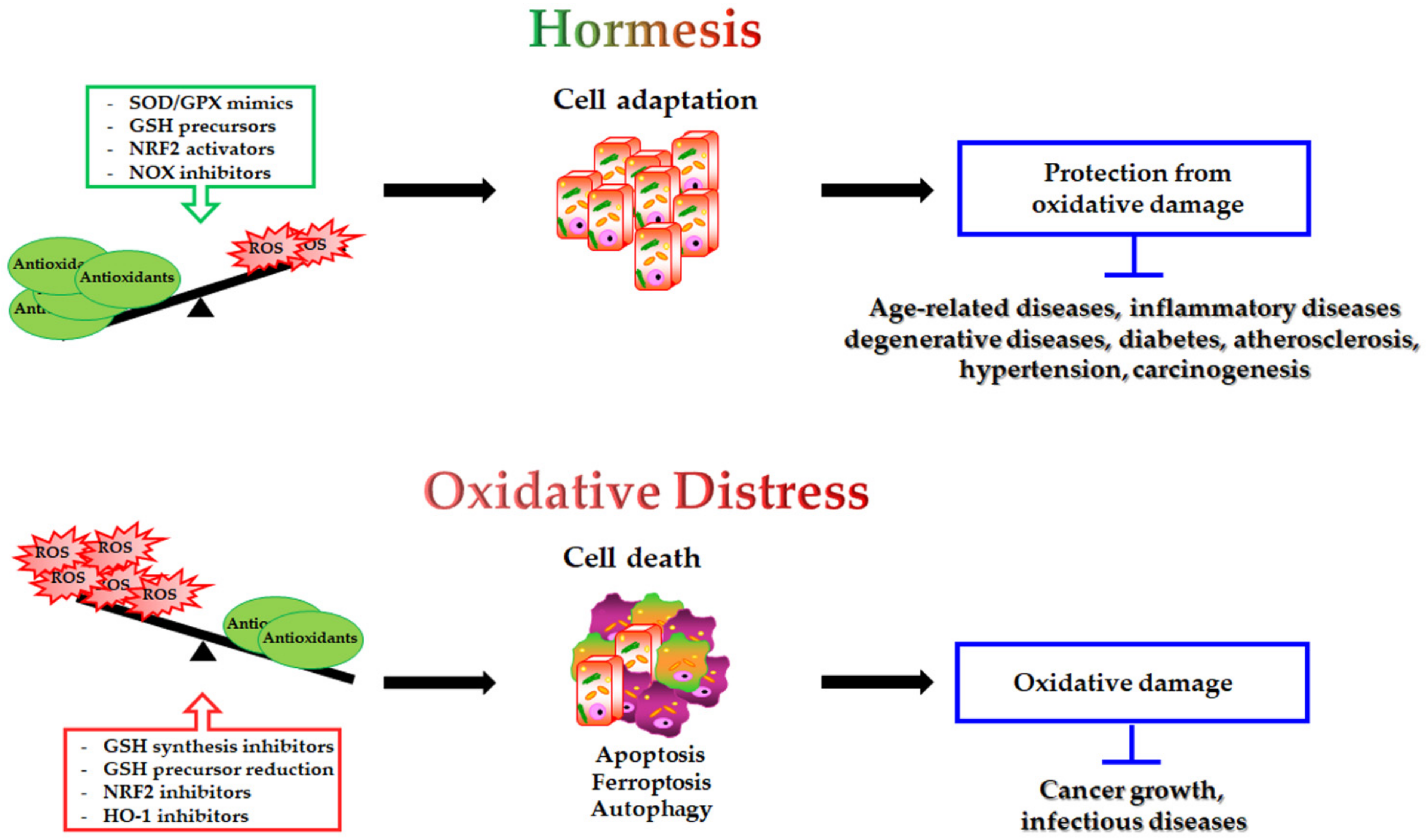
5. Antioxidants Used for Therapy
5.1. SOD Mimics, GPX Mimics and NOX Inhibitors
5.2. Modulation of GSH Levels
5.2.1. Strategies to Maintain GSH Homeostasis
5.2.2. Decreasing GSH Levels to Increase Sensitivity of Cancer Cells to Therapy
5.3. Modulation of NRF2 Activity
5.3.1. NRF2 Inducers
5.3.2. NRF2 Inhibitors to Improve Therapy Susceptibility in Cancer
5.4. Vitamins
5.5. Dietary Supplementation with Antioxidants: Lights and Shadows of Their Effects
6. Conclusions
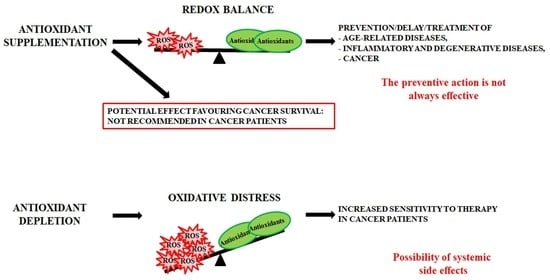
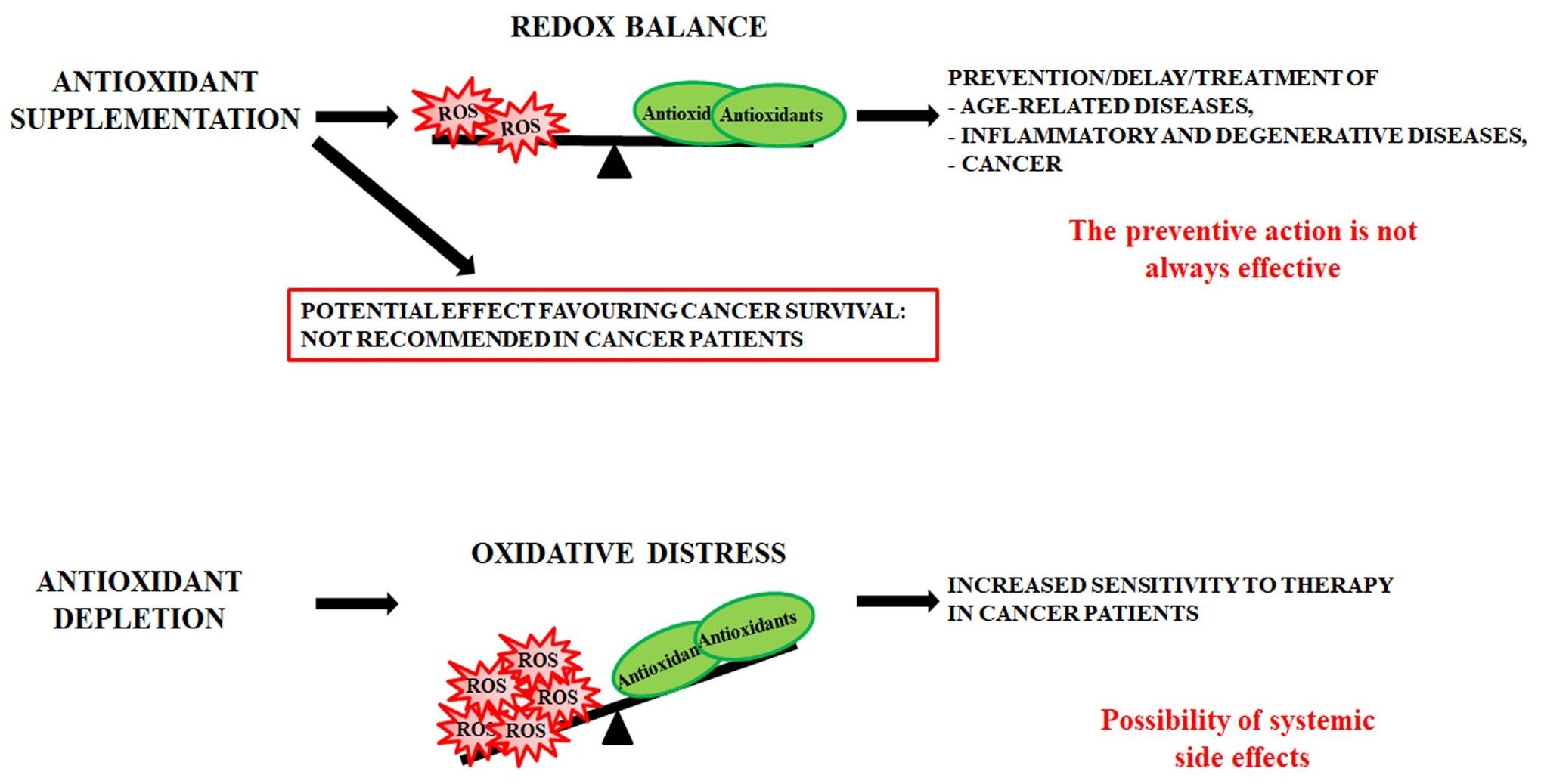
- antioxidant supplementation is utilised to prevent the onset of many age-related diseases even though in most cases it does not appear effective and sometimes can produce harmful effects [293]. Notably, caloric restriction and intermitting fasting by increasing the threshold of stress adaptive resistance has been proposed as alternative strategy able to preserve the human body from diseases and to increase the life expectancy [306,307];
- antioxidant depletion can be employed in association with therapy in order to fight infections [308] or especially cancer [309,310]. In fact, as herein reported, several studies demonstrate that more malignant tumour cells display high levels of endogenous antioxidants [211,212] and become resistant to oxidative stress generated by long term-treatment with anticancer drugs [202,203,204,205,218]. Unfortunately, the sensitising therapy based on the reduction of the antioxidant defence could affect also healthy cells, leading to systemic side effects.
