
Monocarboxylate Transporter-1 (MCT1)-Mediated Lactate Uptake Protects Pancreatic Adenocarcinoma Cells from Oxidative Stress during Glutamine Scarcity Thereby Promoting Resistance against Inhibitors of Glutamine Metabolism

 , and
, and 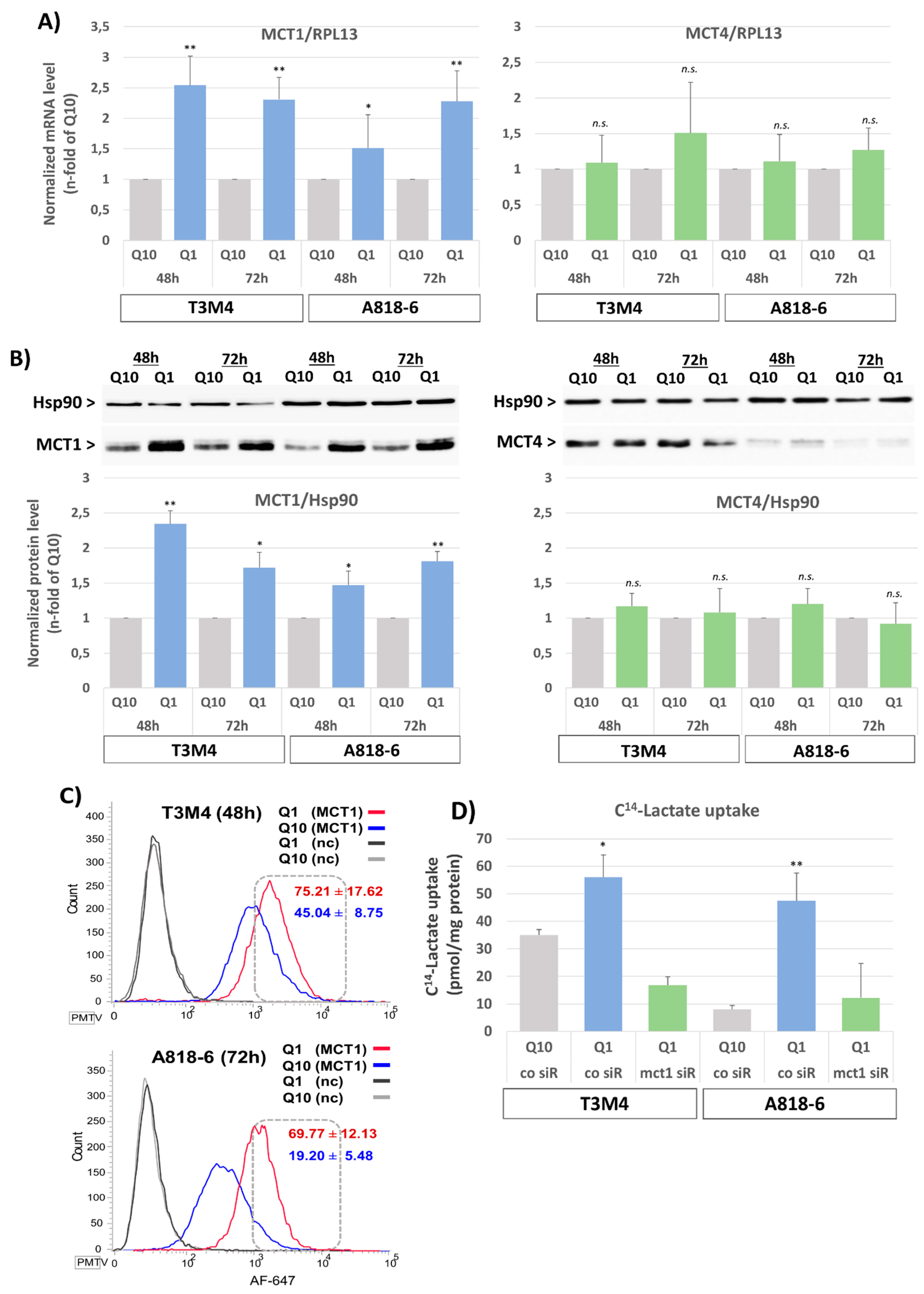{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. RNA Preparation and Real-Time PCR
2.3. Western Blotting
2.4. siRNA Transfection
2.5. MCT1 Surface Immunostaining and Flow Cytometry
2.6. Lactate Uptake Assay
2.7. Propidium Iodide Staining
2.8. Glutathione (GSH) Colorimetric Assay
2.9. ROS Measurement/DCFDA Staining
2.10. Luciferase Assay
2.11. Caspase-3/7 and MTS Assay
2.12. Patients and Tissues
2.13. Immunohistochemical Staining
2.14. Statistical Analysis
3. Results
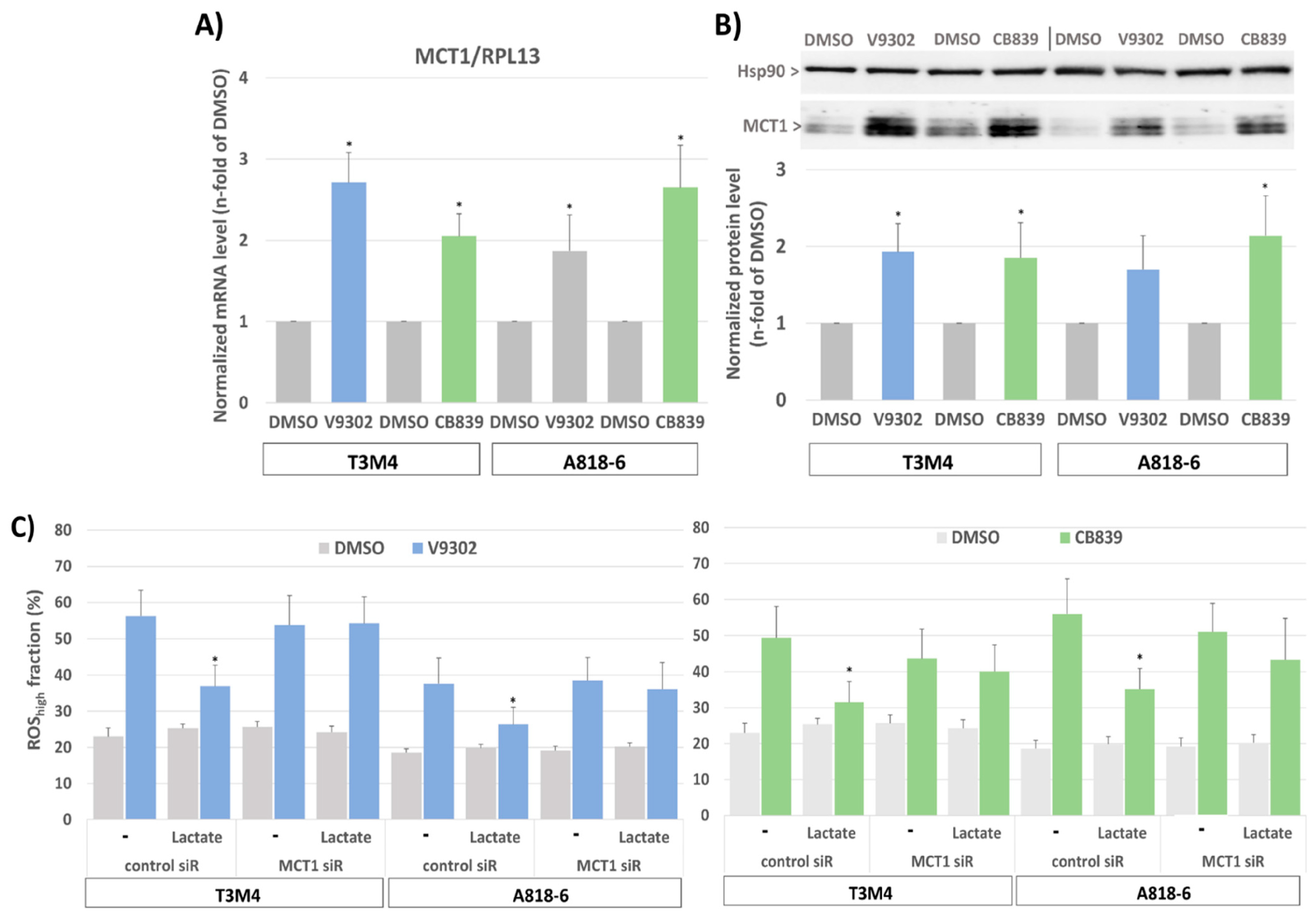
3.1. Upregulation of MCT1 Expression and Lactate Import by Glutamine Depletion in PDAC Cell Lines
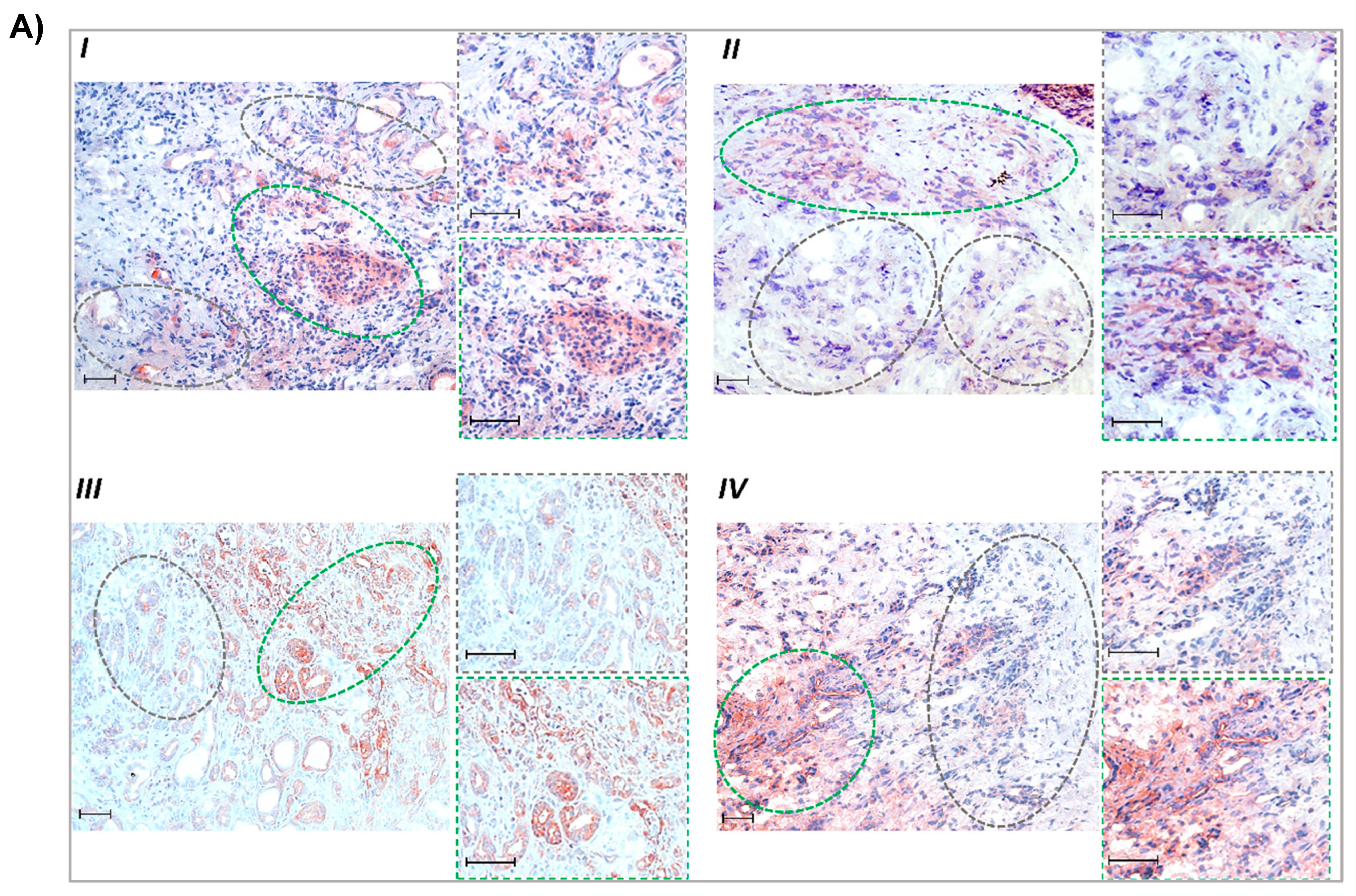
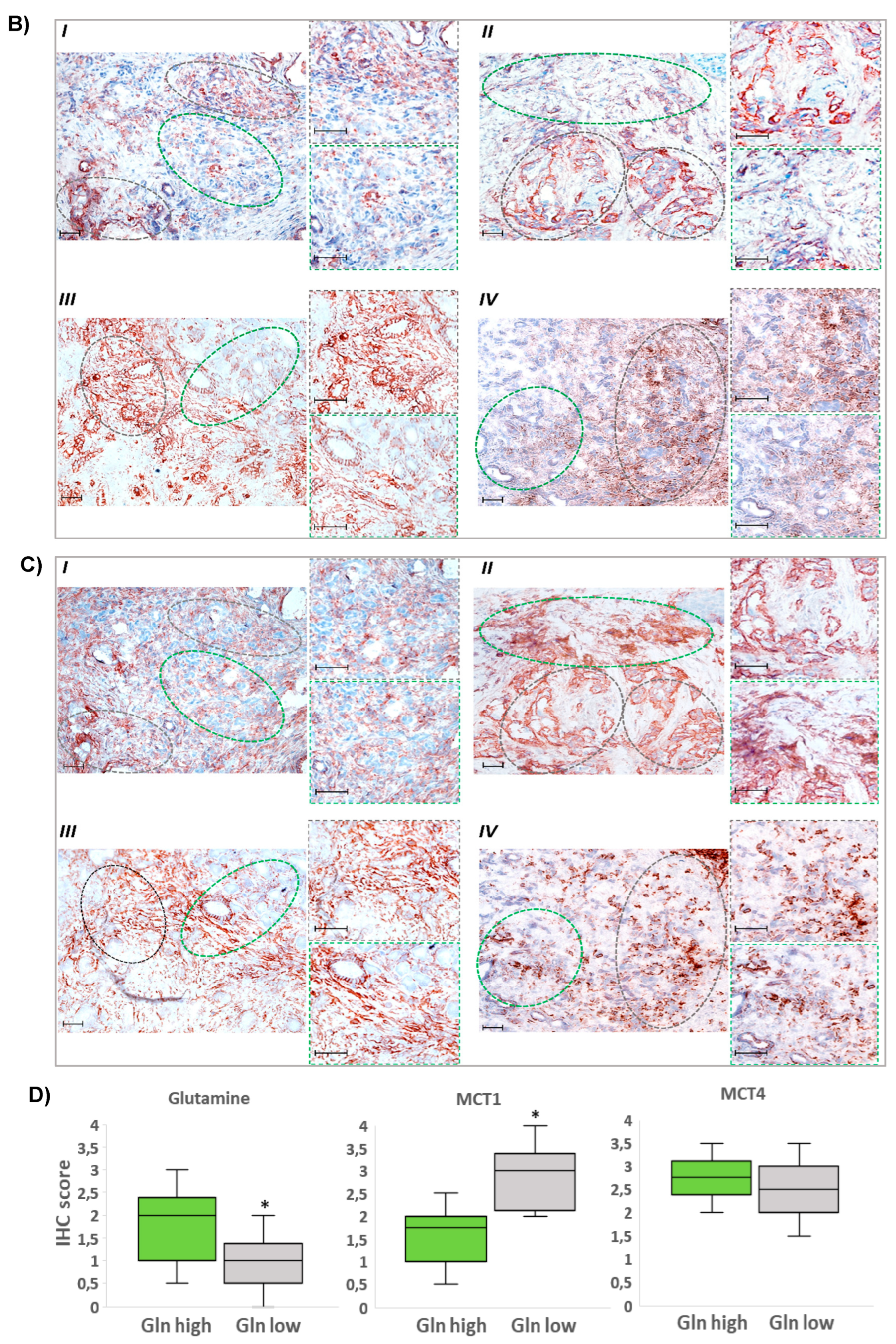
3.2. Glutamine-Scarce Regions in Human PDAC Tissue Exhibit Greater MCT1 Expression
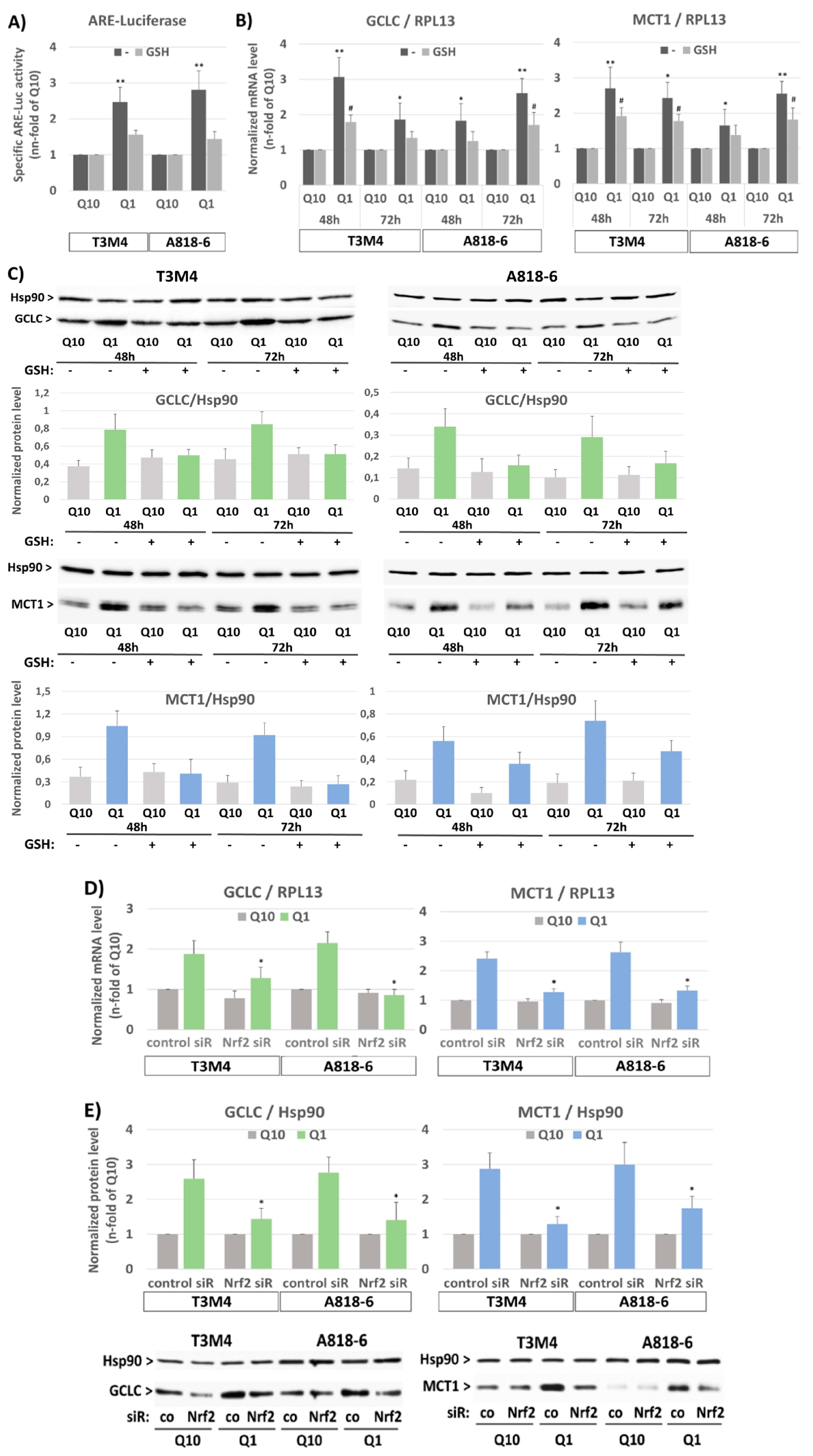
3.3. The Inducing Effect of Glutamine Depletion on MCT1 Expression in T3M4 and A818-6 Cells Depends on Oxidative Stress and Nrf2 Activation
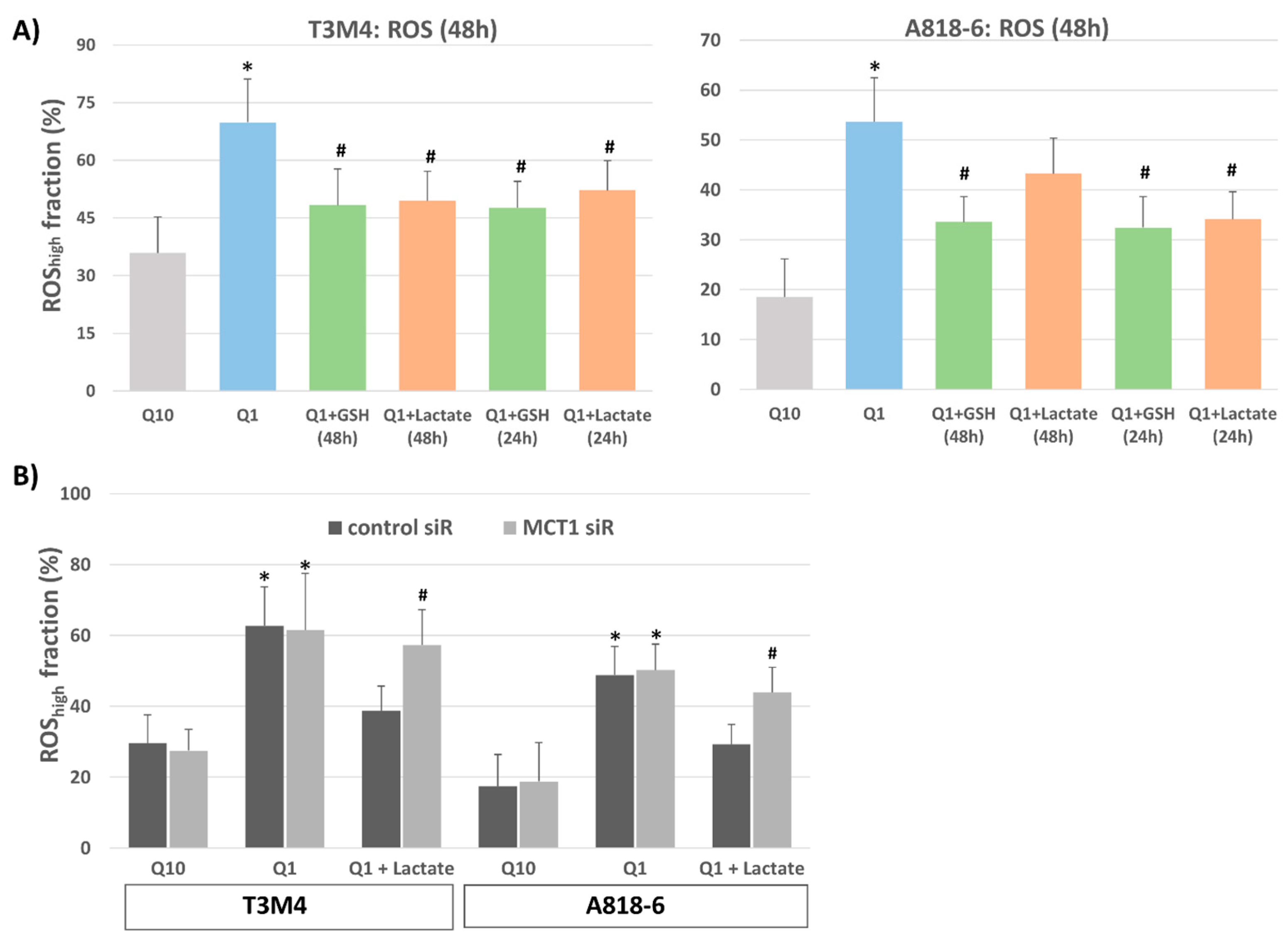
3.4. Lactate Protects T3M4 and A818-6 Cells from Glutamine-Depletion-Induced ROS Stress Depending on MCT1 Expression
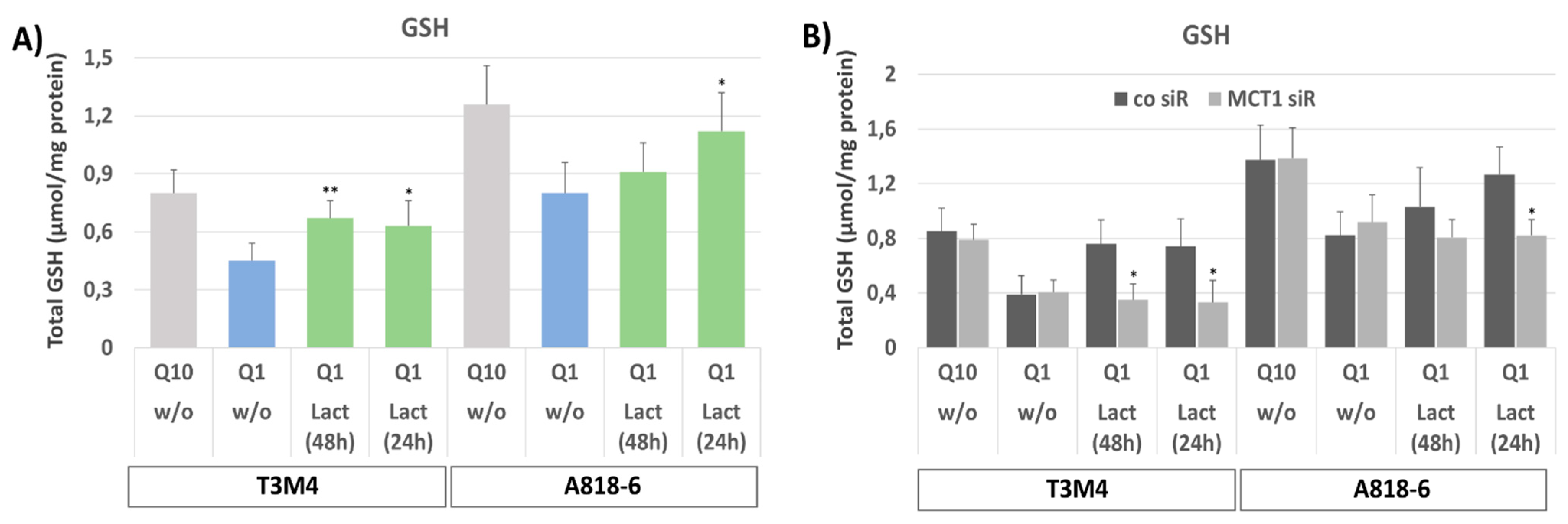
3.5. MCT1-Driven Lactate Uptake Reconstitutes GSH Content in Glutamine-Starved PDAC Cells
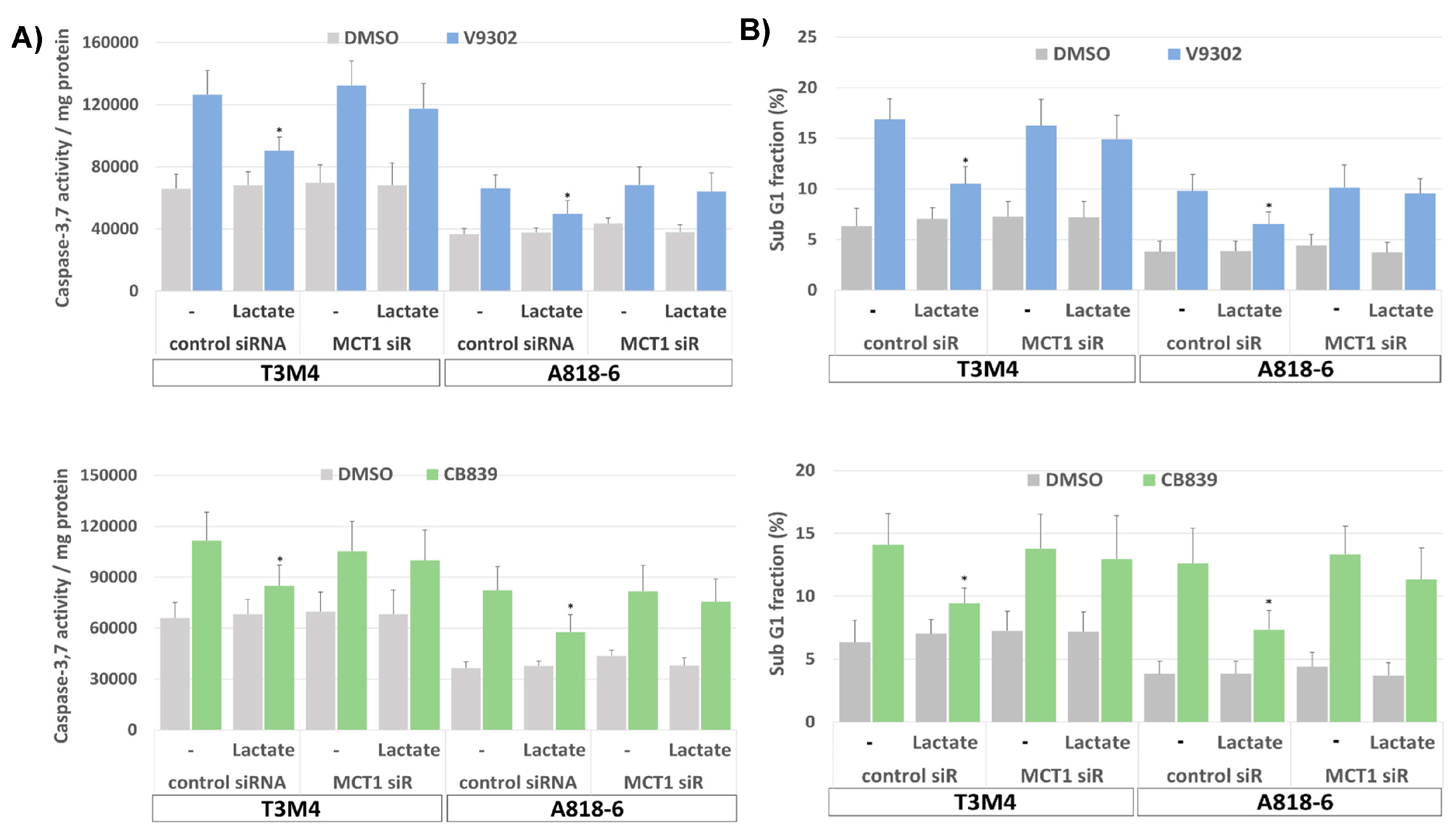
3.6. Lactate Protects T3M4 and A818-6 Cells from Glutamine Depletion-Induced Cell Death Depending on MCT1 Expression
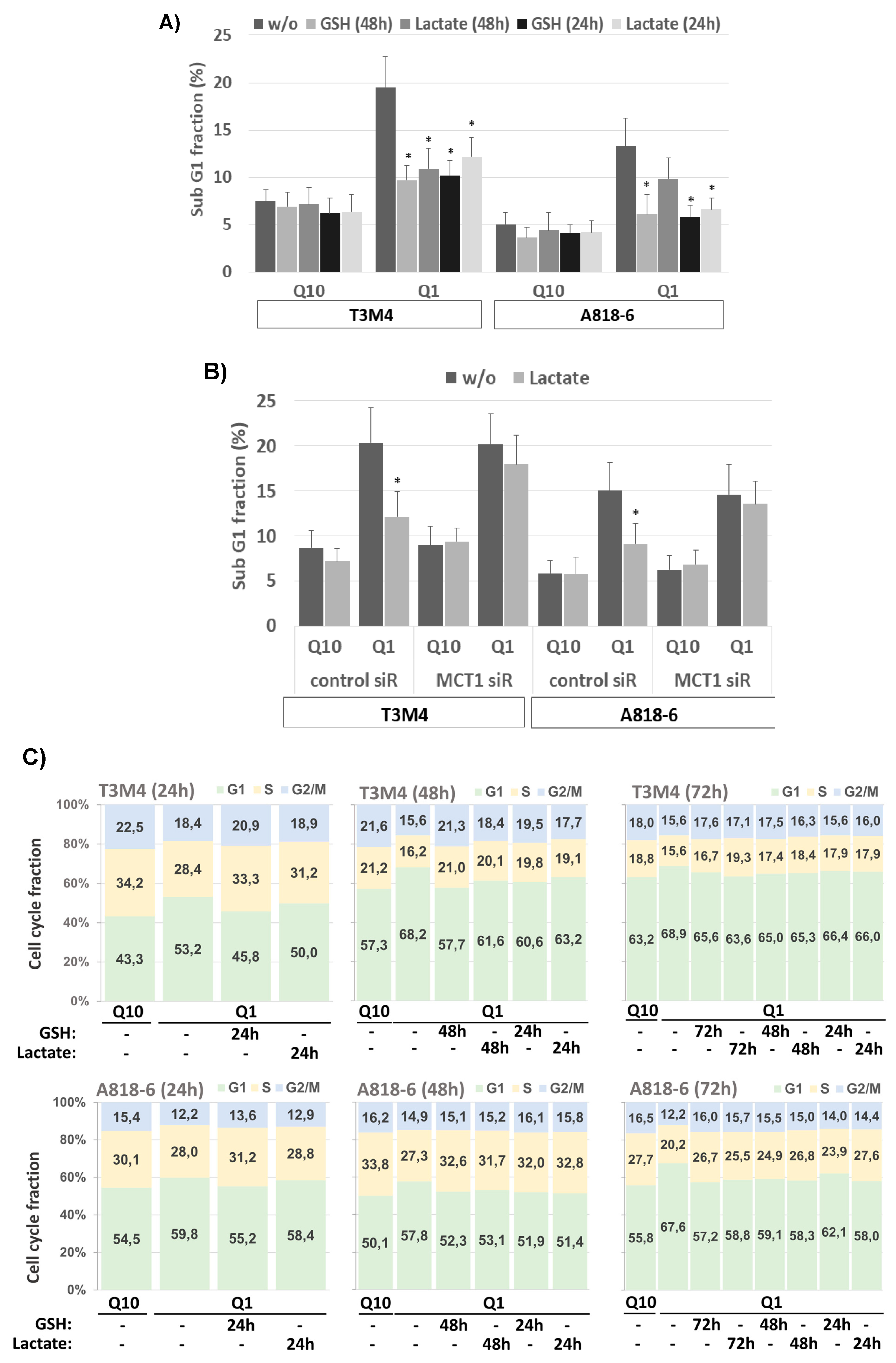
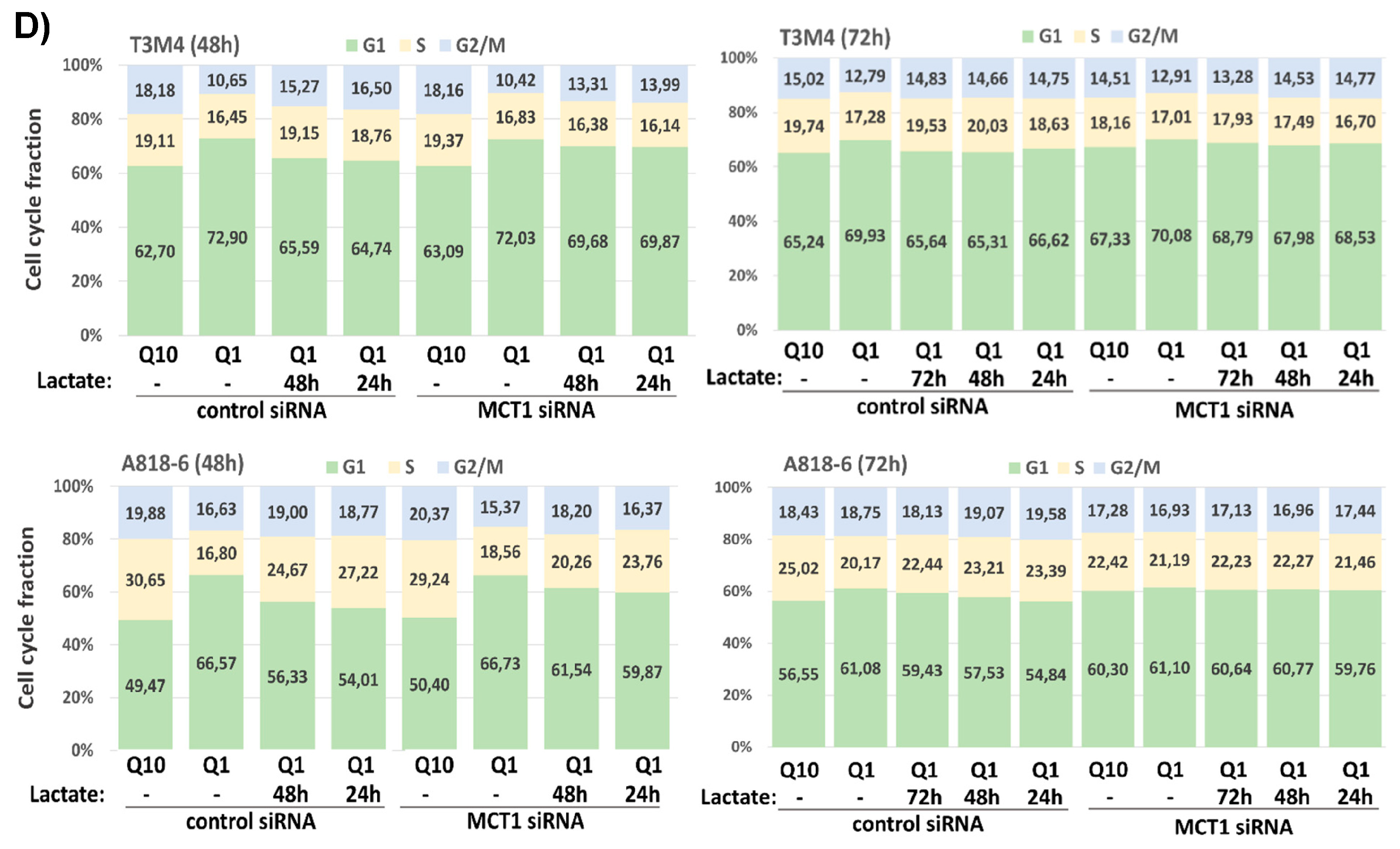
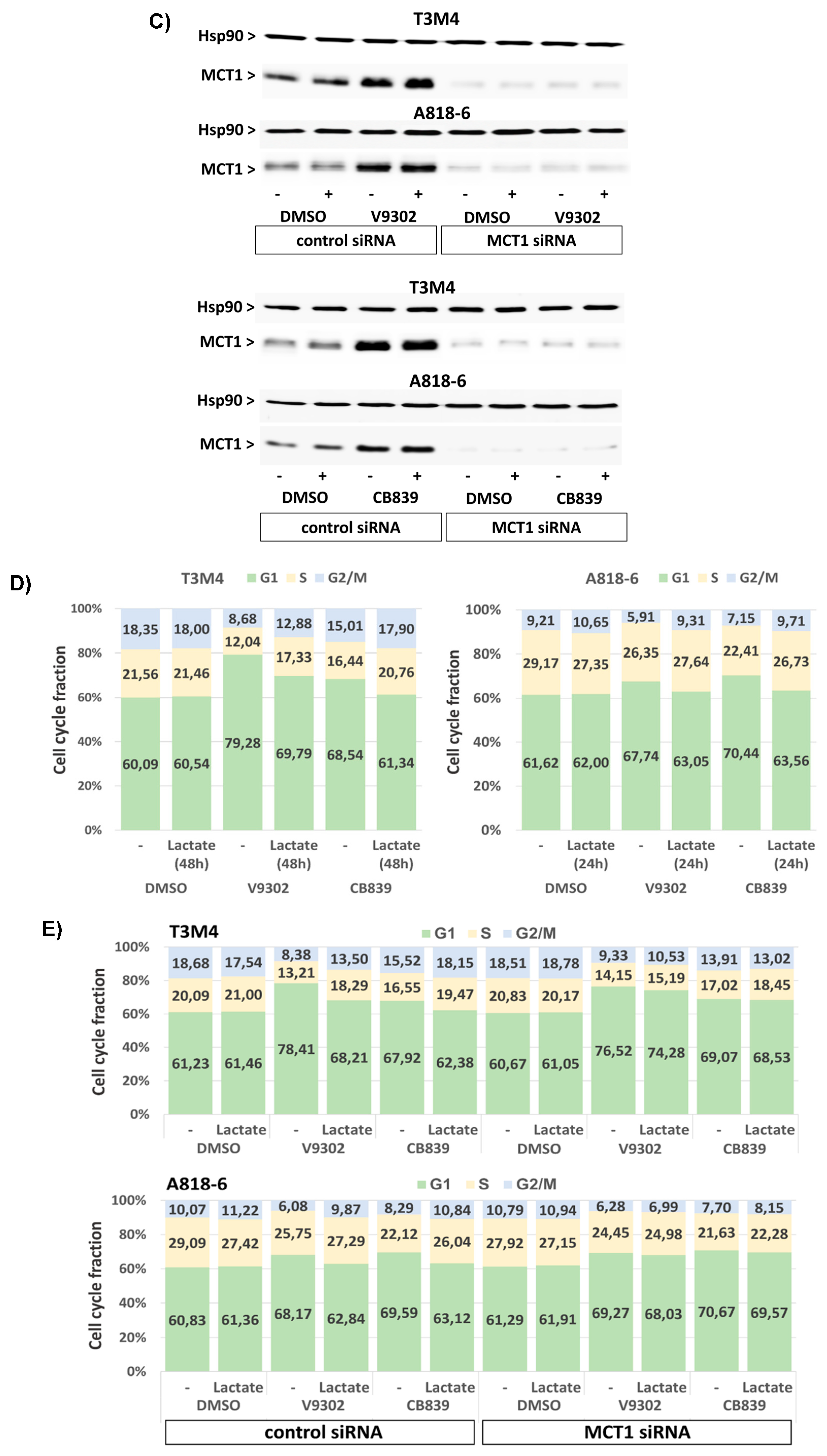
3.7. Lactate Protects T3M4 and A818-6 Cells from Glutamine Depletion-Induced Cell Cycle Arrest Depending on MCT1 Expression
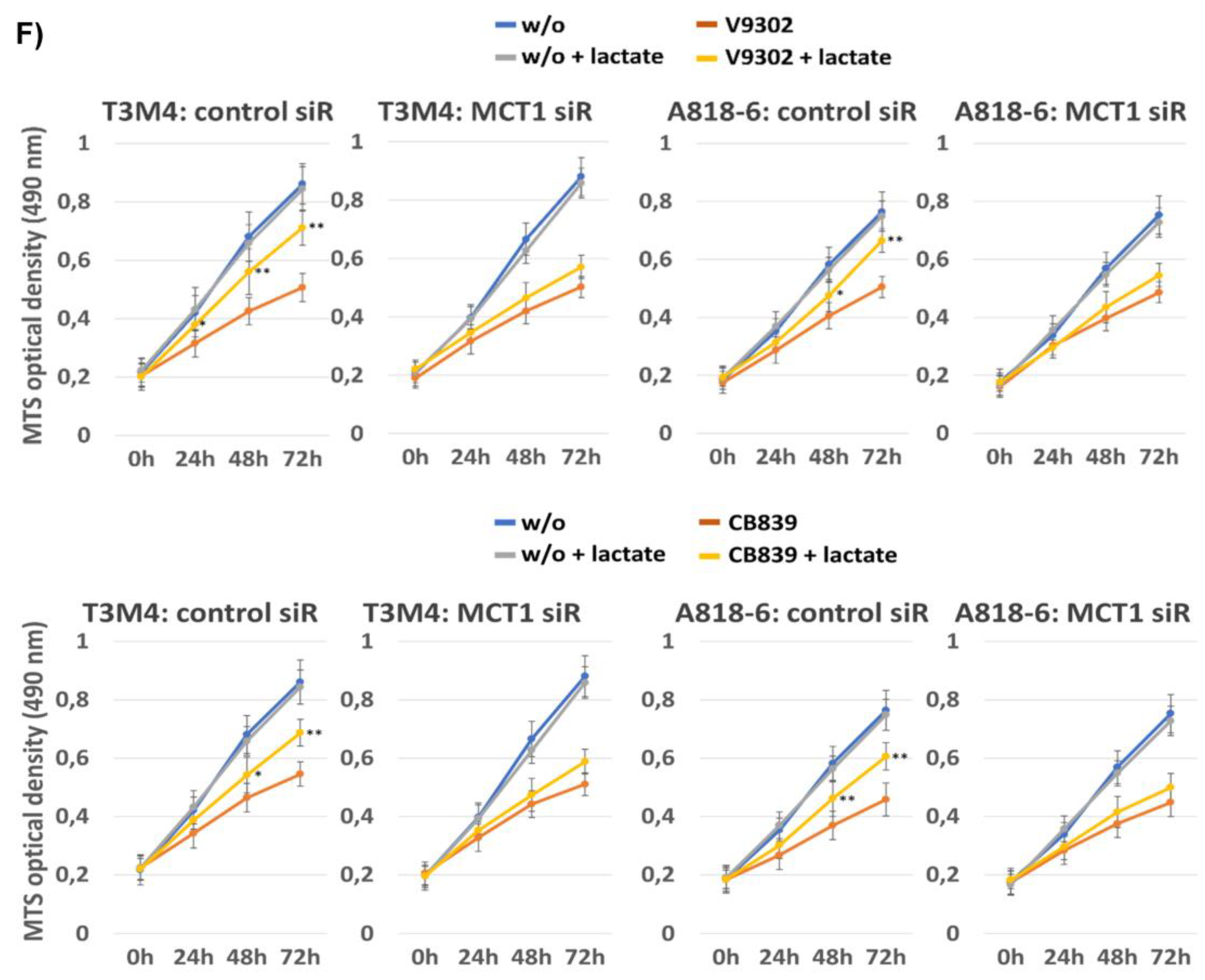
