
Genetic Modulation of HPV Infection and Cervical Lesions: Role of Oxidative Stress-Related Genes

 ,
,  , ,
, ,  ,
,  and
and
Abstract
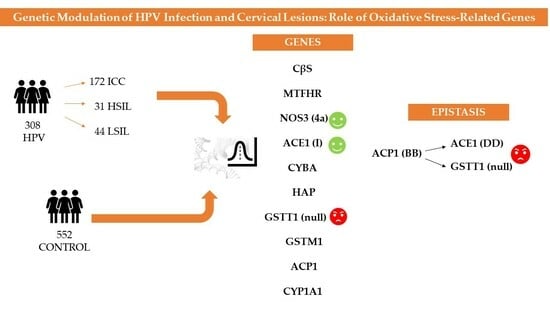
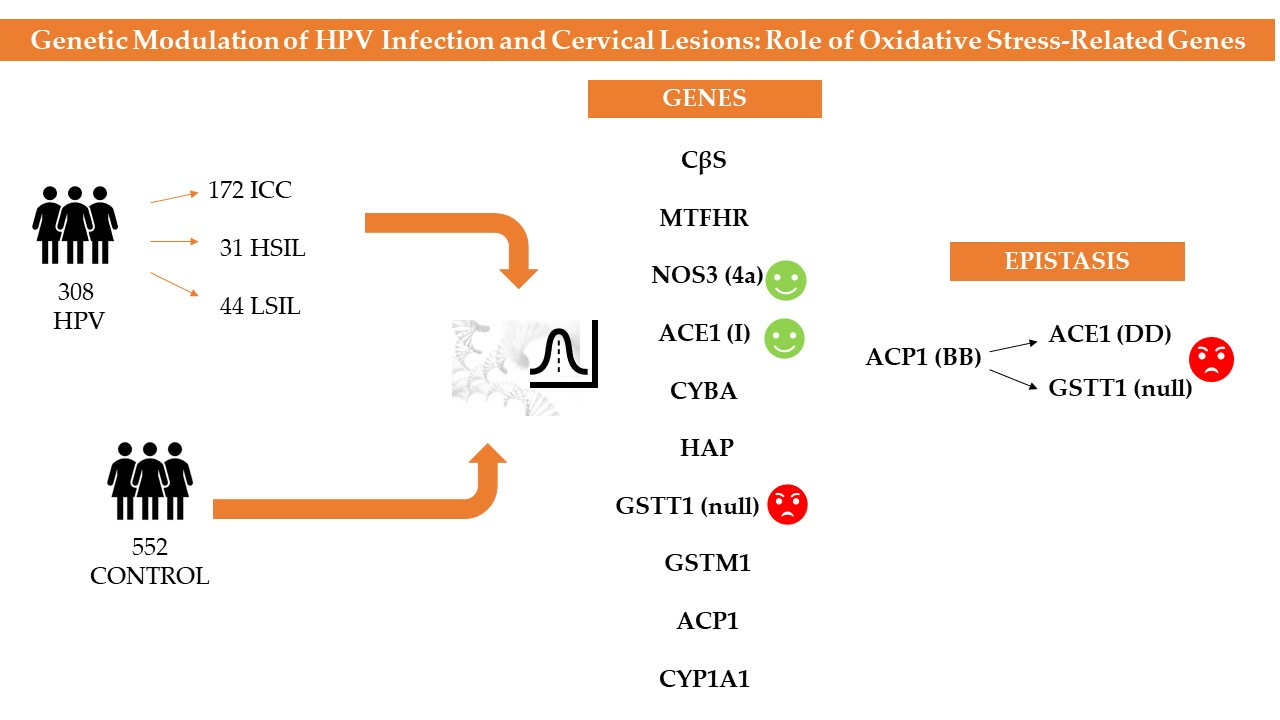
:
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Sample Collection and Ethics
2.3. DNA Extraction
2.4. Genotyping
| Gene | PCR Primers | PCR Conditions |
| CβS | 5′GCAGTTGTTAACGGCGGTATTG3′ 5′GCCGGGCTCTGGACTCGACCTA3′ | 40 cycles 94 °C–30 s 60 °C–30 s 72 °C–40 s |
| MTHFR | 5′TGAAGGAGAAGGTGTCTGCGGGA3′ 5′AGGACGGTGCGGTGAGAGTG3′ | 35 cycles 94 °C–30 s 61 °C–30 s 72 °C–45 s |
| NOS3 | 5′AGGCCCTATGGTAGTGCCTTT3′ 5′TCTCTTAGTGCTGTGGTCAC3′ | 30 cycles 94 °C–30 s 55 °C–30 s 72 °C–45 s |
| ACE1 | 5′GCCCTGCAGGTGTCTGCAGCATGT3′ 5′ GGATGGCTCTCCCCGCCTTCTCTC3′ | 30 cycles 94 °C–60 s 60 °C–30 s 72 °C–30 s |
| CYBA | 5′TGCTTGTGGGTAAACCAAGGCCGGTG3′ 5′AACACTGAGGTAAGTGGGGGTGGCTCCGT3′ | 35 cycles 94 °C–43 s 54 °C–60 s 72 °C–30 s |
| ACP1 | 5′CGATCACCCATTGCAGAAG3′ 5′CCATGATTTCTTAGGCAGCTC3′ | 35 cycles 94 °C–30 s 54 °C–45 s 72 °C–45 s |
| GSTT1 and GSTM1 | 5′GCCATCTTGTGCTACATTGCCCG3′, 5′ATCTTCTCCTCTTCTGTCTCCCC3′, 5′TTCTGGATTGTAGCAGATCATGCCC3′, 5′TTCCTTACTGGTCCTCACATCTC3′ 5′TCACCGGATCATGGCCAGCA3′ | 40 cycles 94 °C–45 s 58 °C–45 s 72 °C–45 s |
| CYP1A1 rs4986884 | 5′CAGTGAAGAGGTGTAGCCGC3′ 5′TAGGAGTCTTGTCTCATGCC 3′ | 30 cycles 94 °C–60 s 61 °C–60 s 72 °C–60 s |
| CYP1A1 rs1048943 rs1799814 | 5′CTGTCTCCCTCTGGTTACAGGAAGC3′ 5′TTCCACCCGTTGCAGCAGGATAGCC3′ | 35 cycles 94 °C–45 s 64 °C–45 s 72 °C–75 s |
- CβS Genotyping—−/+
- MTHFR Genotyping—rs1801133
- NOS3 Genotyping—4b/4a
- ACE1 Genotyping—rs4646994
- CYBA Genotyping—rs4673
- HAP Genotyping—1/2
- ACP1 Genotyping—A/B/C
- GSTT1 and GSTM1 Genotyping—1/0
- CYP1A1 Genotyping—rs4986884|rs1048943|rs1799814
2.5. Statistical Analysis
3. Results
3.1. Analysis Using the Codominant Model
3.2. Analysis Using the Allelic Model
3.3. Analysis Using Dominant, Overdominant, and Recessive Models, or Functional Interest
3.4. Analysis Involving the Most Severe Cytological Phenotypes
3.5. Epistatic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Updates Recommendations on HPV Vaccination Schedule. 2022. Available online: https://www.who.int/news/item/20-12-2022-WHO-updates-recommendations-on-HPV-vaccination-schedule (accessed on 1 March 2023).
