1. Introduction
Melasma, a common facial skin pigmentation disorder, poses ongoing challenges in unravelling its complex pathogenesis. While genetic background and female sex hormones are recognized contributors, chronic exposure to ultraviolet (UV) radiation is considered a key trigger [
1]. UV exposure generates reactive oxygen species (ROS), resulting in oxidative stress when the body’s antioxidant defenses are overpowered [
2]. Studies have pointed to the role of oxidative stress in melasma development [
3,
4,
5,
6].
The impact of oxidative stress on ciliogenesis disruption has been explored in several reports [
7,
8], yet there has been no skin-specific investigations. The role of primary cilia in the negative regulation of melanogenesis was investigated in a study using cultured melanocytes [
9]. The factors leading to ciliogenesis inhibition and the precise mechanisms governing the interplay between primary cilia and skin pigmentation remain elusive. However, the evidence implicating oxidative stress in ciliogenesis disruption and the inhibitory role of primary cilia in melanogenesis suggest a compelling link between oxidative stress, ciliogenesis inhibition, and enhanced melanogenesis.
Primary cilia, antenna-like organelles projecting from the apical surfaces of most eukaryotic cells, serve as sensory organelles that receive and transduce environmental signals into coordinated cellular responses, influencing various cellular processes [
10]. Structural alterations in cilia, such as shortening and/or loss, can compromise their signaling capabilities, resulting in defects that contribute to developmental issues, degenerative diseases, and cancer progression. The core of the primary cilium contains an axoneme composed of a ring of nine microtubule doublets that extend from the basal body at the base of the cilium, derived from the mother centriole [
11]. Among the numerous genes involved in ciliogenesis and ciliary functions [
12], intraflagellar transport (IFT), a bi-directional transport system, plays a critical role in elongating the cilium axoneme during ciliary assembly [
13]. The hedgehog (Hh) pathway represents a central main mechanism for cilium-based signaling [
14,
15], requiring key proteins such as patched (PTCH), smoothened (SMO), and glioma-associated oncogene homologs (GLIs). The PTCH homolog, a transmembrane receptor for the secreted Sonic hedgehog (Shh) protein, triggers SMO homologs to release the suppressor of the fused homolog–zinc finger protein-GLI complex, enabling the GLIs’ nuclear translocation and activation of Hh target gene transcription [
16,
17,
18].
This study aimed to explore the potential link among oxidative stress, primary cilia, and skin hyperpigmentation in melasma. While it is important to acknowledge that approximately 10% of melasma cases occur in men, making gender-based comparisons challenging, the study primarily focused on understanding the inter-individual variations in melasma pathogenesis [
1]. To do so, we compared the expression levels of nuclear factor E2-related factor 2 (NRF2) and the number of primary cilia, along with the related molecule expression, including IFT88, GLIs, and PTCH homologs, between lesional and non-lesional skin specimens from the same melasma patients. Furthermore, we investigated the roles of NRF2, IFT88, and GLI1 in ciliogenesis and melanogenesis using primary cultured adult human keratinocytes and keratinocyte–melanocyte cocultures.
2. Materials and Methods
2.1. Patients
This study population comprised 16 female patients diagnosed with melasma, aged 29–62 years (mean age: 51.7 years). Ethical approval was obtained from the Institutional Review Board of the Dongguk University Ilsan Hospital, and the study adhered to the principles outlined in the Declaration of Helsinki. Written informed consent was acquired from each patient. Paired sets of hyperpigmented and adjacent normally pigmented skin specimens were retrieved through biopsy for direct comparisons. The specimens were subjected to real-time polymerase chain reaction (PCR) and immunohistochemistry.