3.8. MCT1-Driven Lactate Uptake Favors Resistance of PDAC Cells against ASCT2 and GLS Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Gao, W.Q.; Liu, Y. Metabolic heterogeneity in cancer: An overview and therapeutic implications. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188421. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Vander Linden, C.; Corbet, C. Reconciling environment-mediated metabolic heterogeneity with the oncogene-driven cancer paradigm in precision oncology. Semin. Cell Dev. Biol. 2020, 98, 202–210. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707.e6. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. In Advances in Experimental Medicine and Biology; Le, A., Ed.; The Heterogeneity of Cancer Metabolism; Springer: Cham, Switzerland, 2018; Volume 1063, pp. 13–32. [Google Scholar]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Kloog, Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014, 5, e1065. [Google Scholar] [CrossRef] [PubMed]
- Still, E.R.; Yuneva, M.O. Hopefully devoted to Q: Targeting glutamine addiction in cancer. Br. J. Cancer 2017, 116, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. Embo J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef]
- Amores-Sanchez, M.I.; Medina, M.A. Glutamine, as a precursor of glutathione, and oxidative stress. Mol. Genet. Metab. 1999, 67, 100–105. [Google Scholar] [CrossRef]
- Vučetić, M.; Cormerais, Y.; Parks, S.K.; Pouysségur, J. The Central Role of Amino Acids in Cancer Redox Homeostasis: Vulnerability Points of the Cancer Redox Code. Front. Oncol. 2017, 7, 319. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef]
- Nachef, M.; Ali, A.K.; Almutairi, S.M.; Lee, S.H. Targeting SLC1A5 and SLC3A2/SLC7A5 as a Potential Strategy to Strengthen Anti-Tumor Immunity in the Tumor Microenvironment. Front. Immunol. 2021, 12, 624324. [Google Scholar] [CrossRef]
- Méndez-Lucas, A.; Lin, W.; Driscoll, P.C.; Legrave, N.; Novellasdemunt, L.; Xie, C.; Charles, M.; Wilson, Z.; Jones, N.P.; Rayport, S.; et al. Identifying strategies to target the metabolic flexibility of tumours. Nat. Metab. 2020, 2, 335–350. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Sanità, P.; Capulli, M.; Teti, A.; Galatioto, G.P.; Vicentini, C.; Chiarugi, P.; Bologna, M.; Angelucci, A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer 2014, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Moir, J.A.; Mann, J.; White, S.A. The role of pancreatic stellate cells in pancreatic cancer. Surg. Oncol. 2015, 24, 232–238. [Google Scholar] [CrossRef]