- Muñoz, N.; Castellsagué, X.; de González, A.B.; Gissmann, L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006, 24 (Suppl. 3), S1–S10. [Google Scholar] [CrossRef] [PubMed]
- Small, W.J.; Bacon, M.A.; Bajaj, A.; Chuang, L.T.; Fisher, B.J.; Harkenrider, M.M.; Jhingran, A.; Kitchener, H.C.; Mileshkin, L.R.; Viswanathan, A.N.; et al. Cervical cancer: A global health crisis. Cancer 2017, 123, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Jhingran, A.; Oaknin, A.; Denny, L. Cervical cancer. Lancet 2019, 393, 169–182. [Google Scholar] [CrossRef]
- De Marco, F. Oxidative stress and HPV carcinogenesis. Viruses 2013, 5, 708–731. [Google Scholar] [CrossRef]
- Visalli, G.; Riso, R.; Facciolà, A.; Mondello, P.; Caruso, C.; Picerno, I.; Di Pietro, A.; Spataro, P.; Bertuccio, M.P. Higher levels of oxidative DNA damage in cervical cells are correlated with the grade of dysplasia and HPV infection. J. Med. Virol. 2016, 88, 336–344. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Fu, Q.; Kao, Y.-H.; Tseng, T.-S.; Reiss, K.; Cameron, J.E.; Ronis, M.J.; Su, J.; Nair, N.; Chang, H.M.; et al. Antioxidants Associated With Oncogenic Human Papillomavirus Infection in Women. J. Infect. Dis. 2021, 224, 1520–1528. [Google Scholar] [CrossRef]
- Wang, X.; Huang, X.; Zhang, Y. Involvement of Human Papillomaviruses in Cervical Cancer. Front. Microbiol. 2018, 9, 2896. [Google Scholar] [CrossRef]
- Georgescu, S.R.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Sarbu, M.I.; Matei, C.; Nicolae, I.; Tocut, S.M.; Popa, M.I.; Tampa, M. New Insights in the Pathogenesis of HPV Infection and the Associated Carcinogenic Processes: The Role of Chronic Inflammation and Oxidative Stress. J. Immunol. Res. 2018, 2018, 5315816. [Google Scholar] [CrossRef]
- Borges, B.E.S.; de Brito, E.B.; Fuzii, H.T.; Baltazar, C.S.; Sá, A.B.; da Silva, C.I.M.; Santos, G.d.F.S.; Pinheiro, M.d.C.N. Human papillomavirus infection and cervical cancer precursor lesions in women living by Amazon rivers: Investigation of relations with markers of oxidative stress. Einstein 2018, 16, eAO4190. [Google Scholar] [CrossRef]
- Nayki, C.; Gunay, M.; Kulhan, M.; Nayki, U.; Cankaya, M.; Kulhan, N.G. Serum levels of soluble interleukin-2 receptor in association with oxidative stress index in patients with different types of HPV. Ginekol Pol. 2017, 88, 355–359. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K. Redox-sensitive signalling pathways regulated by human papillomavirus in HPV-related cancers. Rev. Med. Virol. 2021, 31, e2230. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Lizano, M. Cellular redox, cancer and human papillomavirus. Virus Res. 2018, 246, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Porter, V.L.; Marra, M.A. The Drivers, Mechanisms, and Consequences of Genome Instability in HPV-Driven Cancers. Cancers 2022, 14, 4623. [Google Scholar] [CrossRef] [PubMed]
- Preci, D.P.; Almeida, A.; Weiler, A.L.; Franciosi, M.L.M.; Cardoso, A.M. Oxidative damage and antioxidants in cervical cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2021, 31, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Pavão, M.L.; Ferin, R.; Lima, A.; Baptista, J. Cysteine and related aminothiols in cardiovascular disease, obesity and insulin resistance. Adv. Clin. Chem. 2022, 109, 75–127. [Google Scholar]
- Tsai, M.Y.; Bignell, M.; Schwichtenberg, K.; Hanson, N.Q. High prevalence of a mutation in the cystathionine beta-synthase gene. Am. J. Hum. Genet. 1996, 59, 1262–1267. [Google Scholar]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- van der Put, N.M.; Gabreëls, F.; Stevens, E.M.; Smeitink, J.A.; Trijbels, F.J.; Eskes, T.K.; van den Heuvel, L.P.; Blom, H.J. A second common mutation in the methylenetetrahydrofolate reductase gene: An additional risk factor for neural-tube defects? Am. J. Hum. Genet. 1998, 62, 1044–1051. [Google Scholar] [CrossRef]
- Tsang, B.L.; Devine, O.J.; Cordero, A.M.; Marchetta, C.M.; Mulinare, J.; Mersereau, P.; Guo, J.; Qi, Y.P.; Berry, R.J.; Rosenthal, J.; et al. Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: A systematic review and meta-analysis of trials and observational studies. Am. J. Clin. Nutr. 2015, 101, 1286–1294. [Google Scholar] [CrossRef]
- Summers, C.M.; Hammons, A.L.; Mitchell, L.E.; Woodside, J.V.; Yarnell, J.W.; Young, I.S.; Evans, A.; Whitehead, A.S. Influence of the cystathionine beta-synthase 844ins68 and methylenetetrahydrofolate reductase 677C>T polymorphisms on folate and homocysteine concentrations. Eur. J. Hum. Genet. 2008, 16, 1010–1013. [Google Scholar] [CrossRef]
- Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? Int. J. Mol. Sci. 2020, 21, 9393. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; El Yazbi, A.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci 2022, 27, 105. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Handy, D.E.; Loscalzo, J. Role of oxidative stress and nitric oxide in atherothrombosis. Front. Biosci. 2008, 13, 5323–5344. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, T.; Yokoyama, K.; Arai, T.; Takemoto, F.; Hara, S.; Yamada, A.; Kawaguchi, Y.; Hosoya, T.; Igari, J. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans. Biochem. Biophys. Res. Commun. 1998, 245, 190–193. [Google Scholar] [CrossRef]
