
Association of Mitochondrial Variants with the Joint Occurrence of Polycystic Ovary Syndrome and Hashimoto’s Thyroiditis

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Population
2.3. Nucleic Acid Extraction
2.4. Mitochondrial DNA Sequencing and Variant Calling
2.5. Statistical Analyses
3. Results
3.1. Identified mtDNA Haplogroups
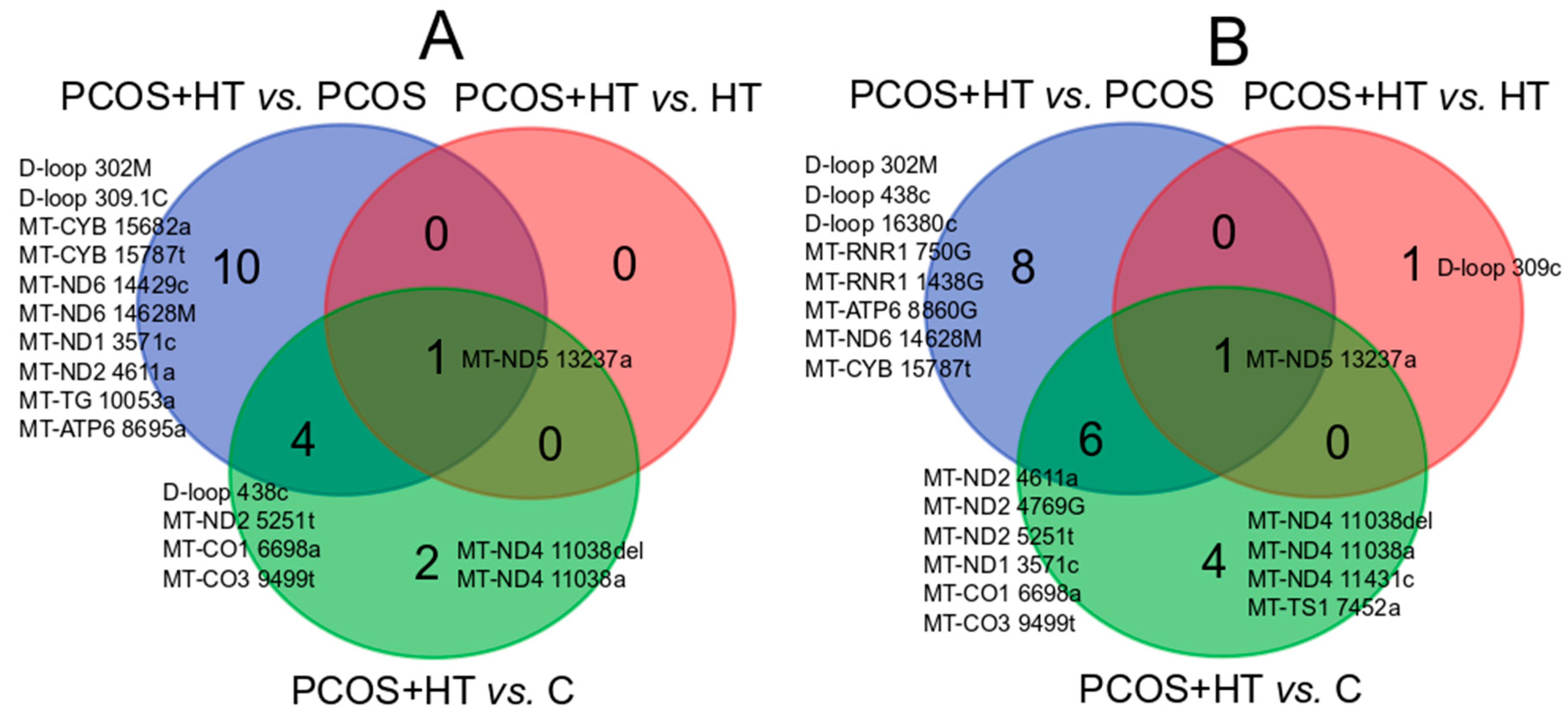
3.2. Significantly Differentiating Mitochondrial Variants
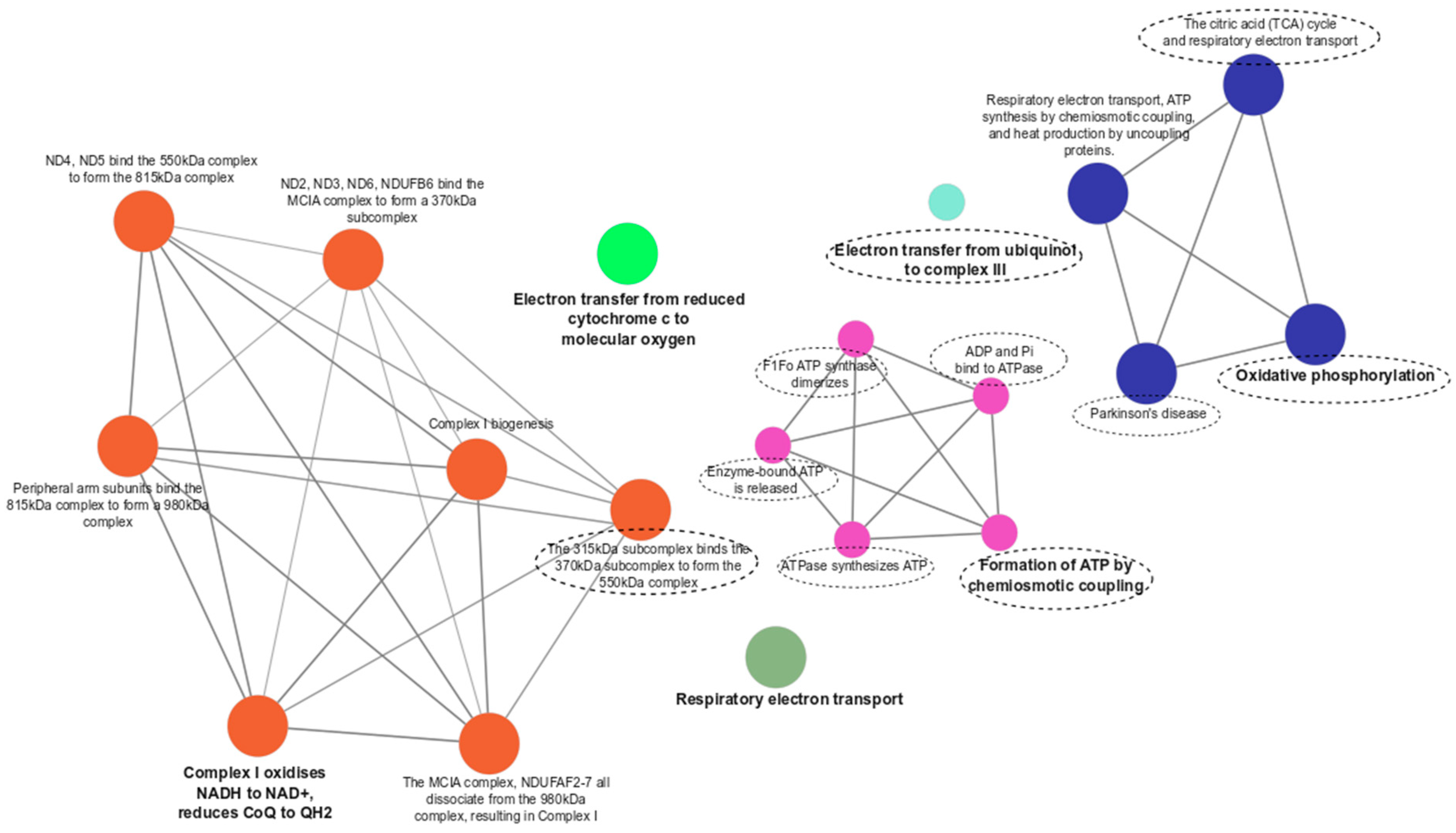
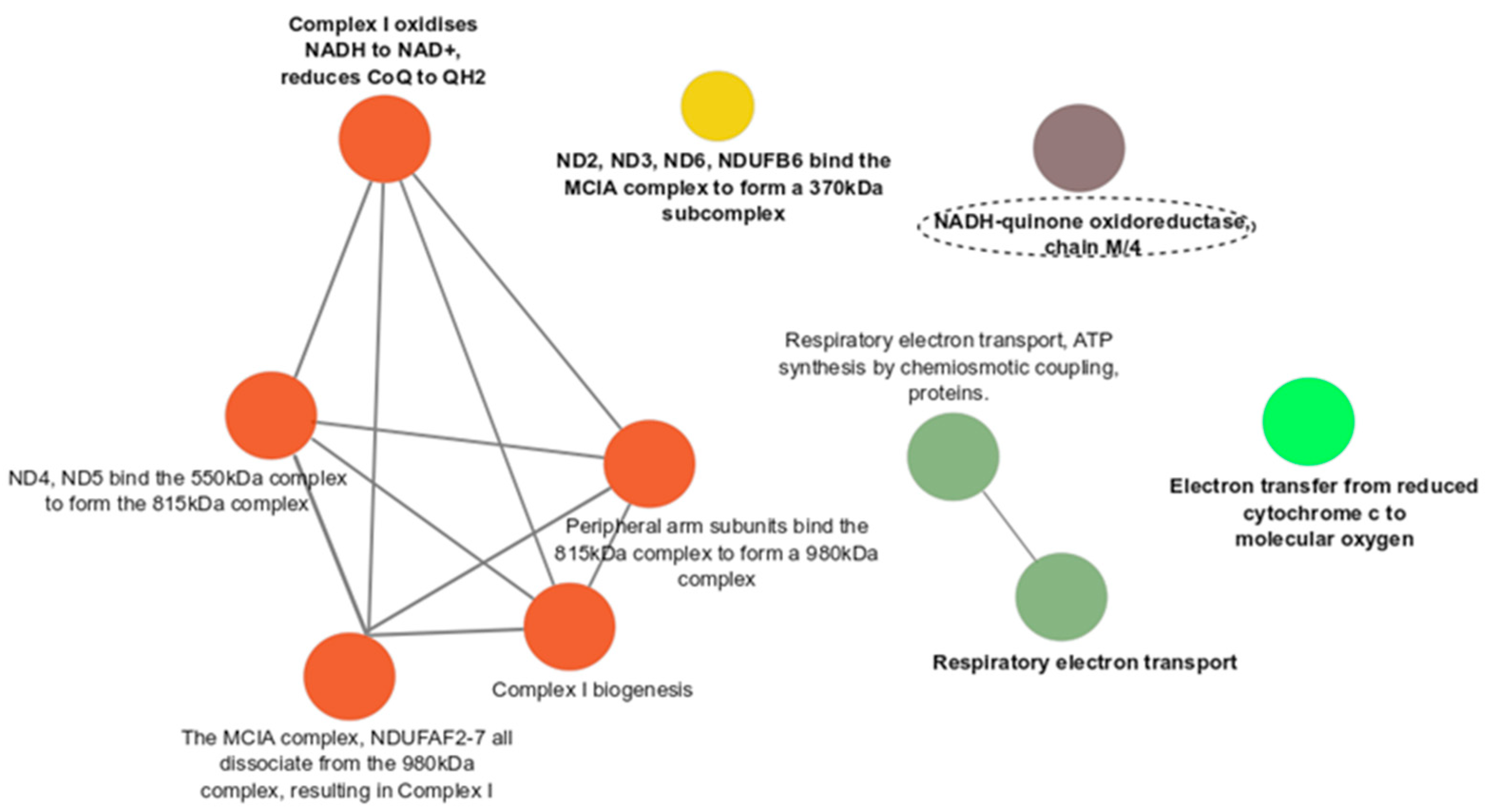
3.3. Functional Enrichment Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zeber-Lubecka, N.; Hennig, E.E. Genetic Susceptibility to Joint Occurrence of Polycystic Ovary Syndrome and Hashimoto’s Thyroiditis: How Far Is Our Understanding? Front. Immunol. 2021, 12, 606620. [Google Scholar] [CrossRef] [PubMed]
- Aversa, A.; La Vignera, S.; Rago, R.; Gambineri, A.; Nappi, R.E.; Calogero, A.E.; Ferlin, A. Fundamental Concepts and Novel Aspects of Polycystic Ovarian Syndrome: Expert Consensus Resolutions. Front. Endocrinol. 