Funding
Conflicts of Interest
References
- IUPAC. The IUPAC Compendium of Chemical Terminology: The Gold Book, 4th ed.; Gold, V., Ed.; International Union of Pure and Applied Chemistry (IUPAC): Research Triangle Park, NC, USA, 2019. [Google Scholar]
- Rubio, C.P.; Cerón, J.J. Spectrophotometric assays for evaluation of Reactive Oxygen Species (ROS) in serum: General concepts and applications in dogs and humans. BMC Vet. Res. 2021, 17, 226. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.C.; Halliwell, B. Mini-review: Oxidative stress, redox stress or redox success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, K.H.; Slater, T.F. An introduction to free radical biochemistry. Br. Med. Bull. 1993, 49, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Perez-Campo, R.; López-Torres, M.; Cadenas, S.; Rojas, C.; Barja, G. The rate of free radical production as a determinant of the rate of aging: Evidence from the comparative approach. J. Comp. Physiol. B 1998, 168, 149–158. [Google Scholar] [CrossRef]
- Cadenas, E. Mitochondrial free radical production and cell signaling. Mol. Asp. Med. 2004, 25, 17–26. [Google Scholar] [CrossRef]
- Nordmann, R.; Ribière, C.; Rouach, H. Implication of free radical mechanisms in ethanol-induced cellular injury. Free Radic. Biol. Med. 1992, 12, 219–240. [Google Scholar] [CrossRef]
- Gutierrez, P.L. The metabolism of quinone-containing alkylating agents: Free radical production and measurement. Front. Biosci. 2000, 5, D629–D638. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Nauseef, W.M. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004, 122, 277–291. [Google Scholar] [CrossRef]
- Leto, T.L.; Morand, S.; Hurt, D.; Ueyama, T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sheppard, O.R.; Shah, A.M.; Brewer, A.C. Positive regulation of the NADPH oxidase NOX4 promoter in vascular smooth muscle cells by E2F. Free Radic. Biol. Med. 2008, 45, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase-mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Free-radical production and oxidative reactions of hemoglobin. Environ. Health Perspect. 1985, 64, 321–330. [Google Scholar] [CrossRef]
- Wolff, S.P.; Dean, R.T. Glucose autoxidation and protein modification. The potential role of “autoxidative glycosylation” in diabetes. Biochem. J. 1987, 245, 243–250. [Google Scholar] [CrossRef]
- Brown, H.A.; Marnett, L.J. Introduction to Lipid Biochemistry, Metabolism, and Signaling. Chem. Rev. 2011, 111, 5817–5820. [Google Scholar] [CrossRef]
- Le Caër, S. Water Radiolysis: Influence of Oxide Surfaces on H2 Production under Ionizing Radiation. Water 2011, 3, 235–253. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015; ISBN 978-0-19-871747-8. [Google Scholar]
- Miller, A.-F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Poole, L.B.; Hall, A.; Nelson, K.J. Overview of peroxiredoxins in oxidant defense and redox regulation. Curr. Protoc. Toxicol. 2011, 7, 7–9. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. Thioredoxin system in cell death progression. Antioxid. Redox Signal. 2012, 17, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Anderson, M.E. Glutathione: An overview of biosynthesis and modulation. Chem. Biol. Interact. 1998, 111–112, 1–14. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Glutathione reductase. Methods Enzymol. 1985, 113, 484–490. [Google Scholar] [CrossRef]
- Leopold, J.A.; Cap, A.; Scribner, A.W.; Stanton, R.C.; Loscalzo, J. Glucose-6-phosphate dehydrogenase deficiency promotes endothelial oxidant stress and decreases endothelial nitric oxide bioavailability. FASEB J. 2001, 15, 1771–1773. [Google Scholar] [CrossRef]
- Manganelli, G.; Masullo, U.; Passarelli, S.; Filosa, S. Glucose-6-phosphate dehydrogenase deficiency: Disadvantages and possible benefits. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Colombo, G.; Garavaglia, M.L.; Astori, E.; Portinaro, N.M.; Reggiani, F.; Badalamenti, S.; Aloisi, A.M.; Santucci, A.; Rossi, R.; et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic. Biol. Med. 2017, 112, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Morel, Y.; Barouki, R. Repression of gene expression by oxidative stress. Biochem. J. 1999, 342 Pt 3, 481–496. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Salinas, A.E.; Wong, M.G. Glutathione S-transferases—A review. Curr. Med. Chem. 1999, 6, 279–309. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Kaur, A. Glutathione Degradation. Antioxid. Redox Signal. 2017, 27, 1200–1216. [Google Scholar] [CrossRef]
- Garibotto, G.; Sofia, A.; Saffioti, S.; Russo, R.; Deferrari, G.; Rossi, D.; Verzola, D.; Gandolfo, M.T.; Sala, M.R. Interorgan exchange of aminothiols in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E757–E763. [Google Scholar] [CrossRef]
- Jansen, T.; Daiber, A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Furfaro, A.L.; Mann, G.E. Heme oxygenase dependent bilirubin generation in vascular cells: A role in preventing endothelial dysfunction in local tissue microenvironment? Front. Physiol. 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.K.; Farrar, J.E.; Gaucher, E.A.; Miner, J.N. Coevolution of URAT1 and uricase during primate evolution: Implications for serum urate homeostasis and gout. Mol. Biol. Evol. 2016, 33, 2193–2200. [Google Scholar] [CrossRef]
- Sies, H.; Stahl, W.; Sundquist, A.R. Antioxidant functions of vitamins. Vitamins E and C, beta-carotene, and other carotenoids. Ann. N. Y. Acad. Sci. 1992, 669, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Duarte, T.L.; Lunec, J. Review: When is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic. Res. 2005, 39, 671–686. [Google Scholar] [CrossRef]
- Frei, B.; England, L.; Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 6377–6381. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R. Vitamin E research: Past, now and future. Free Radic. Biol. Med. 2021, 177, 381–390. [Google Scholar] [CrossRef]
- Azzi, A. Reflections on a century of vitamin E research: Looking at the past with an eye on the future. Free Radic. Biol. Med. 2021, 175, 155–160. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Aggarwal, B.B. Resveratrol: A multitargeted agent for age-associated chronic diseases. Cell Cycle 2008, 7, 1020–1035. [Google Scholar] [CrossRef]
- Monostori, P.; Wittmann, G.; Karg, E.; Túri, S. Determination of glutathione and glutathione disulfide in biological samples: An in-depth review. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 3331–3346. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative Stress, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1985; Available online: https://www.elsevier.com/books/oxidative-stress/sies/978-0-12-642760-8 (accessed on 28 June 2022).