- Zhang, M.-X.; Ou, H.; Shen, Y.H.; Wang, J.; Wang, J.; Coselli, J.; Wang, X.L. Regulation of endothelial nitric oxide synthase by small RNA. Proc. Natl. Acad. Sci. USA 2005, 102, 16967–16972. [Google Scholar] [CrossRef]
- Zhao, S.; Sun, W.; Jiang, P. Role of the ACE2/Ang-(1-7)/Mas axis in glucose metabolism. Rev. Cardiovasc. Med. 2021, 22, 769–777. [Google Scholar] [CrossRef]
- Thakur, S.; Sharma, V.; Kaur, D.; Purkait, P. Angiotensin-Converting Enzyme (ACE) Insertion/Deletion (I/D) Polymorphism as a Conjoint Regulator of Coagulation, Fibrinolytic, and RAAS Pathway in Infertility and Associated Pregnancy Complications. J. Renin-Angiotensin-Aldosterone Syst. 2022, 2022, 1695769. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Role of Reactive Species on Innate Immunity. Vaccines 2022, 10, 1735. [Google Scholar] [CrossRef]
- Szanto, I. NADPH Oxidase 4 (NOX4) in Cancer: Linking Redox Signals to Oncogenic Metabolic Adaptation. Int. J. Mol. Sci. 2022, 23, 2702. [Google Scholar] [CrossRef]
- Paclet, M.-H.; Laurans, S.; Dupré-Crochet, S. Regulation of Neutrophil NADPH Oxidase, NOX2: A Crucial Effector in Neutrophil Phenotype and Function. Front. Cell Dev. Biol. 2022, 10, 945749. [Google Scholar] [CrossRef]
- Stasia, M.J. CYBA encoding p22(phox), the cytochrome b558 alpha polypeptide: Gene structure, expression, role and physiopathology. Gene 2016, 586, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M. The antioxidant activity of haptoglobin towards haemoglobin-stimulated lipid peroxidation. Biochim. Biophys. Acta 1987, 917, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.H. Oxidative effects of heme and porphyrins on proteins and lipids. Semin. Hematol. 1989, 26, 105–113. [Google Scholar] [PubMed]
- Schreiber, R.; Ferreira-Sae, M.C.; Ronchi, J.A.; Pio-Magalhães, J.A.; Cipolli, J.A.; Matos-Souza, J.R.; Mill, J.G.; Vercesi, A.E.; Krieger, J.E.; Franchini, K.G.; et al. The C242T polymorphism of the p22-phox gene (CYBA) is associated with higher left ventricular mass in Brazilian hypertensive patients. BMC Med. Genet. 2011, 12, 114. [Google Scholar] [CrossRef]
- Dai, X.; Dharmage, S.C.; Lodge, C.J. Interactions between glutathione S-transferase genes and household air pollution on asthma and lung function. Front. Mol. Biosci. 2022, 9, 955193. [Google Scholar] [CrossRef] [PubMed]
- Apelt, N.; da Silva, A.P.; Ferreira, J.; Alho, I.; Monteiro, C.; Marinho, C.; Teixeira, P.; Sardinha, L.; Laires, M.J.; Mascarenhas, M.R.; et al. ACP1 genotype, glutathione reductase activity, and riboflavin uptake affect cardiovascular risk in the obese. Metabolism 2009, 58, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- DeSouza, S.R.; Olson, M.C.; Tinucci, S.L.; Sinner, E.K.; Flynn, R.S.; Marshall, Q.F.; Jakubowski, H.V.; McIntee, E.J. SAR of non-hydrolysable analogs of pyridoxal 5′-phosphate against low molecular weight protein tyrosine phosphatase isoforms. Bioorganic Med. Chem. Lett. 2020, 30, 127342. [Google Scholar] [CrossRef]
- Steinert, J.R.; Amal, H. The contribution of an imbalanced redox signalling to neurological and neurodegenerative conditions. Free Radic. Biol. Med. 2023, 194, 71–83. [Google Scholar] [CrossRef]
- Abbas, M.; Kushwaha, V.S.; Srivastava, K.; Banerjee, M. Understanding Role of DNA Repair and Cytochrome p-450 Gene Polymorphisms in Cervical Cancer Patient Treated With Concomitant Chemoradiation. Br. J. Biomed. Sci. 2022, 79, 10120. [Google Scholar] [CrossRef]
- Fankhouser, R.W.; Murrell, D.E.; Anane, Y.Y.; Hurley, D.L.; Mamudu, H.M.; Harirforoosh, S. Type 2 diabetes: An exploratory genetic association analysis of selected metabolizing enzymes and transporters and effects on cardiovascular and renal biomarkers. Drug Metab. Pers. Ther. 2022, 37, 375–382. [Google Scholar] [CrossRef]
- Cheng, T.; Gamage, S.M.K.; Lu, C.-T.; Aktar, S.; Gopalan, V.; Lam, A.K.-Y. Polymorphisms in PAH metabolising enzyme CYP1A1 in colorectal cancer and their clinicopathological correlations. Pathol. Res. Pract. 2022, 231, 153801. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Banerjee, S.; Mukhopadhyay, P.; Guha, U.; Ganguly, K.; Bhattacharjee, S.; Sengupta, M. A meta-analysis and in silico analysis of polymorphic variants conferring breast cancer risk in the Indian subcontinent. Future Oncol. 2020, 16, 2121–2142. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E.; Peyton, C.L.; Alessi, T.Q.; Wheeler, C.M.; Coutlée, F.; Hildesheim, A.; Schiffman, M.H.; Scott, D.R.; Apple, R.J. Improved amplification of genital human papillomaviruses. J. Clin. Microbiol. 2000, 38, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.P. Typing and subtyping of haptoglobin from native serum using disc gel electrophoresis in alkaline buffer: Application to routine screening. Anal. Biochem. 1984, 141, 55–61. [Google Scholar] [CrossRef]