2.2. Normal Human Epidermal Cell Culture
Human epidermal keratinocytes and melanocytes were sourced from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). The keratinocytes were suspended in an EpiLife medium (Thermo Fisher Scientific) supplemented with bovine pituitary extract (BPE, 0.2%), recombinant human insulin-like growth factor-1 (rhIGF-1, 0.01 ug/mL), hydrocortisone (0.18 ug/mL), human epidermal growth factor (0.2 ng/mL), and bovine transferrin (BT, 5 ug/mL) (Thermo Fisher Scientific). The melanocytes were suspended in Medium 254 (Thermo Fisher Scientific) supplemented with BPE (0.2%), fetal bovine serum (0.5%), rhIGF-1 (0.01 ug/mL), hydrocortisone (0.18 ug/mL), basic fibroblast growth factor (3 ng/mL), BT (5 ug/mL), heparin (3 ug/mL), and phorbol 12-myristate 13-acetate (10 ng/m) (Thermo Fisher Scientific). For the experiments, keratinocytes and melanocytes from passages 3–6 and 10–20 were utilized. For the coculture of keratinocytes and melanocytes, keratinocytes were seeded at 2 × 105 cells/well to six-well plates. Four hours later, 1 × 105 melanocytes/well in 2 mL of EpiLife media were added to the keratinocytes. After 24 h, the EpiLife medium was replaced with the supplement-free medium, and GANT61 (5–10 μM; Sigma Aldrich, St. Louis, MO, USA) or CaCl2 (1 mM) treatment was administered by adding these agents to the cell culture for 48 h.
2.3. H2O2 Treatment or UVB Radiation
Primary cultured normal human keratinocytes were seeded at 1.5 × 105 cells/well to six-well plates and incubated for 24 h. The cells in each six-well plate were subjected to different concentrations (50, 100, 200, and 500 mM) and durations (4, 24, and 48 h) of H2O2 or irradiated with 200 mJ of UVB either once or once daily for 3 consecutive days, employing a WL 20 W lamp emitting 305–314 nm with a peak of 311 nm (Royal Philips, Amsterdam, The Netherlands). Cell harvesting for NRF2 expression level evaluation was immediately conducted after the corresponding durations of H2O2 treatment or 2, 4, and 24 h after the final irradiation. The EpiLife medium was replaced with a supplement-free EpiLife medium or phosphate-buffered saline (PBS) during the H2O2 treatment or the irradiation, respectively.
2.4. Knockdown of IFT88, GLI1, and NRF2
Melanocytes and keratinocytes were seeded at 1.5 × 105 cells/well to six-well plates and incubated for 24 h. The cells were transfected with 25 nM CRISPR-CAS9 sgRNA targeting human IFT88, GLI1, NRF2, or negative control sgRNA (Integrated DNA Technologies, San Diego, CA, USA) using the CRISPRMAX transfection reagent (Thermo Fisher Scientific). The cells were used for experiments 48 h after transfection. For the coculture of keratinocytes and melanocytes, 24 h later, 1 × 105 melanocytes/well in 2 mL of EpiLife medium were added to transfected keratinocytes and incubated for another 24 h. Treatment with the SMO agonist (SAG; 1 µM; Sigma Aldrich) or Shh protein (100 nM; R&D Systems, Minneapolis, MN, USA) involved adding these substances to the transfected cells for 24 h after replacing the EpiLife medium with a supplement-free EpiLife medium. All collected cells were used for Western blot analysis, immunohistochemistry, confocal microscopy, tyrosinase activity assay, and melanin content assay.
2.5. Real-Time PCR Analysis
cDNA was synthesized from total RNA using a cDNA Synthesis Kit for RT-PCR (Promega, Fitchburg, WI, USA). The mRNA levels relative to GAPDH were measured using qPCR with a Light Cycler Real-Time PCR (Roche, Mannheim, Germany). The primer sequences used were as follows: IFT88 (NM 001353565) 5′-ATTGCCAATAGTTGTGGAGACTT-3′ (forward) and 5′-CTCGCTGTCTCACCAGGACT-3′ (reverse); PTCH1 (NM 001083605) 5′-TGGATGTCATGGCTTATCCAG-3′ (forward) and 5′-CATTAACTGGAACATGGTCTGC-3′ (reverse); GLI1 (NM_001160045) 5′-ATCAACTCGCGATGCACA-3′ (forward) and 5′-ATTCATCTGGGCTGGGAAT-3′ (reverse); GLI2 (NM_001374354) 5′-ACGGCACTGGATGACACAC-3′ (forward) and 5′-AGTGCTGGACACCTGGTTG-3′ (reverse); GLI3 (NM_000168) 5′-ACATGGAATATCTTCATGCTATGG-3′ (forward) and 5′-GGTGATATGGACAGTGTACGTTTT-3′ (reverse); and GAPDH (NM_001357943) 5′-TCCACTGGCGTCTTCACC-3′ (forward) and 5′-GCAGAGATGATGACCCTTT-3′ (reverse).