- Niki, E. Do free radicals play causal role in atherosclerosis? Low density lipoprotein oxidation and vitamin E revisited. J. Clin. Biochem. Nutr. 2011, 48, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free. Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Reactive oxygen-derived free radicals are key to the endothelial dysfunction of diabetes. J. Diabetes 2009, 1, 151–162. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R.; Shenk, J.C.; Nunomura, A.; Smith, M.A.; Zhu, X.; Perry, G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol. Disord. Drug Targets 2008, 7, 3–10. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox signaling by reactive electrophiles and oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Biological production, detection, and fate of hydrogen peroxide. Antioxid. Redox Signal. 2018, 29, 541–551. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Lorenzen, I.; Eble, J.A.; Hanschmann, E.-M. Thiol switches in membrane proteins—Extracellular redox regulation in cell biology. Biol. Chem. 2021, 402, 253–269. [Google Scholar] [CrossRef]
- Kaya, A.; Lee, B.C.; Gladyshev, V.N. Regulation of protein function by reversible methionine oxidation and the role of selenoprotein MsrB1. Antioxid. Redox Signal. 2015, 23, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Ivanova-Radkevich, V.I.; Chernov, N.N. Role of MicroRNAs in the regulation of redox-dependent processes. Biochemistry 2019, 84, 1233–1246. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox homeostasis and cellular antioxidant systems: Crucial players in cancer growth and therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Dianzani, M.U. 4-Hydroxynonenal and cell signalling. Free Radic. Res. 1998, 28, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, P.A.; Margison, G.P.; Tisdale, M.J. Enzymatic methylation of cytosine in DNA is prevented by adjacent O6-methylguanine residues. J. Biol. Chem. 1991, 266, 7985–7987. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, F.P.; Sparaneo, A.; Trombetta, D.; Muscarella, L.A. Epigenetic versus genetic deregulation of the KEAP1/NRF2 axis in solid tumors: Focus on methylation and noncoding RNAs. Oxid Med. Cell. Longev. 2018, 2018, 2492063. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Sparaneo, A.; Muscarella, L.A. NRF2 regulation by noncoding RNAs in cancers: The present knowledge and the way forward. Cancers 2020, 12, 3621. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Davudian, S.; Mansoori, B.; Shajari, N.; Mohammadi, A.; Baradaran, B. BACH1, the master regulator gene: A novel candidate target for cancer therapy. Gene 2016, 588, 30–37. [Google Scholar] [CrossRef]
- Davies, K.J.A.; Forman, H.J. Does Bach1 & c-Myc dependent redox dysregulation of Nrf2 & adaptive homeostasis decrease cancer risk in ageing? Free Radic. Biol. Med. 2019, 134, 708–714. [Google Scholar] [CrossRef]
- Levy, S.; Forman, H.J. C-Myc is a Nrf2-interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB Life 2010, 62, 237–246. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Kozumbo, W.J. The hormetic dose-response mechanism: Nrf2 activation. Pharmacol. Res. 2021, 167, 105526. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Nguyen, N.; Syal, A.; Dreher, L.A.; Dodson, M.; Zhang, D.D.; Madhavan, L. NRF2 loss accentuates parkinsonian pathology and behavioral dysfunction in human α-synuclein overexpressing mice. Aging Dis. 2021, 12, 964–982. [Google Scholar] [CrossRef] [PubMed]
- Zazueta, C.; Jimenez-Uribe, A.P.; Pedraza-Chaverri, J.; Buelna-Chontal, M. Genetic variations on redox control in cardiometabolic diseases: The role of Nrf2. Antioxidants 2022, 11, 507. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Shen, X.-D.; Zhang, Y.; Ji, H.; Gao, F.; Yue, S.; Kamo, N.; Zhai, Y.; Yamamoto, M.; Busuttil, R.W.; et al. KEAP1-NRF2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. J. Hepatol. 2013, 59, 1200–1207. [Google Scholar] [CrossRef]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef]
- Matsumaru, D.; Motohashi, H. The KEAP1-NRF2 System in healthy aging and longevity. Antioxidants 2021, 10, 1929. [Google Scholar] [CrossRef]
- Ruiz, G.P.; Camara, H.; Fazolini, N.P.B.; Mori, M.A. Extracellular miRNAs in redox signaling: Health, disease and potential therapies. Free Radic. Biol. Med. 2021, 173, 170–187. [Google Scholar] [CrossRef]
- Fierro-Fernández, M.; Miguel, V.; Lamas, S. Role of redoximiRs in fibrogenesis. Redox Biol. 2016, 7, 58–67. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Potential roles of microRNAs in redox state: An update. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Leisegang, M.S.; Schröder, K.; Brandes, R.P. Redox regulation and noncoding RNAs. Antioxid. Redox Signal. 2018, 29, 793–812. [Google Scholar] [CrossRef] [PubMed]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and oxidative stress in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef]
- Shen, W.; Lu, Y.; Hu, J.; Le, H.; Yu, W.; Xu, W.; Yu, W.; Zheng, J. Mechanism of miR-320 in regulating biological characteristics of ischemic cerebral neuron by mediating Nox2/ROS pathway. J. Mol. Neurosci. 2020, 70, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Zuo, M.-L.; Wang, A.-P.; Song, G.-L.; Yang, Z.-B. miR-652 protects rats from cerebral ischemia/reperfusion oxidative stress injury by directly targeting NOX2. Biomed. Pharmacother. 2020, 124, 109860. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Maidana, D.E.; Dib, B.; Miller, J.B.; Bouzika, P.; Miller, J.W.; Vavvas, D.G.; Lin, H. miR-17-3p Exacerbates oxidative damage in human retinal pigment epithelial cells. PLoS ONE 2016, 11, e0160887. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Cuvelliez, M.; Fiedler, J.; Charrier, H.; Mulder, P.; Hebbar, E.; Pfanne, A.; Beseme, O.; Chwastyniak, M.; Amouyel, P.; et al. MicroRNAs regulating superoxide dismutase 2 are new circulating biomarkers of heart failure. Sci. Rep. 2017, 7, 14747. [Google Scholar] [CrossRef]
- Song, Y.-H.; Wang, J.; Nie, G.; Chen, Y.-J.; Li, X.; Jiang, X.; Cao, W.-H. MicroRNA-509-5p functions as an anti-oncogene in breast cancer via targeting SOD2. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3617–3625. [Google Scholar]
- Zhang, X.; Ng, W.-L.; Wang, P.; Tian, L.; Werner, E.; Wang, H.; Doetsch, P.; Wang, Y. MicroRNA-21 modulates the levels of reactive oxygen species by targeting SOD3 and TNFα. Cancer Res. 2012, 72, 4707–4713. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Fierro-Fernández, M.; Sánchez-Gómez, F.; Rodríguez-Pascual, F.; Alique, M.; Ruiz-Ortega, M.; Beraza, N.; Martínez-Chantar, M.L.; Fernández-Hernando, C.; Lamas, S. Targeting of gamma-glutamyl-cysteine ligase by miR-433 Reduces glutathione biosynthesis and promotes TGF-β-dependent fibrogenesis. Antioxid. Redox Signal. 2015, 23, 1092–1105. [Google Scholar] [CrossRef]