- Jang, G.; Oh, J.; Jun, E.; Lee, J.; Kwon, J.Y.; Kim, J.; Lee, S.H.; Kim, S.C.; Cho, S.Y.; Lee, C. Direct cell-to-cell transfer in stressed tumor microenvironment aggravates tumorigenic or metastatic potential in pancreatic cancer. npj Genom. Med. 2022, 7, 63. [Google Scholar] [CrossRef]
- Curry, J.M.; Tuluc, M.; Whitaker-Menezes, D.; Ames, J.A.; Anantharaman, A.; Butera, A.; Leiby, B.; Cognetti, D.M.; Sotgia, F.; Lisanti, M.P.; et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle 2013, 12, 1371–1384. [Google Scholar] [CrossRef]
- Latif, A.; Chadwick, A.L.; Kitson, S.J.; Gregson, H.J.; Sivalingam, V.N.; Bolton, J.; McVey, R.J.; Roberts, S.A.; Marshall, K.M.; Williams, K.J.; et al. Monocarboxylate Transporter 1 (MCT1) is an independent prognostic biomarker in endometrial cancer. BMC Clin. Pathol. 2017, 17, 27. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Johnson, J.M.; Cotzia, P.; Fratamico, R.; Mikkilineni, L.; Chen, J.; Colombo, D.; Mollaee, M.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Lin, Z.; et al. MCT1 in Invasive Ductal Carcinoma: Monocarboxylate Metabolism and Aggressive Breast Cancer. Front. Cell Dev. Biol. 2017, 5, 27. [Google Scholar] [CrossRef]
- Kubelt, C.; Peters, S.; Ahmeti, H.; Huhndorf, M.; Huber, L.; Cohrs, G.; Hövener, J.B.; Jansen, O.; Synowitz, M.; Held-Feindt, J. Intratumoral Distribution of Lactate and the Monocarboxylate Transporters 1 and 4 in Human Glioblastoma Multiforme and Their Relationships to Tumor Progression-Associated Markers. Int. J. Mol. Sci. 2020, 21, 6254. [Google Scholar] [CrossRef] [PubMed]
- Pértega-Gomes, N.; Vizcaíno, J.R.; Attig, J.; Jurmeister, S.; Lopes, C.; Baltazar, F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 2014, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, Y.; Dong, D.; Wang, F.; Ma, X.; Guan, F.; Sun, L. MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. J. Cancer 2018, 9, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Whitaker-Menezes, D.; Dasgupta, A.; Philp, N.J.; Lin, Z.; Gandara, R.; Sneddon, S.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: Stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle 2012, 11, 1108–1117. [Google Scholar] [CrossRef]