2.6. Western Blot Analysis
Equal amounts of extracted proteins (20 μg) were resolved via 8–12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. The membranes were probed with antibodies against various proteins, including IFT88 (rabbit polyclonal; Proteintech, Chicago, IL, USA), tyrosinase, PMEL17, GLI1, K14, K10, PAR2, heme oxygenase-1 (HO-1) (mouse monoclonal; Santa Cruz Biotechnology, Dallas, TX, USA), involucrin, PTCH1, GLI2 (goat; Santa Cruz Biotechnology), Shh (rabbit polyclonal; Cell Signaling Technology, Beverly, MA, USA), and NRF2 (rabbit polyclonal; Abcam, Cambridge, UK). The membranes were further incubated with anti-rabbit, anti-mouse, or anti-goat horseradish peroxidase-conjugated antibodies (Santa Cruz Biotechnology) and then treated with an enhanced chemiluminescence solution (Thermo, Rockford, IL, USA). Signals were captured using an image reader (LAS-3000; Fuji Photo Film, Tokyo, Japan). The membranes were re-probed with a mouse monoclonal anti-β-actin antibody (Sigma Aldrich) and processed as described above. The protein bands were analyzed via densitometry.
2.7. Immunohistochemistry and Confocal Microscopy
For the immunofluorescence staining of the biopsied skin specimens, the sections were pre-incubated with 3% bovine serum albumin after deparaffinization and rehydration. The sections were stained as follows: anti-NRF2 antibody (1:200 dilution), followed by Alexa-Fluor-labeled goat anti-rabbit IgG (1:200, 488; Molecular Probes, Eugene, OR, USA), anti-HO-1 antibodies (1:100) followed by Alexa-Fluor-labeled goat anti-mouse IgG (1:200, 594; Molecular Probes), or anti-involucrin antibodies (1:200) followed by Alexa-Fluor-labeled donkey anti-goat IgG (1:200, 594; Molecular Probes). For the staining of the cultured cells, the cells were fixed in 2% paraformaldehyde. The fixed cells were sequentially double-stained as follows: anti-NRF2 antibody followed by Alexa-Fluor-labeled goat anti-rabbit IgG and anti-ARL13 antibodies (1:200, mouse monoclonal: Santa Cruz Biotechnology), followed by Alexa-Fluor-labeled goat anti-mouse IgG and anti-acetylated-tubulin (1:500, mouse monoclonal; Sigma-Aldrich), followed by Alexa-Fluor-labeled goat anti-mouse IgG and anti-ARL13B antibodies (1:200, rabbit polyclonal; Proteintech), followed by Alexa-Fluor-labeled goat anti-rabbit IgG. The nuclei were counterstained with Hoechst 33258 (Sigma Aldrich). Fluorescence images were obtained and evaluated using an image analysis system (Dp Manager 2.1; Olympus Optical Co., Tokyo, Japan) and Wright Cell Imaging Facility ImageJ software version 1.54d (
https://imagej.net/ij/download.html, accessed on 26 March 2023). Confocal microscopy images were obtained using EZ-C1 3.8 software (Nikon, Tokyo, Japan) and evaluated using NIS-Elements AR 3.2 (Nikon).