- Włodarski, A.; Strycharz, J.; Wróblewski, A.; Kasznicki, J.; Drzewoski, J.; Śliwińska, A. The role of microRNAs in metabolic syndrome-related oxidative stress. Int. J. Mol. Sci. 2020, 21, 6902. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Mena, S.; Ortega, A.; Estrela, J.M. Oxidative stress in environmental-induced carcinogenesis. Mutat. Res. 2009, 674, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative stress-mediated atherosclerosis: Mechanisms and therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative stress in human pathology and aging: Molecular mechanisms and perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef]
- Srivastava, S. The mitochondrial basis of aging and age-related disorders. Genes 2017, 8, E398. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. The interplay between respiratory supercomplexes and ROS in aging. Antioxid. Redox Signal. 2015, 23, 208–238. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Viña, J.; Borras, C.; Abdelaziz, K.M.; Garcia-Valles, R.; Gomez-Cabrera, M.C. The free radical theory of aging revisited: The cell signaling disruption theory of aging. Antioxid. Redox Signal. 2013, 19, 779–787. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Zhang, Y.; Ikeno, Y.; Qi, W.; Chaudhuri, A.; Li, Y.; Bokov, A.; Thorpe, S.R.; Baynes, J.W.; Epstein, C.; Richardson, A.; et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Dyer, D.G.; Blackledge, J.A.; Katz, B.M.; Hull, C.J.; Adkisson, H.D.; Thorpe, S.R.; Lyons, T.J.; Baynes, J.W. The Maillard reaction in vivo. Z. Ernahrungswiss. 1991, 30, 29–45. [Google Scholar] [CrossRef]
- Meli, M.; Frey, J.; Perier, C. Native protein glycoxidation and aging. J. Nutr. Health Aging 2003, 7, 263–266. [Google Scholar]
- Odetti, P.; Pronzato, M.A.; Noberasco, G.; Cosso, L.; Traverso, N.; Cottalasso, D.; Marinari, U.M. Relationships between glycation and oxidation related fluorescences in rat collagen during aging. An in vivo and in vitro study. Lab. Investig. 1994, 70, 61–67. [Google Scholar]
- Traverso, N.; Menini, S.; Cottalasso, D.; Odetti, P.; Marinari, U.M.; Pronzato, M.A. Mutual interaction between glycation and oxidation during non-enzymatic protein modification. Biochim. Biophys. Acta 1997, 1336, 409–418. [Google Scholar] [CrossRef]
- Peppa, M.; Uribarri, J.; Vlassara, H. Advanced glycoxidation. A new risk factor for cardiovascular disease? Cardiovasc. Toxicol. 2002, 2, 275–287. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Sanguineti, R.; Storace, D.; Monacelli, F.; Puzzo, A.; Pronzato, M.A.; Odetti, P.; Traverso, N. Metalloproteinases and advanced glycation end products: Coupled navigation in atherosclerotic plaque pathophysiology? Exp. Clin. Endocrinol. Diabetes 2012, 120, 586–590. [Google Scholar] [CrossRef]
- Reddy, V.P.; Obrenovich, M.E.; Atwood, C.S.; Perry, G.; Smith, M.A. Involvement of Maillard reactions in Alzheimer disease. Neurotox. Res. 2002, 4, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Phagocyte-derived reactive species: Salvation or suicide? Trends Biochem. Sci. 2006, 31, 509–515. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tse, H.M. The role of NADPH oxidases in infectious and inflammatory diseases. Redox Biol. 2021, 48, 102159. [Google Scholar] [CrossRef] [PubMed]
- Pollock, J.D.; Williams, D.A.; Gifford, M.A.C.; Li, L.L.; Du, X.; Fisherman, J.; Orkin, S.H.; Doerschuk, C.M.; Dinauer, M.C. Mouse model of X–linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat. Genet. 1995, 9, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.E.; Heimall, J.R. A review of chronic granulomatous disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z. NADPH Oxidases are essential for macrophage differentiation. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef]
- Marengo, B.; Bellora, F.; Ricciarelli, R.; De Ciucis, C.; Furfaro, A.; Leardi, R.; Colla, R.; Pacini, D.; Traverso, N.; Moretta, A.; et al. Oxysterol mixture and, in particular, 27-hydroxycholesterol drive M2 polarization of human macrophages. Biofactors 2016, 42, 80–92. [Google Scholar] [CrossRef]
- Barisione, C.; Garibaldi, S.; Furfaro, A.L.; Nitti, M.; Palmieri, D.; Passalacqua, M.; Garuti, A.; Verzola, D.; Parodi, A.; Ameri, P.; et al. Moderate increase of indoxyl sulfate promotes monocyte transition into profibrotic macrophages. PLoS ONE 2016, 11, e0149276. [Google Scholar] [CrossRef]
- Latz, E. NOX-free inflammasome activation. Blood 2010, 116, 1393–1394. [Google Scholar] [CrossRef]
- Trevelin, S.C.; Shah, A.M.; Lombardi, G. Beyond bacterial killing: NADPH oxidase 2 is an immunomodulator. Immunol. Lett. 2020, 221, 39–48. [Google Scholar] [CrossRef]
- Xu, T.; Sun, L.; Shen, X.; Chen, Y.; Yin, Y.; Zhang, J.; Huang, D.; Li, W.; Li, W. NADPH oxidase 2-mediated NLRP1 inflammasome activation involves in neuronal senescence in hippocampal neurons in vitro. Int. Immunopharmacol. 2019, 69, 60–70. [Google Scholar] [CrossRef]
- Pan, L.-L.; Ren, Z.; Liu, Y.; Zhao, Y.; Li, H.; Pan, X.; Fang, X.; Liang, W.; Wang, Y.; Yang, J.; et al. A novel danshensu derivative ameliorates experimental colitis by modulating NADPH oxidase 4-dependent NLRP3 inflammasome activation. J. Cell. Mol. Med. 2020, 24, 12955–12969. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Nakahira, K.; Chung, K.-P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.-H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.-T.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 307–319. [Google Scholar] [CrossRef]
- Chong, Y.K.; Tartey, S.; Yoshikawa, Y.; Imami, K.; Li, S.; Yoshinaga, M.; Hirabayashi, A.; Liu, G.; Vandenbon, A.; Hia, F.; et al. Cyclin J–CDK complexes limit innate immune responses by reducing proinflammatory changes in macrophage metabolism. Sci. Signal. 2022, 15, eabm5011. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, P.; Hussain, M.A.; Mazumder, S. mtROS Induced via TLR-2-SOCE Signaling Plays Proapoptotic and Bactericidal Role in Mycobacterium fortuitum-Infected Head Kidney Macrophages of Clarias gariepinus. Front. Immunol. 2021, 12, 748758. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, C.; Cao, X.; Jia, X.; Chen, X.; Wang, Z.; Xu, W.; Dai, F.; Zhang, S. Mitochondria-targeted supramolecular coordination container encapsulated with exogenous itaconate for synergistic therapy of joint inflammation. Theranostics 2022, 12, 3251–3272. [Google Scholar] [CrossRef]