- Parida, P.K.; Marquez-Palencia, M.; Nair, V.; Kaushik, A.K.; Kim, K.; Sudderth, J.; Quesada-Diaz, E.; Cajigas, A.; Vemireddy, V.; Gonzalez-Ericsson, P.I.; et al. Metabolic diversity within breast cancer brain-tropic cells determines metastatic fitness. Cell Metab. 2022, 34, 90–105.e7. [Google Scholar] [CrossRef]
- Weng, Y.S.; Tseng, H.Y.; Chen, Y.A.; Shen, P.C.; Al Haq, A.T.; Chen, L.M.; Tung, Y.C.; Hsu, H.L. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer 2019, 18, 42. [Google Scholar] [CrossRef]
- Sandforth, L.; Ammar, N.; Dinges, L.A.; Röcken, C.; Arlt, A.; Sebens, S.; Schäfer, H. Impact of the Monocarboxylate Transporter-1 (MCT1)-Mediated Cellular Import of Lactate on Stemness Properties of Human Pancreatic Adenocarcinoma Cells. Cancers 2020, 12, 581. [Google Scholar] [CrossRef]
- Lagarde, D.; Jeanson, Y.; Portais, J.-C.; Galinier, A.; Ader, I.; Casteilla, L.; Carrière, A. Lactate Fluxes and Plasticity of Adipose Tissues: A Redox Perspective. Front. Physiol. 2021, 12, 689747. [Google Scholar] [CrossRef]
- Molina, J.; Morlacchi, P.; Silva, L.; Dennison, J.; Mills, G. Extracellular lactate cooperates with limited glucose and glutamine to sustain breast cancer cell survival by providing ATP, NADPH, amino acids, and glutathione. Cancer Metab. 2014, 2 (Suppl. S1), P49. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. eLife 2019, 8, e50663. [Google Scholar] [CrossRef]
- Halford, S.; Veal, G.J.; Wedge, S.R.; Payne, G.S.; Bacon, C.M.; Sloan, P.; Dragoni, I.; Heinzmann, K.; Potter, S.; Salisbury, B.M.; et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2023, 29, 1429–1439. [Google Scholar] [CrossRef]
- Winterhoff, B.J.; Arlt, A.; Duttmann, A.; Ungefroren, H.; Schäfer, H.; Kalthoff, H.; Kruse, M.L. Characterisation of FAP-1 expression and CD95 mediated apoptosis in the A818-6 pancreatic adenocarcinoma differentiation system. Differentiation 2012, 83, 148–157. [Google Scholar] [CrossRef]