2.8. ROS Assay
ROS were detected using the Total ROS Detection Kit (Enzo Life, Frmingdale, NY, USA) according to the manufacturer’s instructions. Keratinocytes were seeded at 1.5 × 105 cells/well to six-well plates and incubated for 24 h. An EpiLife medium was replaced with phosphate-buffered saline (PBS) during the irradiation. After irradiation with 200 mJ of UVB, the keratinocytes were incubated with a ROS detection solution for 1 h at 37 °C in the dark, according to the time schedule. The ROS levels were immediately measured using a fluorescence/multimode microplate reader (Spark; TECAN, Männedorf, Switzerland).
2.9. Tyrosinase Activity Assay
Tyrosinase activity was assayed in keratinocyte–melanocyte cocultures based on DOPA oxidase activity, using a modified version of the described method [
19,
20]. For coculture of keratinocytes and melanocytes, 1.5 × 10
5 keratinocytes/well were transfected with indicated genes for 24 h and 1 × 10
5 melanocytes/well were added to transfected keratinocytes and incubated for another 48 h. The cells were suspended and lysed in a phosphate buffer containing 1% Triton X-100. Cell-free extracts were obtained by centrifuging cell lysates at 10,000×
g for 10 min. The protein concentrations of the supernatants were measured with Bradford assays and adjusted using the lysis buffer. We placed 90 μL of each lysate in a well of a 96-well plate and added 10 μL of L-DOPA. After incubation at 37 °C, absorbance was measured every 10 min for at least 1 h at 475 nm using a fluorescence/multimode microplate reader (Spark).
2.10. Melanin Content Assay
The melanin content of the keratinocyte–melanocyte cocultures was determined with minor modifications to the described method [
21,
22]. For the coculture of keratinocytes and melanocytes, 1.5 × 10
5 keratinocytes/well were transfected with indicated genes for 24 h and 1 × 10
5 melanocytes/well were added to the transfected keratinocytes and incubated for another 48 h. Briefly, after being washed with PBS, cell pellets were dissolved and solubilized with 1 N of NaOH at 80 °C for 2 h. After centrifugation at 12,000×
g for 10 min, the absorbance of the supernatants was measured at a wavelength of 475 nm. To determine the actual melanin formation from the same number of cells, the total melanin content of each pellet was divided by the number of melanocytes.
2.11. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). The significance threshold was set at p < 0.05, with specific significance levels denoted as * p < 0.05, ** p < 0.01, and *** p < 0.001. Statistical comparisons between pairs of groups were performed using a two-tailed Student’s unpaired t-test (parametrical data). A one-way analysis of variance was used to compare multiple groups and parameters. Mean ± standard deviation (SD) values were calculated for the in vitro experimental data. For the human sample data, differences between the non-lesions and lesions were assessed using the Mann–Whitney U test and were expressed as the mean ± standard error of the mean.
4. Discussion
NRF2 is a central regulator of cellular antioxidant defenses. Under conditions of oxidative stress, NRF2 relocates to the cell nucleus and activates the expressions of a wide array of target genes, including HO-1 [
30]. The diminished levels of HO-1 proteins following
NRF2 knockdown (
Figure 1C) confirmed HO-1 as a primary target of NRF2 within keratinocytes. UV radiation, a known source of ROS [
2], elevated ROS levels and NRF2 protein expression in keratinocytes after a single UVB exposure (
Figure 1A,B). In response to H
2O
2-induced oxidative stress, the relative expressions of NRF2 and HO-1 in keratinocytes were increased (
Figure 1C), reflecting an antioxidant response aimed at combating oxidative stress. However, repeated UVB irradiation downregulated NRF2 (
Figure 1B), suggesting that oxidative stress was hardly overcome under repeated UVB irradiation. Given that chronic UV exposure is a significant contributor to melasma development [
1], it is reasonable to expect NRF2 and HO-1 downregulation in the lesional epidermis of melasma patients (
Figure 1E,F). Nevertheless, melasma development is also associated with factors unrelated to UV exposure [
1]. In addition to UV radiation, ROS can be generated by endogenous processes such as cellular metabolism and exogenous factors other than UV radiation [
31]. While the role of UV-independent melasma triggers in oxidative stress requires further investigation, the results from the H
2O
2 treatment suggested that decreased cellular antioxidant capacity may cause oxidative stress in melasma, regardless of its association with UV.