- Wu, K.K.L.; Long, K.; Lin, H.; Siu, P.M.F.; Hoo, R.L.C.; Ye, D.; Xu, A.; Cheng, K.K.Y. The APPL1-Rab5 axis restricts NLRP3 inflammasome activation through early endosomal-dependent mitophagy in macrophages. Nat. Commun. 2021, 12, 6637. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, R.; Munari, F.; Angioni, R.; Venegas, F.; Agnellini, A.; Castro-Gil, M.P.; Castegna, A.; Luisetto, R.; Viola, A.; Canton, M. Targeting monoamine oxidase to dampen NLRP3 inflammasome activation in inflammation. Cell Mol. Immunol. 2021, 18, 1311–1313. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.-J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Schönrich, G.; Raftery, M.J.; Samstag, Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.J.; Choi, H.; Ko, E.H.; Kim, J. Reactive Oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 2009, 284, 10601–10609. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y.; Hinata, T.; Kang, S.W.; Watanabe, Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci. 2011, 89, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef]
- Raut, S.K.; Khullar, M. Oxidative stress in metabolic diseases: Current scenario and therapeutic relevance. Mol. Cell. Biochem. 2022. Advanced online publication. [Google Scholar] [CrossRef]
- Choromańska, B.; Myśliwiec, P.; Łuba, M.; Wojskowicz, P.; Myśliwiec, H.; Choromańska, K.; Dadan, J.; Żendzian-Piotrowska, M.; Zalewska, A.; Maciejczyk, M. Bariatric surgery normalizes protein glycoxidation and nitrosative stress in morbidly obese patients. Antioxidants 2020, 9, 1087. [Google Scholar] [CrossRef]
- Chetboun, M.; Abitbol, G.; Rozenberg, K.; Rozenfeld, H.; Deutsch, A.; Sampson, S.R.; Rosenzweig, T. Maintenance of redox state and pancreatic beta-cell function: Role of leptin and adiponectin. J. Cell. Biochem. 2012, 113, 1966–1976. [Google Scholar] [CrossRef]
- Rehman, K.; Akash, M.S.H. Mechanisms of inflammatory responses and development of insulin resistance: How are they interlinked? J. Biomed. Sci. 2016, 23, 87. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Bathish, B.; Robertson, H.; Dillon, J.F.; Dinkova-Kostova, A.T.; Hayes, J.D. Nonalcoholic steatohepatitis and mechanisms by which it is ameliorated by activation of the CNC-bZIP transcription factor Nrf2. Free Radic. Biol. Med. 2022, 188, 221–261. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial dysfunction in cardiovascular diseases: Potential targets for treatment. Front. Cell Dev. Biol. 2022, 10, 841523. [Google Scholar] [CrossRef]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Kitching, M.; Redmond, E.M.; Megson, I.L.; Cahill, P.A. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front. Cardiovasc. Med. 2019, 6, 89. [Google Scholar] [CrossRef]
- Gill, P.S.; Wilcox, C.S. NADPH oxidases in the kidney. Antioxid. Redox Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Rummel, N.G.; Butterfield, D.A. Altered metabolism in Alzheimer disease brain: Role of oxidative stress. Antioxid. Redox Signal. 2022, 36, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Fão, L.; Rego, A.C. Mitochondrial and redox-based therapeutic strategies in Huntington’s disease. Antioxid. Redox Signal. 2021, 34, 650–673. [Google Scholar] [CrossRef]
- Fang, J.; Sheng, R.; Qin, Z.-H. NADPH Oxidases in the central nervous system: Regional and cellular localization and the possible link to brain diseases. Antioxid. Redox Signal. 2021, 35, 951–973. [Google Scholar] [CrossRef]
- Nitti, M.; Furfaro, A.L.; Cevasco, C.; Traverso, N.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. PKC delta and NADPH oxidase in retinoic acid-induced neuroblastoma cell differentiation. Cell Signal. 2010, 22, 828–835. [Google Scholar] [CrossRef]
- Piras, S.; Furfaro, A.L.; Piccini, A.; Passalacqua, M.; Borghi, R.; Carminati, E.; Parodi, A.; Colombo, L.; Salmona, M.; Pronzato, M.A.; et al. Monomeric Abeta1-42 and RAGE: Key players in neuronal differentiation. Neurobiol. Aging 2014, 35, 1301–1308. [Google Scholar] [CrossRef]
- Nitti, M.; Furfaro, A.L.; Traverso, N.; Odetti, P.; Storace, D.; Cottalasso, D.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. PKC delta and NADPH oxidase in AGE-induced neuronal death. Neurosci. Lett. 2007, 416, 261–265. [Google Scholar] [CrossRef]
- Piras, S.; Furfaro, A.L.; Domenicotti, C.; Traverso, N.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. RAGE Expression and ROS Generation in Neurons: Differentiation versus Damage. Oxid. Med. Cell. Longev. 2016, 2016, 9348651. [Google Scholar] [CrossRef]
- Nitti, M.; d’Abramo, C.; Traverso, N.; Verzola, D.; Garibotto, G.; Poggi, A.; Odetti, P.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A.; et al. Central role of PKCdelta in glycoxidation-dependent apoptosis of human neurons. Free Radic. Biol. Med. 2005, 38, 846–856. [Google Scholar] [CrossRef]
- Alonso-Piñeiro, J.A.; Gonzalez-Rovira, A.; Sánchez-Gomar, I.; Moreno, J.A.; Durán-Ruiz, M.C. Nrf2 and heme oxygenase-1 involvement in atherosclerosis related oxidative stress. Antioxidants 2021, 10, 1463. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Nitti, M.; Marengo, B.; Domenicotti, C.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N. Impaired synthesis contributes to diabetes-induced decrease in liver glutathione. Int. J. Mol. Med. 2012, 29, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Tramutola, A.; Pagnotta, S.; Barone, E.; Butterfield, D.A. The BACH1/Nrf2 axis in brain in down syndrome and transition to Alzheimer disease-like neuropathology and dementia. Antioxidants 2020, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: Insights for transition to Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef]
- Jia, M.; Li, Q.; Guo, J.; Shi, W.; Zhu, L.; Huang, Y.; Li, Y.; Wang, L.; Ma, S.; Zhuang, T.; et al. Deletion of BACH1 attenuates atherosclerosis by reducing endothelial inflammation. Circ. Res. 2022, 130, 1038–1055. [Google Scholar] [CrossRef]
- Piras, S.; Furfaro, A.L.; Brondolo, L.; Passalacqua, M.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. Differentiation impairs Bach1 dependent HO-1 activation and increases sensitivity to oxidative stress in SH-SY5Y neuroblastoma cells. Sci. Rep. 2017, 7, 7568. [Google Scholar] [CrossRef]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding cellular redox homeostasis: A challenge for precision medicine. Int. J. Mol. Sci. 2021, 23, 106. [Google Scholar] [CrossRef]
- Korkmaz, K.S.; Butuner, B.D.; Roggenbuck, D. Detection of 8-OHdG as a diagnostic biomarker. J. Lab. Precis. Med. 2018, 3, 1–6. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Bouyahya, A.; El Menyiy, N.; Oumeslakht, L.; El Allam, A.; Balahbib, A.; Rauf, A.; Muhammad, N.; Kuznetsova, E.; Derkho, M.; Thiruvengadam, M.; et al. Preclinical and clinical antioxidant effects of natural compounds against oxidative stress-induced epigenetic instability in tumor cells. Antioxidants 2021, 10, 1553. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Jiang, B.-H. Interplay between reactive oxygen species and MicroRNAs in cancer. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; Pulliero, A.; Corrias, M.V.; Leardi, R.; Farinini, E.; Fronza, G.; Menichini, P.; Monti, P.; Monteleone, L.; Valenti, G.E.; et al. Potential role of miRNAs in the acquisition of chemoresistance in neuroblastoma. J. Pers. Med. 2021, 11, 107. [Google Scholar] [CrossRef]