- Sebens Müerköster, S.; Werbing, V.; Koch, D.; Sipos, B.; Ammerpohl, O.; Kalthoff, H.; Tsao, M.S.; Fölsch, U.R.; Schäfer, H. Role of myofibroblasts on innate chemoresistance in pancreatic carcinoma-epigenetic down-regulation of caspases. Int. J. Cancer 2008, 123, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Müerköster, S.; Arlt, A.; Sipos, B.; Witt, M.; Großmann, M.; Klöppel, G.; Kalthoff, H.; Fölsch, U.R.; Schäfer, H. Increased expression of the E3-ubiquitin ligase receptor subunit ßTRCP1 relates to constitutive NF-kappaB activation and chemoresistance in pancreatic carcinoma cells. Cancer Res. 2005, 65, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Helm, O.; Mennrich, R.; Petrick, D.; Göbel, L.; Freitag-Wolf, S.; Roeder, C.; Kalthoff, H.; Röcken, C.; Sipos, B.; Kabelitz, D.; et al. Comparative characterization of stromal cells and ductal epithelium in chronic pancreatitis and pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e94357. [Google Scholar] [CrossRef] [PubMed]
- Diehl, K.; Dinges, L.A.; Helm, O.; Ammar, N.; Plundrich, D.; Arlt, A.; Röcken, C.; Sebens, S.; Schäfer, H. Nuclear factor E2 related factor-2 differentially impacts on MCT1 and MCT4 lactate carrier expression in colonic epithelial cells—A condition favouring metabolic symbiosis between colorectal cancer and stromal cells. Oncogene 2018, 37, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Chen, R.; Lai, L.A.; Sullivan, Y.; Wong, M.; Wang, L.; Riddell, J.; Jung, L.; Pillarisetty, V.G.; Brentnall, T.A.; Pan, S. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci. Rep. 2017, 7, 7950. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB839 in Triple- Negative Breast Cancer. Mol. Cancer Therap. 2014, 13, 890–901. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef]
- Lee, S.W.; Zhang, Y.; Jung, M.; Cruz, N.; Alas, B.; Commisso, C. EGFR-Pak Signaling Selectively Regulates Glutamine Deprivation-Induced Macropinocytosis. Dev. Cell 2019, 50, 381–392.e5. [Google Scholar] [CrossRef] [PubMed]