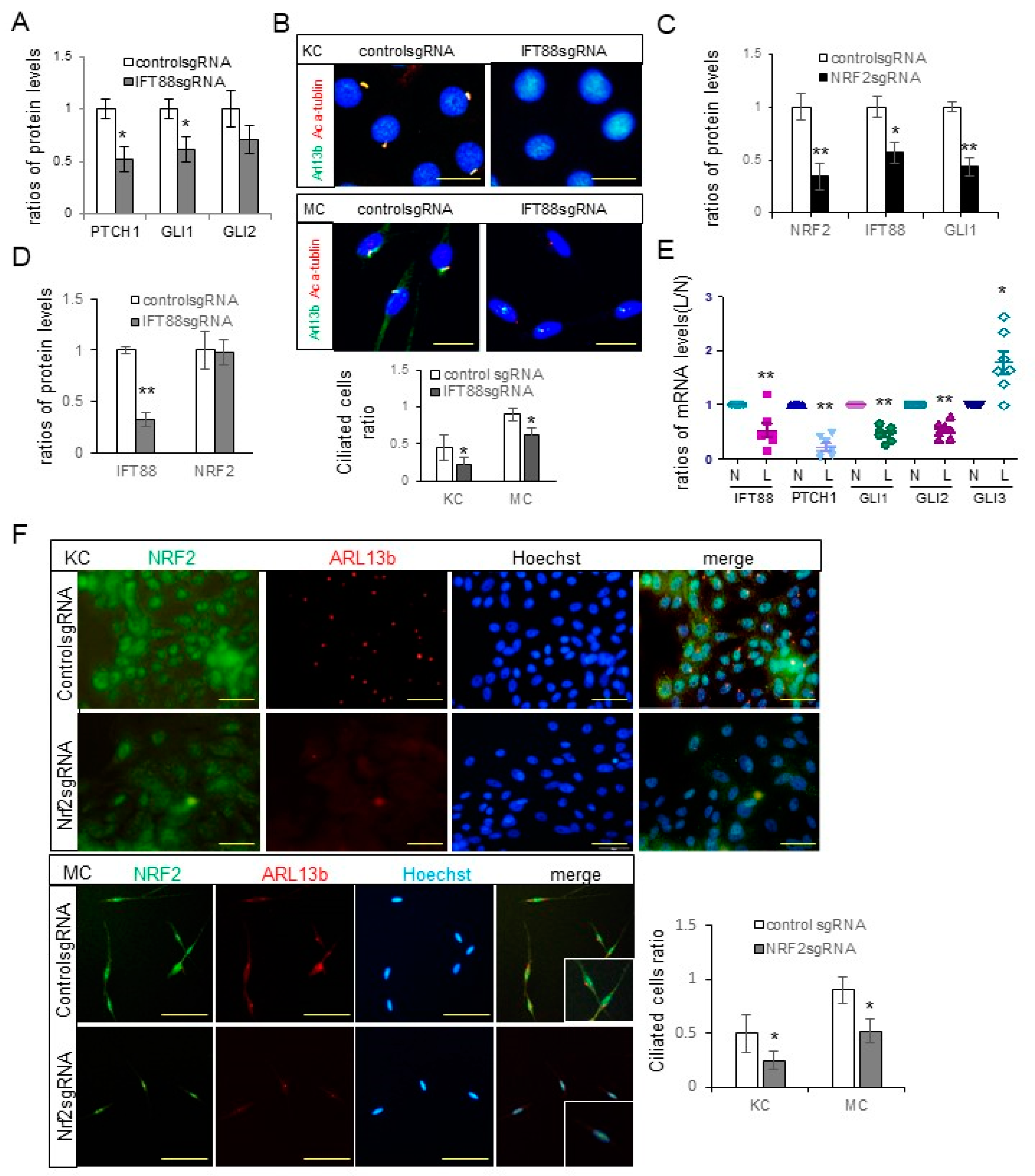
The downregulation of Hh signaling molecules, including PTCH, GLI1, and GLI2 (
Figure 2A), and a decrease in the number of ciliated cells (
Figure 2B) following
IFT88 knockdown, verified the role of IFT88 in ciliogenesis in keratinocytes and melanocytes. The reduced expression levels of IFT88 and GLI1 (
Figure 2C) and number of ciliated cells (
Figure 2F) following
NRF2 knockdown, with no reciprocal impacts on NRF2 expression following
IFT88 knockdown (
Figure 2D), suggested that NRF2 can regulate ciliogenesis via IFT88 and GLI1. The downregulation of IFT88 and GLI1 in the lesional skin of patients with melasma with NRF2 downregulation (
Figure 2E) substantiates the role of oxidative stress in ciliogenesis in melasma. NRF2 has been established as a pivotal regulator of ciliogenesis [
32,
33], with various genes related to ciliogenesis and the Hh signaling pathway identified as NRF2 target genes [
32].