- Marengo, B.; Monti, P.; Miele, M.; Menichini, P.; Ottaggio, L.; Foggetti, G.; Pulliero, A.; Izzotti, A.; Speciale, A.; Garbarino, O.; et al. Etoposide-resistance in a neuroblastoma model cell line is associated with 13q14.3 mono-allelic deletion and miRNA-15a/16-1 down-regulation. Sci. Rep. 2018, 8, 13762. [Google Scholar] [CrossRef]
- Zhang, H.J.; Zhao, W.; Venkataraman, S.; Robbins, M.E.C.; Buettner, G.R.; Kregel, K.C.; Oberley, L.W. Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J. Biol. Chem. 2002, 277, 20919–20926. [Google Scholar] [CrossRef]
- Marengo, B.; De Ciucis, C.G.; Ricciarelli, R.; Furfaro, A.L.; Colla, R.; Canepa, E.; Traverso, N.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. p38MAPK inhibition: A new combined approach to reduce neuroblastoma resistance under etoposide treatment. Cell Death Dis. 2013, 4, e589. [Google Scholar] [CrossRef]
- Meng, J.; Lv, Z.; Zhang, Y.; Wang, Y.; Qiao, X.; Sun, C.; Chen, Y.; Guo, M.; Han, W.; Ye, A.; et al. Precision redox: The key for antioxidant pharmacology. Antioxid. Redox Signal. 2021, 34, 1069–1082. [Google Scholar] [CrossRef]
- Nitti, M.; Ivaldo, C.; Traverso, N.; Furfaro, A.L. Clinical significance of heme oxygenase 1 in tumor progression. Antioxidants 2021, 10, 789. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 induction in cancer progression: A matter of cell adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Piras, S.; Domenicotti, C.; Fenoglio, D.; De Luigi, A.; Salmona, M.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; et al. Role of Nrf2, HO-1 and GSH in neuroblastoma cell resistance to bortezomib. PLoS ONE 2016, 11, e0152465. [Google Scholar]
- Colla, R.; Izzotti, A.; De Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Ricciarelli, R.; Furfaro, A.L.; Pulliero, A.; Passalacqua, M.; et al. Glutathione-mediated antioxidant response and aerobic metabolism: Two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 2016, 7, 70715–70737. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, thioredoxin, and glutathione system in tumorigenesis and anticancer therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Emanuele, S.; D’Anneo, A.; Calvaruso, G.; Cernigliaro, C.; Giuliano, M.; Lauricella, M. The double-edged sword profile of redox signaling: Oxidative events as molecular switches in the balance between cell physiology and cancer. Chem. Res. Toxicol. 2018, 31, 201–210. [Google Scholar] [CrossRef]
- Domenicotti, C.; Marengo, B. Paradox role of oxidative stress in cancer: State of the art. Antioxidants 2022, 11, 1027. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget 2016, 7, 33297–33305. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic heterogeneity in human lung tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, Y.; Zhang, S.; Liu, Y.; Guo, H.; Ahmed, M.; Bell, T.; Zhang, H.; Han, G.; Lorence, E.; et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019, 11, eaau1167. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, L.; Speciale, A.; Valenti, G.E.; Traverso, N.; Ravera, S.; Garbarino, O.; Leardi, R.; Farinini, E.; Roveri, A.; Ursini, F.; et al. PKCα inhibition as a strategy to sensitize neuroblastoma stem cells to etoposide by stimulating ferroptosis. Antioxidants 2021, 10, 691. [Google Scholar] [CrossRef]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef]
- Hofbauer, S.L.; Stangl, K.I.; de Martino, M.; Lucca, I.; Haitel, A.; Shariat, S.F.; Klatte, T. Pretherapeutic gamma-glutamyltransferase is an independent prognostic factor for patients with renal cell carcinoma. Br. J. Cancer 2014, 111, 1526–1531. [Google Scholar] [CrossRef]
- Corti, A.; Belcastro, E.; Dominici, S.; Maellaro, E.; Pompella, A. The dark side of gamma-glutamyltransferase (GGT): Pathogenic effects of an “antioxidant” enzyme. Free Radic. Biol. Med. 2020, 160, 807–819. [Google Scholar] [CrossRef]
- Hiyama, N.; Ando, T.; Maemura, K.; Sakatani, T.; Amano, Y.; Watanabe, K.; Kage, H.; Yatomi, Y.; Nagase, T.; Nakajima, J.; et al. Glutamate-cysteine ligase catalytic subunit is associated with cisplatin resistance in lung adenocarcinoma. Jpn. J. Clin. Oncol. 2018, 48, 303–307. [Google Scholar] [CrossRef]
- Dequanter, D.; VAN DE Velde, M.; Bar, I.; Nuyens, V.; Rousseau, A.; Nagy, N.; Vanhamme, L.; Vanhaeverbeek, M.; Brohée, D.; Delrée, P.; et al. Nuclear localization of glutamate-cysteine ligase is associated with proliferation in head and neck squamous cell carcinoma. Oncol. Lett. 2016, 11, 3660–3668. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Yuan, J.; Zhang, Y.; Jin, X. Altered glutamate cysteine ligase expression and activity in renal cell carcinoma. Biomed. Rep. 2014, 2, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Syu, J.-P.; Chi, J.-T.; Kung, H.-N. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Chang, C.; Worley, B.L.; Phaëton, R.; Hempel, N. Extracellular glutathione peroxidase GPx3 and its role in cancer. Cancers 2020, 12, 2197. [Google Scholar] [CrossRef]
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res. 2013, 73, 1245–1255. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Jyotsana, N.; Ta, K.T.; DelGiorno, K.E. The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front. Oncol. 2022, 12, 858462. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Batinić-Haberle, I.; Rebouças, J.S.; Spasojević, I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef]
- Bonetta, R. Potential therapeutic applications of MnSODs and SOD-mimetics. Chemistry 2018, 24, 5032–5041. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Sonis, S.T.; Lee, C.M.; Adkins, D.; Allen, B.G.; Sun, W.; Agarwala, S.S.; Venigalla, M.L.; Chen, Y.; Zhen, W.; et al. Phase 1b/2a trial of the superoxide dismutase mimetic GC4419 to reduce chemoradiotherapy-induced oral mucositis in patients with oral cavity or oropharyngeal carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Ebselen, a selenoorganic compound as glutathione peroxidase mimic. Free Radic. Biol. Med. 1993, 14, 313–323. [Google Scholar] [CrossRef]
- Kil, J.; Pierce, C.; Tran, H.; Gu, R.; Lynch, E.D. Ebselen treatment reduces noise induced hearing loss via the mimicry and induction of glutathione peroxidase. Hear. Res. 2007, 226, 44–51. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, M.E.; Meijles, D.N.; Pagano, P.J. Nox inhibitors & therapies: Rational design of peptidic and small molecule inhibitors. Curr. Pharm. Des. 2015, 21, 6023–6035. [Google Scholar] [CrossRef]