- Biancur, D.E.; Paulo, J.A.; Małachowska, B.; Quiles Del Rey, M.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Lu, C.; Mancuso, A.; Lemons, J.M.; Ryczko, M.; Dennis, J.W.; Rabinowitz, J.D.; Coller, H.A.; Thompson, C.B. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010, 24, 2784–2799. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.J.; Guo, L.Y.; Li, P.; Zhao, Z.; Zhou, H.; Di, L.J. Molecular link between glucose and glutamine consumption in cancer cells mediated by CtBP and SIRT4. Oncogenesis 2018, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef]
- Ying, M.; You, D.; Zhu, X.; Cai, L.; Zeng, S.; Hu, X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021, 46, 102065. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Tompkins, S.C.; Sheldon, R.D.; Rauckhorst, A.J.; Noterman, M.F.; Solst, S.R.; Buchanan, J.L.; Mapuskar, K.A.; Pewa, A.D.; Gray, L.R.; Oonthonpan, L.; et al. Disrupting Mitochondrial Pyruvate Uptake Directs Glutamine into the TCA Cycle away from Glutathione Synthesis and Impairs Hepatocellular Tumorigenesis. Cell Rep. 2019, 28, 2608–2619.e6. [Google Scholar] [CrossRef]
- Inigo, M.; Deja, S.; Burgess, S.C. Ins and Outs of the TCA Cycle: The Central Role of Anaplerosis. Annu. Rev. Nutr. 2021, 41, 19–47. [Google Scholar] [CrossRef]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Bousamra, M.; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non–small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Bonglack, E.N.; Messinger, J.E.; Cable, J.M.; Ch’ng, J.; Parnell, K.M.; Reinoso-Vizcaíno, N.M.; Barry, A.P.; Russell, V.S.; Dave, S.S.; Christofk, H.R.; et al. Monocarboxylate transporter antagonism reveals metabolic vulnerabilities of viral-driven lymphomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2022495118. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.C.; Dechaume, A.L.; Murray, P.M.; Singleton, D.C.; Dechaume, A.L.; Murray, P.M.; Katt, W.P.; Baguley, B.C.; Leung, E.Y. Pyruvate anaplerosis is a mechanism of resistance to pharmacological glutaminase inhibition in triple-receptor negative breast cancer. BMC Cancer 2020, 20, 470. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Matés, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef] [PubMed]