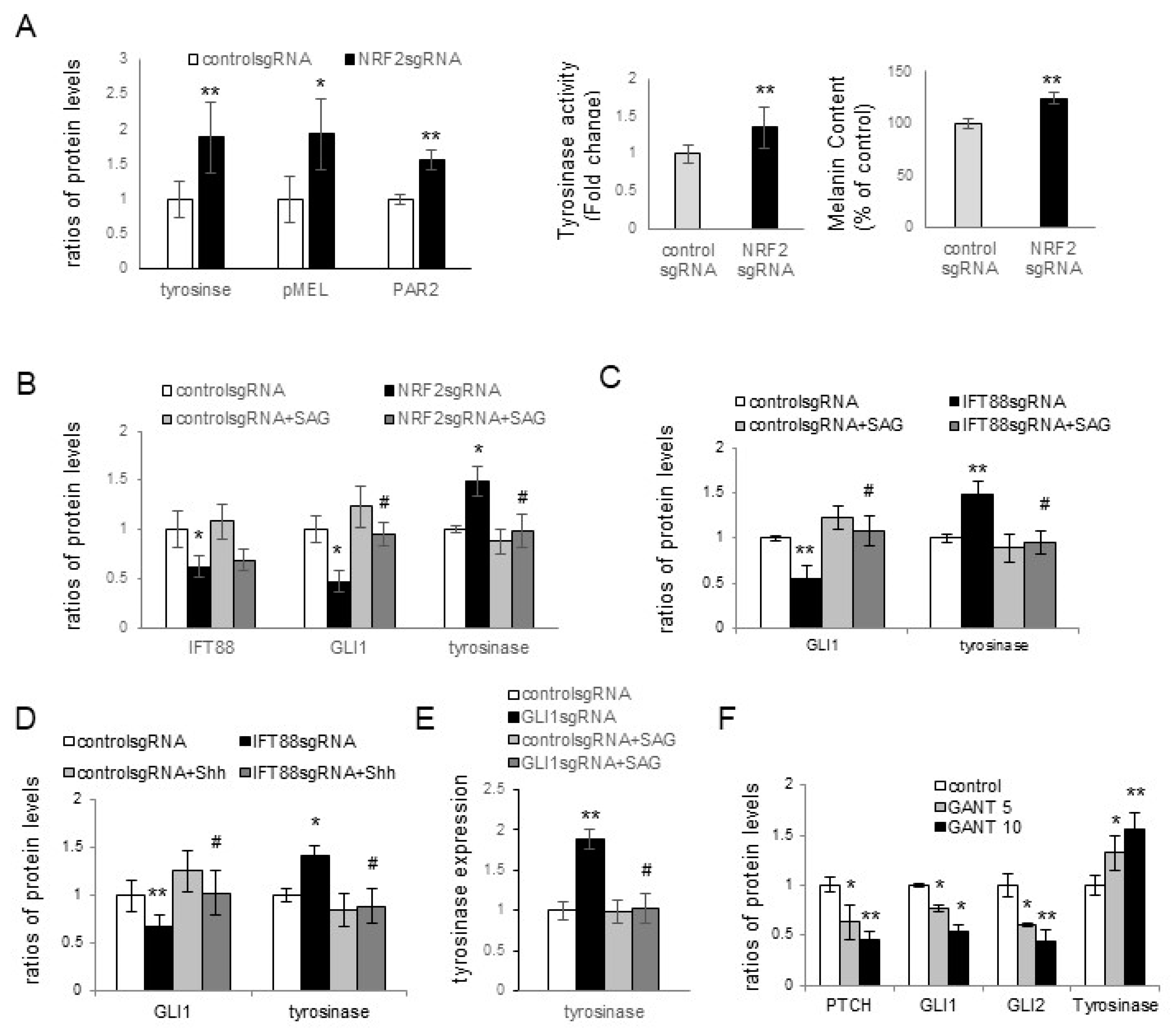
Melasma is a skin disorder marked by hyperpigmentation, which indicates that oxidative-stress-induced inhibition of ciliogenesis contributes to hyperpigmentation. Skin pigmentation is intricately linked to melanin synthesis, the transfer of melanosomes to keratinocytes, and melanosome degradation. Increased tyrosinase activity indicated an upregulation in melanin synthesis with
NRF2 knockdown in keratinocyte–melanocyte cocultures (
Figure 3A). However, other factors, such as stimulated melanosome transfer to keratinocytes and reduced melanosome degradation, may have contributed to the elevation of melanin contents. The role of PAR2 in the transfer of melanosomes to keratinocytes is well known [
29,
34]. The upregulation of tyrosinase, PMEL17, and PAR-2 in keratinocytes with downregulated NRF2, IFT88, or GLI1 (
Figure 3A–E) indicated that the inhibition of oxidative stress-related ciliogenesis enhanced melanin synthesis and melanosome transfer. The restoration of the increased levels of tyrosinase or PAR-2 with GLI1, but not NRF2 or IFT88, with SAG or Shh 200 treatments (