- Dissing, J.; Johnsen, A.H.; Sensabaugh, G.F. Human red cell acid phosphatase (ACP1). The amino acid sequence of the two isozymes Bf and Bs encoded by the ACP1*B allele. J. Biol. Chem. 1991, 266, 20619–20625. [Google Scholar] [CrossRef]
- Modesti, A.; Marzocchini, R.; Raugei, G.; Chiti, F.; Sereni, A.; Magherini, F.; Ramponi, G. Cloning, expression and characterisation of a new human low Mr phosphotyrosine protein phosphatase originating by alternative splicing. FEBS Lett. 1998, 431, 111–115. [Google Scholar] [CrossRef]
- Timoshenko, O.S.; Kugaevskaya, E.V.; Gureeva, T.A.; Zavalishina, L.E.; Andreeva, Y.Y.; Solovyeva, N.I. Matrix metalloproteinases 2 and 9, their endogenous regulators, and angiotensin-converting enzyme in cervical squamous cell carcinoma. Arkh. Patol. 2015, 77, 31–35. [Google Scholar] [CrossRef]
- Solov’Eva, N.I.; Timoshenko, O.S.; Kugaevskaia, E.V.; Andreeva, I.I.; Zavalishina, L.E. Key enzymes of degradation and angiogenesis as a factors of tumor progression in squamous cell carcinoma of the cervix. Bioorg. Khim. 2014, 40, 743–751. [Google Scholar]
- Bueno, V.; Frasca, D. Mini-review: Angiotensin-converting enzyme 1 (ACE1) and the impact for diseases such as Alzheimer’s disease, sarcopenia, cancer, and COVID-19. Front. Aging 2023, 4, 1117502. [Google Scholar] [CrossRef]
- Du, J.; Lan, J.; Yang, H.; Ying, Q.; Huang, G.; Mou, J.; Long, J.; Qiao, Z.; Hu, Q. Association of angiotensin-converting enzyme insertion/deletion (ACE I/D) gene polymorphism with susceptibility to prostate cancer: An updated meta-analysis. World J. Surg. Oncol. 2022, 20, 354. [Google Scholar] [CrossRef]
- Koh, W.-P.; Yuan, J.-M.; Sun, C.-L.; Berg, D.V.D.; Seow, A.; Lee, H.-P.; Yu, M.C. Angiotensin I-converting enzyme (ACE) gene polymorphism and breast cancer risk among Chinese women in Singapore. Cancer Res. 2003, 63, 573–578. [Google Scholar] [PubMed]
- Cozma, A.; Fodor, A.; Orasan, O.H.; Vulturar, R.; Samplelean, D.; Negrean, V.; Muresan, C.; Suharoschi, R.; Sitar-Taut, A. Pharmacogenetic Implications of eNOS Polymorphisms (Glu298Asp, T786C, 4b/4a) in Cardiovascular Drug Therapy. In Vivo 2019, 33, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, L.; Dharmarajan, A.; Samji, P. Regulation of pleiotropic physiological roles of nitric oxide signaling. Cell. Signal. 2023, 101, 110496. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef]
- De Andrea, M.; Mondini, M.; Azzimonti, B.; Dell’Oste, V.; Germano, S.; Gaudino, G.; Musso, T.; Landolfo, S.; Gariglio, M. Alpha- and betapapillomavirus E6/E7 genes differentially modulate pro-inflammatory gene expression. Virus Res. 2007, 124, 220–225. [Google Scholar] [CrossRef]
- Wei, L.; Gravitt, P.E.; Song, H.; Maldonado, A.M.; Ozbun, M.A. Nitric oxide induces early viral transcription coincident with increased DNA damage and mutation rates in human papillomavirus-infected cells. Cancer Res. 2009, 69, 4878–4884. [Google Scholar] [CrossRef]
- Rahkola, P.; Mikkola, T.S.; Ylikorkala, O.; Vaisanen-Tommiska, M. Association between high risk papillomavirus DNA and nitric oxide release in the human uterine cervix. Gynecol. Oncol. 2009, 114, 323–326. [Google Scholar] [CrossRef]
- Hiraku, Y.; Tabata, T.; Ma, N.; Murata, M.; Ding, X.; Kawanishi, S. Nitrative and oxidative DNA damage in cervical intraepithelial neoplasia associated with human papilloma virus infection. Cancer Sci. 2007, 98, 964–972. [Google Scholar] [CrossRef]
- Mazibrada, J.; Rittà, M.; Mondini, M.; De Andrea, M.; Azzimonti, B.; Borgogna, C.; Ciotti, M.; Orlando, A.; Surico, N.; Chiusa, L.; et al. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol. Oncol. 2008, 108, 112–120. [Google Scholar] [CrossRef]
- Rahkola-Soisalo, P.; Savolainen-Peltonen, H.; Väisänen-Tommiska, M.; Butzow, R.; Ylikorkala, O.; Mikkola, T.S. High-risk human papillomavirus-induced expression of endothelial and inducible nitric oxide synthase in human uterine cervix. Ann. Med. 2013, 45, 79–84. [Google Scholar] [CrossRef]
- Vahora, H.; Khan, M.A.; Alalami, U.; Hussain, A. The Potential Role of Nitric Oxide in Halting Cancer Progression Through Chemoprevention. J. Cancer Prev. 2016, 21, 1–12. [Google Scholar] [CrossRef]
- Sundaram, M.K.; Khan, M.A.; Alalami, U.; Somvanshi, P.; Bhardwaj, T.; Pramodh, S.; Raina, R.; Shekfeh, Z.; Haque, S.; Hussain, A. Phytochemicals induce apoptosis by modulation of nitric oxide signaling pathway in cervical cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 11827–11844. [Google Scholar]
- Hayes, J.D.; Pulford, D.J. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–600. [Google Scholar] [CrossRef]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef]
- de Carvalho, C.N.; da Silva, I.G.; Pereira, J.S.; de Souza, N.N.; de Azevedo Focchi, G.R.; Ribalta, J.C.L. Polymorphisms of p53, GSTM1 and GSTT1, and HPV in uterine cervix adenocarcinoma. Eur. J. Gynaecol. Oncol. 2008, 29, 590–593. [Google Scholar]
- Liu, Y.; Xu, L.-Z. Meta-analysis of association between GSTM1 gene polymorphism and cervical cancer. Asian Pac. J. Trop. Med. 2012, 5, 480–484. [Google Scholar] [CrossRef]