- Dao, V.T.-V.; Elbatreek, M.H.; Altenhöfer, S.; Casas, A.I.; Pachado, M.P.; Neullens, C.T.; Knaus, U.G.; Schmidt, H.H.H.W. Isoform-selective NADPH oxidase inhibitor panel for pharmacological target validation. Free. Radic. Biol. Med. 2020, 148, 60–69. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Poljšak, B.; Milisav, I. Medical and dietary uses of n-acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Powrie, F.; Puri, R.N.; Meister, A. Glutathione monoethyl ester: Preparation, uptake by tissues, and conversion to glutathione. Arch. Biochem. Biophys. 1985, 239, 538–548. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Manzino, L.; Sonsalla, P.K.; Bernard, L.P. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: Relevance to Parkinson’s disease. Exp. Neurol. 2007, 203, 512–520. [Google Scholar] [CrossRef]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting glutathione metabolism: Partner in crime in anticancer therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef]
- Villablanca, J.G.; Volchenboum, S.L.; Cho, H.; Kang, M.H.; Cohn, S.L.; Anderson, C.P.; Marachelian, A.; Groshen, S.; Tsao-Wei, D.; Matthay, K.K.; et al. A phase I new approaches to neuroblastoma therapy study of buthionine sulfoximine and melphalan with autologous stem cells for recurrent/refractory high-risk neuroblastoma. Pediatr. Blood Cancer 2016, 63, 1349–1356. [Google Scholar] [CrossRef]
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar] [CrossRef]
- Domenicotti, C.; Marengo, B.; Verzola, D.; Garibotto, G.; Traverso, N.; Patriarca, S.; Maloberti, G.; Cottalasso, D.; Poli, G.; Passalacqua, M.; et al. Role of PKC-delta activity in glutathione-depleted neuroblastoma cells. Free Radic. Biol. Med. 2003, 35, 504–516. [Google Scholar] [CrossRef]
- Marengo, B.; De Ciucis, C.; Verzola, D.; Pistoia, V.; Raffaghello, L.; Patriarca, S.; Balbis, E.; Traverso, N.; Cottalasso, D.; Pronzato, M.A.; et al. Mechanisms of BSO (L-buthionine-S,R-sulfoximine)-induced cytotoxic effects in neuroblastoma. Free Radic. Biol. Med. 2008, 44, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; De Ciucis, C.; Ricciarelli, R.; Passalacqua, M.; Nitti, M.; Zingg, J.-M.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. PKC delta sensitizes neuroblastoma cells to L-buthionine-sulfoximine and etoposide inducing reactive oxygen species overproduction and DNA damage. PLoS ONE 2011, 6, e14661. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef]
- Hamilton, D.; Wu, J.H.; Batist, G. Structure-based identification of novel human gamma-glutamylcysteine synthetase inhibitors. Mol. Pharmacol. 2007, 71, 1140–1147. [Google Scholar] [CrossRef]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Lee, D.-H.; Jeong, S.Y.; Park, S.H.; Oh, S.C.; Park, Y.S.; Yu, J.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J. Cell Biochem. 2019, 120, 928–939. [Google Scholar] [CrossRef]
- Pan, X.; Lin, Z.; Jiang, D.; Yu, Y.; Yang, D.; Zhou, H.; Zhan, D.; Liu, S.; Peng, G.; Chen, Z.; et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol. Lett. 2019, 17, 3001–3008. [Google Scholar] [CrossRef]
- Terzyan, S.S.; Burgett, A.W.G.; Heroux, A.; Smith, C.A.; Mooers, B.H.M.; Hanigan, M.H. Human γ-glutamyl transpeptidase 1: Structures of the free enzyme, inhibitor-bound tetrahedral transition states, and glutamate-bound enzyme reveal novel movement within the active site during catalysis. J. Biol. Chem. 2015, 290, 17576–17586. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-Me, Two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef]
- Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current landscape of NRF2 biomarkers in clinical trials. Antioxidants 2020, 9, 716. [Google Scholar] [CrossRef]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef]
- Thiruvengadam, M.; Venkidasamy, B.; Subramanian, U.; Samynathan, R.; Ali Shariati, M.; Rebezov, M.; Girish, S.; Thangavel, S.; Dhanapal, A.R.; Fedoseeva, N.; et al. Bioactive compounds in oxidative stress-mediated diseases: Targeting the NRF2/ARE signaling pathway and epigenetic regulation. Antioxidants 2021, 10, 1859. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, N.; He, H.; Tang, X. Pharmaceutical strategies of improving oral systemic bioavailability of curcumin for clinical application. J. Control Release 2019, 316, 359–380. [Google Scholar] [CrossRef] [PubMed]
- He, H.-J.; Wang, G.-Y.; Gao, Y.; Ling, W.-H.; Yu, Z.-W.; Jin, T.-R. Curcumin attenuates Nrf2 signaling defect, oxidative stress in muscle and glucose intolerance in high fat diet-fed mice. World J. Diabetes 2012, 3, 94–104. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, F.; Liu, M.; Zhou, X.; Wang, M.; Cao, K.; Jin, S.; Shan, A.; Feng, X. Curcumin mitigates aflatoxin B1-induced liver injury via regulating the NLRP3 inflammasome and Nrf2 signaling pathway. Food Chem. Toxicol. 2022, 161, 112823. [Google Scholar] [CrossRef]
- Lin, X.; Bai, D.; Wei, Z.; Zhang, Y.; Huang, Y.; Deng, H.; Huang, X. Curcumin attenuates oxidative stress in RAW264.7 cells by increasing the activity of antioxidant enzymes and activating the Nrf2-Keap1 pathway. PLoS ONE 2019, 14, e0216711. [Google Scholar] [CrossRef]
- Liu, S.; Tian, L.; Chai, G.; Wen, B.; Wang, B. Targeting heme oxygenase-1 by quercetin ameliorates alcohol-induced acute liver injury via inhibiting NLRP3 inflammasome activation. Food Funct. 2018, 9, 4184–4193. [Google Scholar] [CrossRef]
- Ruhee, R.T.; Ma, S.; Suzuki, K. Protective Effects of sulforaphane on exercise-induced organ damage via inducing antioxidant defense responses. Antioxidants 2020, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Bai, Y.; Jiang, X.; Zhou, S.; Wang, Y.; Wintergerst, K.A.; Cui, T.; Ji, H.; Tan, Y.; Cai, L. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3ß/Fyn pathway. Redox Biol. 2018, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, H.; Chen, X. Protective effects of sulforaphane on diabetic retinopathy: Activation of the Nrf2 pathway and inhibition of NLRP3 inflammasome formation. Exp. Anim. 2019, 68, 221–231. [Google Scholar] [CrossRef]
- Tsai, P.-Y.; Ka, S.-M.; Chang, J.-M.; Chen, H.-C.; Shui, H.-A.; Li, C.-Y.; Hua, K.-F.; Chang, W.-L.; Huang, J.-J.; Yang, S.-S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, M.; Jing, X.; Shi, H.; Ren, M.; Lou, H. (-)-Epigallocatechin gallate protects against cerebral ischemia-induced oxidative stress via Nrf2/ARE signaling. Neurochem. Res. 2014, 39, 1292–1299. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.M.; Martins, L.A.M.; Souza, D.O.; Quincozes-Santos, A. Glioprotective effect of resveratrol: An emerging therapeutic role for oligodendroglial cells. Mol. Neurobiol. 2018, 55, 2967–2978. [Google Scholar] [CrossRef]