Figure 3B–E) suggested that GLI1 is a downstream molecule for the inhibitory role of primary cilia in hyperpigmentation. The elevation of tyrosinase expression with GANT61, a GLI1 inhibitor (
Figure 3F), supported the results obtained with GLI1.
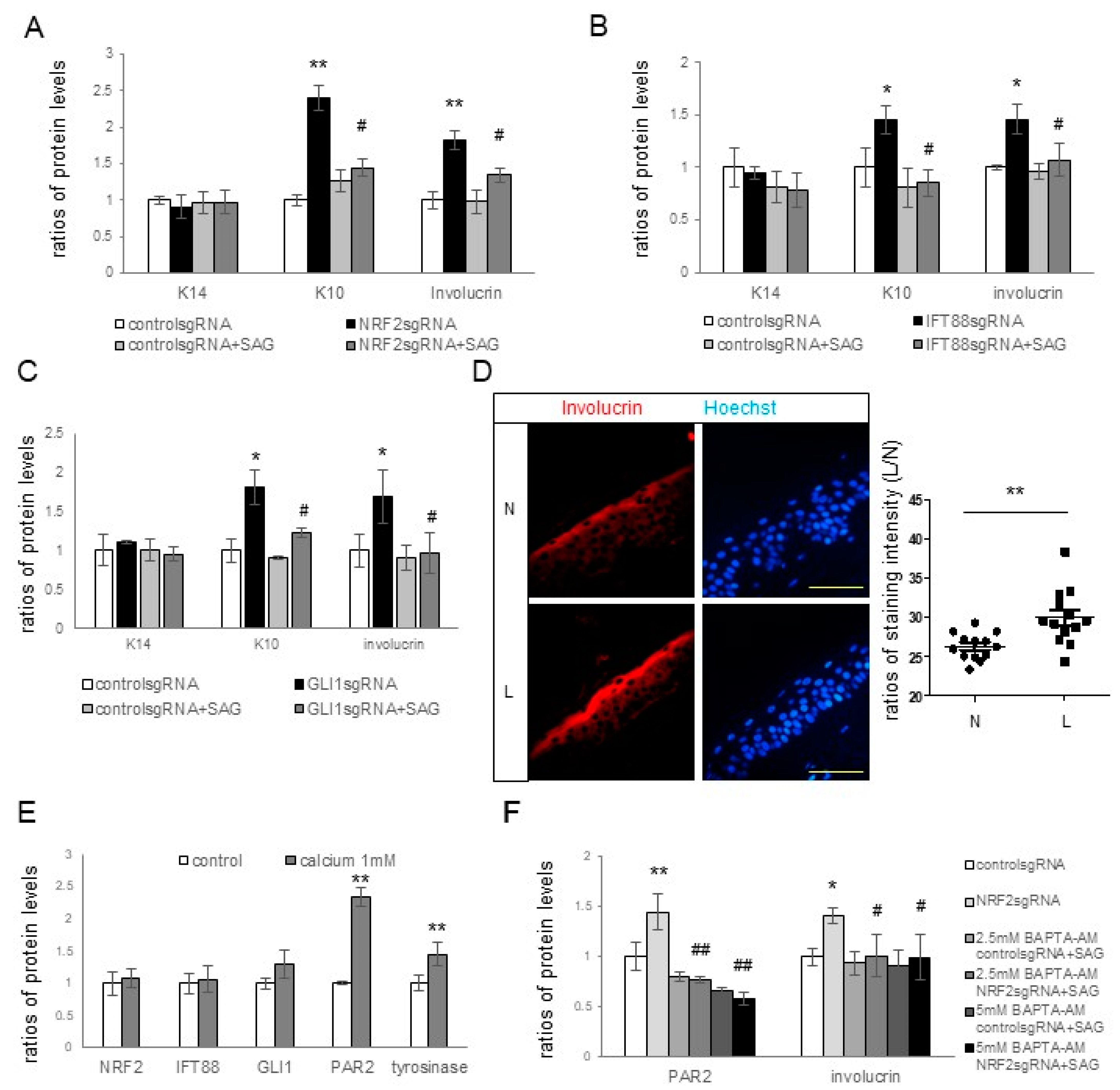
However, the mechanism by which primary cilia suppress melanin synthesis and melanosome transfer remains unclear. Primary cilia and the Hh signaling pathway are known to contribute to epidermal homeostasis, including keratinocyte proliferation or differentiation [
26,
27,
28]. Consequently, markers for keratinocyte differentiation and proliferation, as well as those of tyrosinase and PAR2, were examined in the presence and absence of
NRF2,
IFT88, or
GLI1 knockdown. The expression levels of involucrin and K10, but not of K14, in keratinocytes increased upon the knockdown of
IFT88 or
GLI1 as well as
NRF2 (
Figure 4A–C), suggesting an inhibitory role of primary cilia in keratinocyte differentiation, but not in keratinocyte proliferation. The restoration of upregulated K10 and involucrin in cultured keratinocytes with
NRF2,
IFT88, or
GLI1 knockdown with the SAG (
Figure 4A–C) supported the findings that primary cilia are involved in the inhibition of keratinocyte differentiation. Enhanced keratinocyte differentiation was also detected in the lesional skin of melasma patients with downregulated IFT88 and GLI1 (
Figure 4D). Calcium upregulated involucrin, tyrosinase, and PAR2 (
Figure 4E), whereas calcium chelator restored the upregulation of involucrin and PAR2 with
NRF2 knockdown (
Figure 4F). However, neither calcium nor calcium chelators altered the expression of NRF2, IFT88, or GLI1 (
Figure 4E,F). Although more studies are necessary to reach definitive conclusions, the connection from NRF2 downregulation to IFT88 and GLI1 downregulation may be involved in increasing melanin synthesis and melanosome transfer by stimulating keratinocyte differentiation in melasma.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