- Tian, S.; Yang, X.; Zhang, L.; Zhao, J.; Pei, M.; Yu, Y.; Yang, T. Polymorphic variants conferring genetic risk to cervical lesions support GSTs as important associated loci. Medicine 2019, 98, e17487. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, C.G.; Park, Y.G.; Kim, K.S.; Kim, I.K.; Sohn, Y.W.; Min, H.K.; Lee, J.M.; Namkoong, S.E. Combined analysis of germline polymorphisms of p53, GSTM1, GSTT1, CYP1A1, and CYP2E1: Relation to the incidence rate of cervical carcinoma. Cancer 2000, 88, 2082–2091. [Google Scholar] [CrossRef]
- Nakanishi, G.; Pita-Oliveira, M.; Bertagnolli, L.S.; Torres-Loureiro, S.; Scudeler, M.M.; Cirino, H.S.; Chaves, M.L.; Miwa, B.; Rodrigues-Soares, F. Worldwide Systematic Review of GSTM1 and GSTT1 Null Genotypes by Continent, Ethnicity, and Therapeutic Area. OMICS J. Integr. Biol. 2022, 26, 528–541. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Zhao, F.; Chen, Z.; Yang, X.; Sun, C.; Gao, Y.; Yang, T.; Tian, G.; Chen, Y.; et al. Methylenetetrahydrofolate reductase polymorphisms and colorectal cancer prognosis: A meta-analysis. J. Gene Med. 2019, 21, e3114. [Google Scholar] [CrossRef]
- Chen, X.; Ahamada, H.; Zhang, T.; Bai, Z.; Wang, C. Association of Intake Folate and Related Gene Polymorphisms with Breast Cancer. J. Nutr. Sci. Vitaminol. 2019, 65, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Farshid, S.; Alijanpour, A.; Barahman, M.; Dastgheib, S.A.; Narimani, N.; Shirinzadeh-Dastgiri, Z.; Maleki, H.; Asadian, F.; Mazaheri, M.; Neamatzadeh, H. Associations of MTHFR rs1801133 (677C>T) and rs180113 (1298A>C) Polymorphisms with Susceptibility to Bladder Cancer: A Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 2022, 23, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pei, Y.-X.; Wang, L.-N.; Liang, C.; Tang, Y.-L.; Zhang, X.-L.; Huang, L.-B.; Luo, X.-Q.; Ke, Z.-Y. MTHFR-C677T Gene Polymorphism and Susceptibility to Acute Lymphoblastic Leukemia in Children: A Meta-Analysis. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 125–136. [Google Scholar] [CrossRef]
- Rai, V. Folate pathway gene MTHFR C677T polymorphism and risk of lung cancer in Asian populations. Asian Pac. J. Cancer Prev. 2014, 15, 9259–9264. [Google Scholar] [CrossRef]
- Tanha, K.; Mottaghi, A.; Nojomi, M.; Moradi, M.; Rajabzadeh, R.; Lotfi, S.; Janani, L. Investigation on factors associated with ovarian cancer: An umbrella review of systematic review and meta-analyses. J. Ovarian Res. 2021, 14, 153. [Google Scholar] [CrossRef]
- Wang, Z.; Li, K.; Ouyang, L.; Iko, H.; Safi, A.J.; Gao, S. Effects of methylenetetrahydrofolate reductase single-nucleotide polymorphisms on breast, cervical, ovarian, and endometrial cancer susceptibilities. Chronic Dis. Transl. Med. 2021, 7, 169–181. [Google Scholar] [CrossRef]
- Hajiesmaeil, M.; Tafvizi, F.; Sarmadi, S. The effect of methylenetetrahydrofolate reductase polymorphisms on susceptibility to human papilloma virus infection and cervical cancer. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 1–6. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, L.; Kohlmeier, M.; Ye, F.; Cai, W. Association between MTHFR C677T, MTHFR A1298C and MS A2756G polymorphisms and risk of cervical intraepithelial neoplasia II/III and cervical cancer: A meta-analysis. Mol. Med. Rep. 2013, 8, 919–927. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, J. C677T polymorphism of methylenetetrahydrofolate reductase may contribute to cervical cancer risk in complete over-dominant model. Med. Hypotheses 2013, 80, 679–683. [Google Scholar] [CrossRef]
- Wu, C.Y.; Yang, M.; Lin, M.; Li, L.P.; Wen, X.Z. MTHFR C677T polymorphism was an ethnicity-dependent risk factor for cervical cancer development: Evidence based on a meta-analysis. Arch. Gynecol. Obstet. 2013, 288, 595–605. [Google Scholar] [CrossRef]
- Gong, J.-M.; Shen, Y.; Shan, W.-W.; He, Y.-X. The association between MTHFR polymorphism and cervical cancer. Sci. Rep. 2018, 8, 7244. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.N.T.; Sabino, A.d.P.; Tafuri, A.; Lima, A.A. Lack of association between methylenetetrahydrofolate reductase C677T polymorphism, HPV infection and cervical intraepithelial neoplasia in Brazilian women. BMC Med. Genet. 2019, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, A.; Bassam-Tolami, F.; Imani, M. The Impact of MTHFR 1298 A > C and 677 C > T Gene Polymorphisms as Susceptibility Risk Factors in Cervical Intraepithelial Neoplasia Related to HPV and Sexually Transmitted Infections. J. Obstet. Gynaecol. India. 2020, 70, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Zarchi, M.; Moghimi, M.; Abbasi, H.; Hadadan, A.; Salimi, E.; Morovati-Sharifabad, M.; Akbarian-Bafghi, M.J.; Zare-Shehneh, M.; Mosavi-Jarrahi, A.; Neamatzadeh, H. Association of MTHFR 677C>T Polymorphism with Susceptibility to Ovarian and Cervical Cancers: A Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 2019, 20, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Gallegos-Arreola, M.P.; Figuera-Villanueva, L.E.; Ramos-Silva, A.; Salas-González, E.; Puebla-Pérez, A.M.; Peralta-Leal, V.; García-Ortiz, J.E.; Dávalos-Rodríguez, I.P.; Zúńiga-González, G.M. The association between the 844ins68 polymorphism in the CBS gene and breast cancer. Arch. Med. Sci. 2014, 10, 1214–1224. [Google Scholar] [CrossRef]