- Salloum, G.; Bresnick, A.R.; Backer, J.M. Macropinocytosis: Mechanisms and regulation. Biochem. J. 2023, 480, 335–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Recouvreux, M.V.; Jung, M.; Galenkamp, K.M.O.; Li, Y.; Zagnitko, O.; Scott, D.A.; Lowy, A.M.; Commisso, C. Macropinocytosis in Cancer-Associated Fibroblasts Is Dependent on CaMKK2/ARHGEF2 Signaling and Functions to Support Tumor and Stromal Cell Fitness. Cancer Discov. 2021, 11, 1808–1825. [Google Scholar] [CrossRef]
- Occhiuto, C.J.; Moerland, J.A.; Leal, A.S.; Gallo, K.A.; Liby, K.T. The Multi-Faceted Consequences of NRF2 Activation throughout Carcinogenesis. Mol. Cells 2023, 46, 176–186. [Google Scholar] [CrossRef]
- Su, H.; Yang, F.; Fu, R.; Li, X.; French, R.; Mose, E.; Pu, X.; Trinh, B.; Kumar, A.; Liu, J.; et al. Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell 2021, 39, 678–693.e11. [Google Scholar] [CrossRef]
- Mendes, C.; Serpa, J. Revisiting lactate dynamics in cancer-a metabolic expertise or an alternative attempt to survive? J. Mol. Med. 2020, 98, 1397–1414. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.G.; Cai, B.; Cao, F.; Huang, Q.X.; Mi, P.; Wang, H.P.; Liu, W.H.; Wang, B.G. MCT1 promotes tumor progression through regulating epithelial-mesenchymal transition in pancreatic cancer. J. Clin. Exp. Pathol. 2017, 10, 3243–3250. [Google Scholar]
- Yu, S.; Wu, Y.; Li, C.; Qu, Z.; Lou, G.; Guo, X.; Ji, J.; Li, N.; Guo, M.; Zhang, M.; et al. Comprehensive analysis of the SLC16A gene family in pancreatic cancer via integrated bioinformatics. Sci. Rep. 2020, 10, 7315. [Google Scholar] [CrossRef]
- Rao, Y.; Gammon, S.; Zacharias, N.M.; Liu, T.; Salzillo, T.; Xi, Y.; Wang, J.; Bhattacharya, P.; Piwnica-Worms, D. Hyperpolarized [1-13C]-pyruvate-to-[1-13C]lactate conversion is rate-limited by monocarboxylate transporter-1 in the plasma membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 22378–22389. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Gao, Y.; Wu, H.; Liu, Y.; Yang, Y. Functional heterogeneity of MCT1 and MCT4 in metabolic reprogramming affects osteosarcoma growth and metastasis. J. Orthop. Surg. Res. 2023, 18, 131. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Puri, S.; Juvale, K. Monocarboxylate transporter 1 and 4 inhibitors as potential therapeutics for treating solid tumours: A review with structure-activity relationship insights. Eur. J. Med. Chem. 2020, 199, 112393. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, L.; Chen, W.; Chen, Q.; Sun, J.; Wang, G. Novel strategies to improve tumour therapy by targeting the proteins MCT1, MCT4 and LAT1. Eur. J. Med. Chem. 2021, 226, 113806. [Google Scholar] [CrossRef]