- Hui, Y.; Chengyong, T.; Cheng, L.; Haixia, H.; Yuanda, Z.; Weihua, Y. Resveratrol attenuates the cytotoxicity induced by amyloid-β1-42 in PC12 cells by upregulating heme oxygenase-1 via the PI3K/Akt/Nrf2 pathway. Neurochem. Res. 2018, 43, 297–305. [Google Scholar] [CrossRef]
- Wang, G.; Song, X.; Zhao, L.; Li, Z.; Liu, B. Resveratrol prevents diabetic cardiomyopathy by increasing Nrf2 expression and transcriptional activity. Biomed. Res. Int. 2018, 2018, 2150218. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef]
- Vartanian, S.; Ma, T.P.; Lee, J.; Haverty, P.M.; Kirkpatrick, D.S.; Yu, K.; Stokoe, D. Application of mass spectrometry profiling to establish brusatol as an inhibitor of global protein synthesis. Mol. Cell Proteomics 2016, 15, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Fouzder, C.; Mukhuty, A.; Mukherjee, S.; Malick, C.; Kundu, R. Trigonelline inhibits Nrf2 via EGFR signalling pathway and augments efficacy of Cisplatin and Etoposide in NSCLC cells. Toxicol. In Vitro 2021, 70, 105038. [Google Scholar] [CrossRef] [PubMed]
- Abazeed, M.E.; Adams, D.J.; Hurov, K.E.; Tamayo, P.; Creighton, C.J.; Sonkin, D.; Giacomelli, A.O.; Du, C.; Fries, D.F.; Wong, K.-K.; et al. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 2013, 73, 6289–6298. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Torrente, L.; DeNicola, G.M. Targeting NRF2 and its downstream processes: Opportunities and challenges. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 279–300. [Google Scholar] [CrossRef]
- Ames, B.N. Prolonging healthy aging: Longevity vitamins and proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 10836–10844. [Google Scholar] [CrossRef]
- Fang, Y.-Z.; Yang, S.; Wu, G. Free radicals, antioxidants, and nutrition. Nutrition 2002, 18, 872–879. [Google Scholar] [CrossRef]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.-C.; Trus, C.; Ciobica, A.; Timofte, D. The involvement of the oxidative stress status in cancer pathology: A double view on the role of the antioxidants. Oxidative Med. Cell. Longev. 2021, 2021, e9965916. [Google Scholar] [CrossRef]
- Ozben, T. Antioxidant supplementation on cancer risk and during cancer therapy: An update. Curr. Top. Med. Chem. 2015, 15, 170–178. [Google Scholar] [CrossRef]
- Harvie, M. Nutritional supplements and cancer: Potential benefits and proven harms. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e478–e486. [Google Scholar] [CrossRef]
- Abdali, D.; Samson, S.E.; Grover, A.K. How effective are antioxidant supplements in obesity and diabetes? Med. Princ Pract. 2015, 24, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Ramos, R.; Guadarrama-López, A.L.; Martínez-Carrillo, B.E.; Benítez-Arciniega, A.D. Vitamins and type 2 diabetes mellitus. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.-K.; Ju, W.; Cho, B.; Oh, S.-W.; Park, S.M.; Koo, B.-K.; Park, B.-J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346, f10. [Google Scholar] [CrossRef]
- Cheng, H.M.; Koutsidis, G.; Lodge, J.K.; Ashor, A.; Siervo, M.; Lara, J. Tomato and lycopene supplementation and cardiovascular risk factors: A systematic review and meta-analysis. Atherosclerosis 2017, 257, 100–108. [Google Scholar] [CrossRef]
- Mirahmadi, M.; Azimi-Hashemi, S.; Saburi, E.; Kamali, H.; Pishbin, M.; Hadizadeh, F. Potential inhibitory effect of lycopene on prostate cancer. Biomed. Pharmacother. 2020, 129, 110459. [Google Scholar] [CrossRef]
- Jenkins, D.J.A.; Kitts, D.; Giovannucci, E.L.; Sahye-Pudaruth, S.; Paquette, M.; Blanco Mejia, S.; Patel, D.; Kavanagh, M.; Tsirakis, T.; Kendall, C.W.C.; et al. Selenium, antioxidants, cardiovascular disease, and all-cause mortality: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2020, 112, 1642–1652. [Google Scholar] [CrossRef]
- Tosetti, F.; Noonan, D.M.; Albini, A. Metabolic regulation and redox activity as mechanisms for angioprevention by dietary phytochemicals. Int. J. Cancer 2009, 125, 1997–2003. [Google Scholar] [CrossRef]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, aging and Alzheimer’s disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef]
- Ciofu, O.; Smith, S.; Lykkesfeldt, J. Antioxidant supplementation for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2019, 1–129. [Google Scholar] [CrossRef]
- Liakopoulos, V.; Roumeliotis, S.; Bozikas, A.; Eleftheriadis, T.; Dounousi, E. Antioxidant supplementation in renal replacement therapy patients: Is there evidence? Oxid. Med. Cell. Longev. 2019, 2019, 9109473. [Google Scholar] [CrossRef]
- Saso, L.; Firuzi, O. Pharmacological applications of antioxidants: Lights and shadows. Curr. Drug Targets 2014, 15, 1177–1199. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; Izzo, V.; Corbi, G.; Russomanno, G.; Manzo, V.; De Lise, F.; Di Donato, A.; Filippelli, A. Antioxidant supplementation in the treatment of aging-associated diseases. Front. Pharmacol. 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Savencu, C.E.; Linţa, A.; Farcaş, G.; Bînă, A.M.; Creţu, O.M.; Maliţa, D.C.; Muntean, D.M.; Sturza, A. Impact of dietary restriction regimens on mitochondria, heart, and endothelial function: A brief overview. Front. Physiol. 2021, 12, 768383. [Google Scholar] [CrossRef]
- Longo, V.D.; Anderson, R.M. Nutrition, longevity and disease: From molecular mechanisms to interventions. Cell 2022, 185, 1455–1470. [Google Scholar] [CrossRef]
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, H.; Finkel, T. Unraveling the truth about antioxidants: ROS and disease: Finding the right balance. Nat. Med. 2014, 20, 711–713. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C.; Traverso, N. Hormesis and Oxidative Distress: Pathophysiology of Reactive Oxygen Species and the Open Question of Antioxidant Modulation and Supplementation. Antioxidants 2022, 11, 1613. https://doi.org/10.3390/antiox11081613
Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C, Traverso N. Hormesis and Oxidative Distress: Pathophysiology of Reactive Oxygen Species and the Open Question of Antioxidant Modulation and Supplementation. Antioxidants. 2022; 11(8):1613. https://doi.org/10.3390/antiox11081613
Chicago/Turabian StyleNitti, Mariapaola, Barbara Marengo, Anna Lisa Furfaro, Maria Adelaide Pronzato, Umberto Maria Marinari, Cinzia Domenicotti, and Nicola Traverso. 2022. "Hormesis and Oxidative Distress: Pathophysiology of Reactive Oxygen Species and the Open Question of Antioxidant Modulation and Supplementation" Antioxidants 11, no. 8: 1613. https://doi.org/10.3390/antiox11081613
APA StyleNitti, M., Marengo, B., Furfaro, A. L., Pronzato, M. A., Marinari, U. M., Domenicotti, C., & Traverso, N. (2022). Hormesis and Oxidative Distress: Pathophysiology of Reactive Oxygen Species and the Open Question of Antioxidant Modulation and Supplementation. Antioxidants, 11(8), 1613. https://doi.org/10.3390/antiox11081613