- Ascenção, K.; Szabo, C. Emerging roles of cystathionine β-synthase in various forms of cancer. Redox Biol. 2022, 53, 102331. [Google Scholar] [CrossRef]
- Alho, I.; Bicho, M.C.; Carvalho, R.; Da Silva, A.P.; Costa, L.; Bicho, M. Low molecular weight protein tyrosine phosphatase genetic polymorphism and susceptibility to cancer development. Cancer Genet. Cytogenet. 2008, 181, 20–24. [Google Scholar] [CrossRef]
- Alho, I.; Costa, L.; Bicho, M.; Coelho, C. Characterization of low molecular weight protein tyrosine phosphatase isoforms in human breast cancer epithelial cell lines. Anticancer Res. 2013, 33, 1983–1987. [Google Scholar]
- Faria, A.V.S.; Fonseca, E.M.B.; Cordeiro, H.G.; Clerici, S.P.; Ferreira-Halder, C.V. Low molecular weight protein tyrosine phosphatase as signaling hub of cancer hallmarks. Cell. Mol. Life Sci. 2021, 78, 1263–1273. [Google Scholar] [CrossRef]
- Castaldo, S.A.; da Silva, A.P.; Matos, A.; Inácio, Â.; Bicho, M.; Medeiros, R.; Alho, I.; Bicho, M.C. The role of CYBA (p22phox) and catalase genetic polymorphisms and their possible epistatic interaction in cervical cancer. Tumor Biol. 2015, 36, 909–914. [Google Scholar] [CrossRef]
- Wyche, K.E.; Wang, S.S.; Griendling, K.K.; Dikalov, S.I.; Austin, H.; Rao, S.; Fink, B.; Harrison, D.G.; Zafari, A.M. C242T CYBA polymorphism of the NADPH oxidase is associated with reduced respiratory burst in human neutrophils. Hypertens 2004, 43, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Li, S.; Liu, H.; Bai, H.; Liu, Q.; Hu, K.; Guan, L.; Fan, P. Myeloperoxidase and CYBA genetic variants in polycystic ovary syndrome. Eur. J. Clin. Investig. 2021, 51, e13438. [Google Scholar] [CrossRef] [PubMed]
- Tupurani, M.A.; Padala, C.; Puranam, K.; Galimudi, R.K.; Kupsal, K.; Shyamala, N.; Gantala, S.; Kummari, R.; Chinta, S.K.; Hanumanth, S.R. Association of CYBA gene (-930 A/G and 242 C/T) polymorphisms with oxidative stress in breast cancer: A case-control study. PeerJ 2018, 6, e5509. [Google Scholar] [CrossRef] [PubMed]
- Naryzny, S.N.; Legina, O.K. Haptoglobin as a Biomarker. Biochem. Suppl. Ser. B Biomed. Chem. 2021, 15, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.M.; Su, B.T.; May, S.Y.; Huang, S.C.; Wen, H.K.; Ouyang, P.C. Haptoglobin typing and quantitation in normal Chinese females and gynecologic cancer patients. Zhonghua Minguo Wei Sheng Wu Ji Mian Yi Xue Za Zhi = Chin. J. Microbiol. Immunol. 1980, 13, 149–157. [Google Scholar]
- Mahmud, S.M.; Koushik, A.; Duarte-Franco, E.; Costa, J.; Fontes, G.; Bicho, M.; Coutlée, F.; Franco, E.L. Haptoglobin phenotype and risk of cervical neoplasia: A case-control study. Clin. Chim. Acta. 2007, 385, 67–72. [Google Scholar] [CrossRef]
- Quaye, I.K.; Agbolosu, K.; Ibrahim, M.; Bannerman-Williams, P. Haptoglobin phenotypes in cervical cancer: Decreased risk for Hp2-2 individuals. Clin. Chim. Acta Int. J. Clin. Chem. 2009, 403, 267–268. [Google Scholar] [CrossRef]
- Delanghe, J.R.; Langlois, M.R. Haptoglobin polymorphism and body iron stores. Clin. Chem. Lab. Med. 2002, 40, 212–216. [Google Scholar] [CrossRef]
- Nath, N.; Mishra, P.; Panda, A.K.; Mishra, R. Polymorphisms and haplotypes of TLR4, TLR9 and CYP1A1 genes possibly interfere with high-risk human papillomavirus infection and cervical cancer susceptibility in Jharkhand, India. Int. Immunopharmacol. 2020, 88, 106925. [Google Scholar] [CrossRef]
- Helaoui, A.; Sfar, S.; Boudhiba, N.; Dehghanian, F.; Dehbashi, M.; Bouchahda, H.; Hojati, Z.; Kenani, A. Association of xenobiotic-metabolizing genes polymorphisms with cervical cancer risk in the Tunisian population. Mol. Biol. Rep. 2023, 50, 949–959. [Google Scholar] [CrossRef]
- Wongpratate, M.; Ishida, W.; Phuthong, S.; Natphopsuk, S.; Ishida, T. Genetic Polymorphisms of the Human Cytochrome P450 1A1 (CYP1A1) and Cervical Cancer Susceptibility among Northeast Thai Women. Asian Pac. J. Cancer Prev. 2020, 21, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Guha, U.; Mitra, S.; Ghosh, S.; Bhattacharjee, S.; Sengupta, M. Meta-Analysis of Polymorphic Variants Conferring Genetic Risk to Cervical Cancer in Indian Women Supports CYP1A1 as an Important Associated Locus. Asian Pac. J. Cancer Prev. 2018, 19, 2071–2081. [Google Scholar] [PubMed]
- Jain, V.; Ratre, Y.K.; Amle, D.; Mishra, P.K.; Patra, P.K. Polymorphism of CYP1A1 gene variants rs4646903 and rs1048943 relation to the incidence of cervical cancer in Chhattisgarh. Environ. Toxicol. Pharmacol. 2017, 52, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, G.; Kong, F.; Liu, Z.; Li, N.; Li, Y.; Guo, X. The Association of CYP1A1 Gene With Cervical Cancer and Additional SNP-SNP Interaction in Chinese Women. J. Clin. Lab. Anal. 2016, 30, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.H.; Sidik, S.M.; Husain, S.N.A.S.; Lye, M.S.; Chong, P.P. CYP1A1 MspI Polymorphism and Cervical Carcinoma Risk in the Multi-Ethnic Population of Malaysia: A Case-Control Study. Asian Pac. J. Cancer Prev. 2016, 17, 57–64. [Google Scholar] [CrossRef]