- Singh, M.; Afonso, J.; Sharma, D.; Gupta, R.; Kumar, V.; Rani, R.; Baltazar, F.; Kumar, V. Targeting monocarboxylate transporters (MCTs) in cancer: How close are we to the clinics? Semin. Cancer Biol. 2023, 90, 1–14. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ammar, N.; Hildebrandt, M.; Geismann, C.; Röder, C.; Gemoll, T.; Sebens, S.; Trauzold, A.; Schäfer, H. Monocarboxylate Transporter-1 (MCT1)-Mediated Lactate Uptake Protects Pancreatic Adenocarcinoma Cells from Oxidative Stress during Glutamine Scarcity Thereby Promoting Resistance against Inhibitors of Glutamine Metabolism. Antioxidants 2023, 12, 1818. https://doi.org/10.3390/antiox12101818
Ammar N, Hildebrandt M, Geismann C, Röder C, Gemoll T, Sebens S, Trauzold A, Schäfer H. Monocarboxylate Transporter-1 (MCT1)-Mediated Lactate Uptake Protects Pancreatic Adenocarcinoma Cells from Oxidative Stress during Glutamine Scarcity Thereby Promoting Resistance against Inhibitors of Glutamine Metabolism. Antioxidants. 2023; 12(10):1818. https://doi.org/10.3390/antiox12101818
Chicago/Turabian StyleAmmar, Nourhane, Maya Hildebrandt, Claudia Geismann, Christian Röder, Timo Gemoll, Susanne Sebens, Ania Trauzold, and Heiner Schäfer. 2023. "Monocarboxylate Transporter-1 (MCT1)-Mediated Lactate Uptake Protects Pancreatic Adenocarcinoma Cells from Oxidative Stress during Glutamine Scarcity Thereby Promoting Resistance against Inhibitors of Glutamine Metabolism" Antioxidants 12, no. 10: 1818. https://doi.org/10.3390/antiox12101818
APA StyleAmmar, N., Hildebrandt, M., Geismann, C., Röder, C., Gemoll, T., Sebens, S., Trauzold, A., & Schäfer, H. (2023). Monocarboxylate Transporter-1 (MCT1)-Mediated Lactate Uptake Protects Pancreatic Adenocarcinoma Cells from Oxidative Stress during Glutamine Scarcity Thereby Promoting Resistance against Inhibitors of Glutamine Metabolism. Antioxidants, 12(10), 1818. https://doi.org/10.3390/antiox12101818