- Ding, B.; Sun, W.; Han, S.; Cai, Y.; Ren, M.; Shen, Y. Cytochrome P450 1A1 gene polymorphisms and cervical cancer risk: A systematic review and meta-analysis. Medicine 2018, 97, e0210. [Google Scholar] [CrossRef]
- Wang, L.-N.; Wang, F.; Liu, J.; Jin, Y.-H.; Fang, C.; Ren, X.-Q. CYP1A1 Ile462Val Polymorphism Is Associated with Cervical Cancer Risk in Caucasians Not Asians: A Meta-Analysis. Front. Physiol. 2017, 8, 1081. [Google Scholar] [CrossRef]

{kind=link}
| Genes | HPV | Controls | p-Value | |
|---|---|---|---|---|
| N (%) | N (%) | |||
| CβS | −/− | 46 (93.9%) | 220 (83.0%) | 0.077 a |
| +/− | 2 (4.1%) | 42 (15.8%) | ||
| +/+ | 1 (2.0%) | 3 (1.1%) | ||
| MTHFR | CC | 53 (44.5%) | 200 (48.5%) | 0.546 b |
| CT | 54 (45.4%) | 164 (39.8%) | ||
| TT | 12 (10.1%) | 48 (11.7%) | ||
| NOS3 | 4b4b | 80 (79.2%) | 125 (66.1%) | 0.018 b |
| 4a4b | 20 (19.8%) | 50 (26.5%) | ||
| 4a4a | 1 (1.0%) | 14 (7.4%) | ||
| ACE1 | DD | 70 (64.2%) | 176 (45.7%) | 0.003 b,* |
| ID | 29 (26.6%) | 159 (41.3%) | ||
| II | 10 (9.2%) | 50 (13.0%) | ||
| CYBA | CC | 46 (51.7%) | 34 (38.2%) | 0.190 b |
| CT | 36 (40.4%) | 47 (52.8%) | ||
| TT | 7 (7.9%) | 8 (9.0%) | ||
| HAP | 2.2 | 72 (36.5%) | 129 (38.7%) | 0.163 b |
| 2.1 | 78 (39.6%) | 147 (44.1%) | ||
| 1.1 | 47 (23.9%) | 57 (17.1%) | ||
| ACP1 | AA | 3 (4.9%) | 27 (11.0%) | 0.299 b |
| BB | 30 (49.2%) | 87 (35.4%) | ||
| AB | 21 (34.4%) | 97 (39.4%) | ||
| AC | 3 (4.9%) | 16 (6.5%) | ||
| BC | 4 (6.6%) | 19 (7.7%) | ||
| CYP1A1 T5639C | TT | 41 (70.7%) | 98 (81.0%) | 0.121 b |
| TC | 17 (29.3%) | 23 (19.0%) | ||
| CYP1A1 A4889G | AA | 43 (87.8%) | 117 (92.1%) | 0.387 c |
| AG | 6 (12.2%) | 10 (7.9%) | ||
| CYP1A1 C4887A | CC | 39 (90.7%) | 109 (86.5%) | 0.472 b |
| CA | 4 (9.3%) | 17 (13.5%) |
| Genes | HPV | Controls | p-Value | OR (CI) | ||
|---|---|---|---|---|---|---|
| N (%) | N (%) | |||||
| CβS | Allele − | 94 (95.9%) | 482 (90.9%) | 0.101 a | ||
| Allele + | 4 (4.1%) | 48 (9.1%) | ||||
| MTHFR | Allele C | 160 (67.2%) | 564 (68.4%) | 0.722 a | ||
| Allele T | 78 (32.8%) | 260 (31.6%) | ||||
| NOS3 | Allele 4b | 180 (89.1%) | 300 (79.4%) | 0.003 a,* | 0.470 (0.283–0.781) | |
| Allele 4a | 22 (10.9%) | 78 (20.6%) | ||||
| ACE1 | Allele D | 169 (77.5%) | 511 (66.4%) | 0.002 a,* | 0.572 (0.403–0.813) | |
| Allele I | 49 (22.5%) | 259 (33.6%) | ||||
| CYBA | Allele C | 128 (71.9%) | 115 (64.6%) | 0.139 a | ||
| Allele T | 50 (28.1%) | 63 (35.4%) | ||||
| HAP | Allele 2 | 222 (56.3%) | 405 (60.8%) | 0.153 a | ||
| Allele 1 | 172 (43.7%) | 261 (39.2%) | ||||
| ACP1 | Allele A | 30 (24.6%) | 167 (33.9%) | 0.091 a | ||
| Allele B | 85 (69.7%) | 290 (58.9%) | ||||
| Allele C | 7 (5.7%) | 35 (7.1%) | ||||
| CYP1A1 | T5639C | Allele T | 99 (85.3%) | 219 (90.5%) | 0.148 a | |
| Allele C | 17 (14.7%) | 23 (9.5%) | ||||
| A4889G | Allele A | 92 (93.9%) | 244 (96.1%) | 0.397 b | ||
| Allele G | 6 (6.1%) | 10 (3.9%) | ||||
| C4887A | Allele C | 82 (95.3%) | 235 (93.3%) | 0.487 a | ||
| Allele A | 4 (4.7%) | 17 (6.7%) | ||||
| Genes 1 | HPV | Controls | p-Value | OR (CI) | |||||
|---|---|---|---|---|---|---|---|---|---|
| N (%) | N (%) | ||||||||
| CβS | +/+ and +/− vs. −/− | 3 (6.1%) | 46 (93.9%) | 45 (17.0%) | 220 (83.0%) | 0.052 a | |||
| +/− vs. −/− and +/+ | 2 (4.1%) | 47 (95.9%) | 42 (15.8%) | 223 (84.2%) | 0.029 a | ||||
| +/+ vs. −/+ and +/+ | 1 (2.0%) | 48 (98.0%) | 3 (1.1%) | 262 (98.9%) | 0.495 b | ||||
| MTHFR | TT and TC vs. CC | 66 (55.5%) | 53 (44.5%) | 212 (51.5%) | 200 (48.5%) | 0.441 a | |||
| CT vs. CC and TT | 54 (45.4%) | 65 (54.6%) | 164 (39.8%) | 248 (60.2%) | 0.276 a | ||||
| TT vs. CC and CT | 12 (10.1%) | 107 (89.9%) | 48 (11.7%) | 364 (88.3%) | 0.634 a | ||||
| NOS3 | 4a4a and 4a4b vs. 4b4b | 21 (20.8%) | 80 (79.2%) | 64 (33.9%) | 125 (66.1%) | 0.020 a | |||
| 4a4b vs. 4a4a and 4b4b | 20 (19.8%) | 81 (80.2%) | 50 (26.5%) | 139 (73.5%) | 0.207a | ||||
| 4a4a vs. 4b4b and 4a4b | 1 (1.0%) | 100 (99.0%) | 14 (7.4%) | 175 (92.6%) | 0.019 a | ||||
| ACE1 | II and ID vs. DD | 39 (35.8%) | 70 (64.2%) | 209 (54.3%) | 176 (45.7%) | <0.001 a,* | 0.469 (0.302–0.728) | ||
| ID vs. DD and II | 29 (26.6%) | 80 (73.4%) | 159 (41.3%) | 226 (58.7%) | 0.005 a | ||||
| II vs. DD and ID | 10 (9.2%) | 99 (90.8%) | 50 (13.0%) | 335 (87.0%) | 0.282 a | ||||
| CYBA | TT and TC vs. CC | 43 (48.3%) | 46 (51.7%) | 55 (61.8%) | 34 (38.2%) | 0.071 a | |||
| CT vs. CC and TT | 36 (40.4%) | 53 (59.6%) | 47 (52.8%) | 42 (47.2%) | 0.098 a | ||||
| TT vs. TC and CC | 7 (7.9%) | 82 (92.1%) | 8 (9.0%) | 81 (91.0%) | 0.787 a | ||||
| HAP | 2.1 and 1.1 vs. 2.2 | 125 (63.5%) | 72 (36.5%) | 204 (61.3%) | 129 (38.7%) | 0.615 a | |||
| 2.1 vs. 2.2 and 1.1 | 78 (39.6%) | 119 (60.4%) | 147 (44.1%) | 186 (55.9%) | 0.306 a | ||||
| 1.1 vs. 2.2 and 2.1 | 47 (23.9%) | 150 (76.1%) | 57 (17.1%) | 276 (82.9%) | 0.059 a | ||||
| ACP1 | AA vs. BB and CC and AB and AC and BC | 3 (4.9%) | 58 (95.1%) | 27 (11.0%) | 219 (89.0%) | 0.154 a | |||
| BB vs. AA and CC and AB and AC and BC | 30 (49.2%) | 31 (50.8%) | 87 (35.4%) | 159 (64.6%) | 0.047 a | ||||
| AB vs. AA and BB and CC and AC and BC | 21 (34.4%) | 40 (65.6%) | 97 (39.4%) | 149 (60.6%) | 0.472 a | ||||
| AC vs. AA and BB and CC and AB and BC | 3 (4.9%) | 58 (95.1%) | 16 (6.5%) | 230 (93.5%) | 0.775 b | ||||
| BC vs. AA and BB and CC and AB and AC | 4 (6.6%) | 57 (93.4%) | 19 (7.7%) | 227 (92.3%) | 1 b | ||||
| AA and AB and BB vs. AC and BC | 54 (88.5%) | 7 (11.5%) | 211 (85.8%) | 35 (14.2%) | 0.576 a | ||||
| AA and AC and BC vs. BB and AB | 10 (16.4%) | 51 (83.6%) | 62 (25.2%) | 184 (74.8%) | 0.146 a | ||||
| AA and AC vs. AB and BC vs. BB | 6 (9.8%) | 25 (41.0%) | 30 (49.2%) | 43 (17.5%) | 116 (47.2%) | 87 (35.4%) | 0.098 a | ||
| AA and AB vs. BB and AC vs. BC | 24 (39.3%) | 33 (54.1%) | 4 (6.6%) | 124 (50.4%) | 103 (41.9%) | 19 (7.7%) | 0.225 a | ||
| GSTT1 | 0/0 vs. 1/1 and 1/0 | 35 (36.5%) | 61 (63.5%) | 29 (17.6%) | 136 (82.4%) | <0.001 a,* | 2.691 (1.510–4.794) | ||
| GSTM1 | 0/0 vs. 1/1 and 1/0 | 58 (53.2%) | 51 (46.8%) | 73 (44.0%) | 93 (56.0%) | 0.134 a | |||
| ICC | Controls | p-Value * | OR (CI) | ||
|---|---|---|---|---|---|
| N (%) | N (%) | ||||
| GSTT1 | 20 | 29 | 0.003 | 2.759 (1.394–5.459) | |
| 00 | (37.0%) | (17.6%) | |||
| 34 | 136 | ||||
| 01&11 | (63.0%) | (82.4%) | |||
| HSIL and ICC | Controls | p-Value * | OR (CI) | ||
| N (%) | N (%) | ||||
| GSTT1 | 24 | 29 | 0.003 | 2.759 (1.394–5.459) | |
| 00 | (37.5%) | (17.6%) | |||
| 40 | 136 | ||||
| 01&11 | (62.5%) | (82.4%) | |||
| Gene | LSIL N (%) | HSIL N (%) | ICC N (%) | p-Value | |
|---|---|---|---|---|---|
| −/− | 7 (87.5) | 2 (100.0) | 20 (95.2) | ||
| CβS | +/− | 0 (0.0) | 0 (0.0) | 1 (4.8) | 0.548 a |
| +/+ | 1 (12.5) | 0 (0.0) | 0 (0.0) | ||
| CC | 16 (55.2) | 3 (27.3) | 28 (41.2) | ||
| MTHFR | CT | 8 (27.6) | 7 (63.6) | 34 (50.0) | 0.182 a |
| TT | 5 (17.2) | 1 (9.1) | 6 (8.8) | ||
| 4a4a | 0 (0.0) | 1 (12.5) | 1 (0.0) | ||
| NOS3 | 4a4b | 4 (28.6) | 1 (12.5) | 12 (24.0) | 0.072 b |
| 4b4b | 10 (71.4) | 6 (75.0) | 38 (76.0) | ||
| DD | 20 (62.5) | 11 (68.8) | 31 (63.9) | ||
| ACE1 | ID | 11 (34.4) | 4 (25.0) | 11 (22.4) | 0.424 a |
| II | 1 (3.1) | 1 (6.3) | 7 (14.3) | ||
| CC | 7 (38.9) | 10 (71.4) | 18 (50.0) | ||
| CYBA | CT | 11 (61.1) | 3 (21.4) | 15 (41.7) | 0.190 a |
| TT | 0 (0.0) | 1 (7.1) | 3 (8.3) | ||
| 1.1 | 6 (20.7) | 6 (25.0) | 32 (25.0) | ||
| HAP | 2.1 | 11 (37.9) | 4 (16.7) | 55 (43.0) | 0.101 b |
| 2.2 | 12 (41.4) | 14 (58.3) | 41 (32.0) | ||
| AA | 0 (0.0) | 0 (0.0) | 1 (3.6) | ||
| AB | 10 (52.6) | 3 (33.3) | 7 (25.0) | ||
| ACP1 | AC | 0 (0.0) | 0 (0.0) | 3 (10.7) | 0.406 a |
| BB | 9 (47.4) | 5 (55.6) | 15 (53.6) | ||
| BC | 0 (0.0) | 1 (11.1) | 2 (7.1) | ||
| CYP1A1 T5639C | TT | 11 (64.7) | 7 (87.5) | 21 (70.0) | 0.551 a |
| TC | 6 (35.3) | 1 (12.5) | 9 (29.1) | ||
| CYP1A1 A4889G | AA | 14 (87.5) | 7 (87.5) | 19 (86.4) | 1.000 a |
| AG | 2 (12.5) | 1 (12.5) | 3 (13.6) | ||
| CYP1A1 C4887A | CC | 12 (75.0) | 7 (100.0) | 17 (100.0) | 0.034 a |
| CA | 4 (25.0) | 0 (0.0) | 0 (0.0) | ||
| GSTT1 | 0/0 | 6 (27.3) | 4 (40.0) | 20 (37.0) | 0.034 a |
| 1/1&1/0 | 16 (72.7) | 6 (60.0) | 34 (63.0) | ||
| GSTM1 | 0/0 | 15 (55.6) | 10 (83.3) | 28 (46.7) | 0.675 b |
| 1/1&1/0 | 12 (44.4) | 2 (16.7) | 32 (53.3) |
| Genes | HPV | Controls | p-Value a | OR (CI) | |
|---|---|---|---|---|---|
| N (%) | N (%) | ||||
| ACP1-ACEI | BB-DD Others | 19 | 28 | 0.004 a | 2.643 (1.335–5.232) |
| (33.3%) | (15.9%) | ||||
| 38 | 148 | ||||
| (66.7%) | (84.1%) | ||||
| ACP1-GSTT1 | BB-0.0 | 9 | 4 | 0.004 b | 5.707 (1.665–19.565) |
| (18.0%) | (3.7%) | ||||
| Others | 41 | 104 | |||
| (82.0%) | (96.3%) | ||||
| HSIL and ICC | Controls | ||||
| ACP1-GSTT1 | BB-0.0 | 8 | 4 | <0.001 b | 9.455 (2.615–34.187) |
| (26.7%) | (3.7%) | ||||
| Others | 22 | 104 | |||
| (73.3%) | (96.3%) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inácio, Â.; Aguiar, L.; Rodrigues, B.; Pires, P.; Ferreira, J.; Matos, A.; Mendonça, I.; Rosa, R.; Bicho, M.; Medeiros, R.; et al. Genetic Modulation of HPV Infection and Cervical Lesions: Role of Oxidative Stress-Related Genes. Antioxidants 2023, 12, 1806. https://doi.org/10.3390/antiox12101806
Inácio Â, Aguiar L, Rodrigues B, Pires P, Ferreira J, Matos A, Mendonça I, Rosa R, Bicho M, Medeiros R, et al. Genetic Modulation of HPV Infection and Cervical Lesions: Role of Oxidative Stress-Related Genes. Antioxidants. 2023; 12(10):1806. https://doi.org/10.3390/antiox12101806
Chicago/Turabian StyleInácio, Ângela, Laura Aguiar, Beatriz Rodrigues, Patrícia Pires, Joana Ferreira, Andreia Matos, Inês Mendonça, Raquel Rosa, Manuel Bicho, Rui Medeiros, and et al. 2023. "Genetic Modulation of HPV Infection and Cervical Lesions: Role of Oxidative Stress-Related Genes" Antioxidants 12, no. 10: 1806. https://doi.org/10.3390/antiox12101806
APA StyleInácio, Â., Aguiar, L., Rodrigues, B., Pires, P., Ferreira, J., Matos, A., Mendonça, I., Rosa, R., Bicho, M., Medeiros, R., & Bicho, M. C. (2023). Genetic Modulation of HPV Infection and Cervical Lesions: Role of Oxidative Stress-Related Genes. Antioxidants, 12(10), 1806. https://doi.org/10.3390/antiox12101806