2020, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Wekker, V.; van Dammen, L.; Koning, A.; Heida, K.Y.; Painter, R.C.; Limpens, J.; Laven, J.S.E.; Lennep, J.E.R.v.; Roseboom, T.J.; Hoek, A. Long-Term Cardiometabolic Disease Risk in Women with PCOS: A Systematic Review and Meta-Analysis. Hum. Reprod. Update 2020, 26, 942–960. [Google Scholar] [CrossRef] [PubMed]
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S. Mitochondrial Signal Transduction. Cell Metab. 2022, 34, 1620–1653. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative Stress: The Mitochondria-Dependent and Mitochondria-Independent Pathways of Apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Lax, N.Z.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Mutations. Neuroscientist 2011, 17, 645–658. [Google Scholar] [CrossRef]
- Zhang, J.; Bao, Y.; Zhou, X.; Zheng, L. Polycystic Ovary Syndrome and Mitochondrial Dysfunction. Reprod. Biol. Endocrinol. 2019, 17, 67. [Google Scholar] [CrossRef]
- Shukla, P.; Mukherjee, S. Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Polycystic Ovary Syndrome. Mitochondrion 2020, 52, 24–39. [Google Scholar] [CrossRef]
- Zhuo, G.; Ding, Y.; Feng, G.; Yu, L.; Jiang, Y. Analysis of Mitochondrial DNA Sequence Variants in Patients with Polycystic Ovary Syndrome. Arch. Gynecol. Obstet. 2012, 286, 653–659. [Google Scholar] [CrossRef]
- Ding, Y.; Zhuo, G.; Zhang, C.; Leng, J. Point Mutation in Mitochondrial TRNA Gene Is Associated with Polycystic Ovary Syndrome and Insulin Resistance. Mol. Med. Rep. 2016, 13, 3169–3172. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhuo, G.; Zhang, C. The Mitochondrial TRNALeu(UUR) A3302G Mutation May Be Associated with Insulin Resistance in Woman with Polycystic Ovary Syndrome. Reprod. Sci. 2016, 23, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Hu, B.; Shi, W.; Guo, F.; Xu, C.; Li, S. Mitochondrial DNA 4977 Bp Deletion in Peripheral Blood Is Associated with Polycystic Ovary Syndrome. Front. Endocrinol. 2021, 12, 675581. [Google Scholar] [CrossRef]
- Ding, Y.; Xia, B.-H.; Zhang, C.-J.; Zhuo, G.-C. Mutations in Mitochondrial TRNA Genes May Be Related to Insulin Resistance in Women with Polycystic Ovary Syndrome. Am. J. Transl. Res. 2017, 9, 2984–2996. [Google Scholar]
- Esfahanian, F.; Hazaveh, M.M.; Garehbagh, L.H.; Falahati, K.; Ataei, M.; Sanati, M.H.; Jadali, Z. Increased Mitochondrial DNA Copy Number and Oxidative Damage in Patients with Hashimoto’s Thyroiditis. Iran. J. Public Health 2021, 50, 1697–1704. [Google Scholar] [CrossRef]
- The Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 Consensus on Diagnostic Criteria and Long-Term Health Risks Related to Polycystic Ovary Syndrome. Fertil. Steril. 2004, 81, 19–25. [Google Scholar] [CrossRef]
- Parson, W.; Gusmão, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA Commission of the International Society for Forensic Genetics: Revised and Extended Guidelines for Mitochondrial DNA Typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Attimonelli, M.; Bai, R.; Lott, M.T.; Wallace, D.C.; Falk, M.J.; Gai, X. MSeqDR MvTool: A Mitochondrial DNA Web and API Resource for Comprehensive Variant Annotation, Universal Nomenclature Collation, and Reference Genome Conversion. Hum. Mutat. 2018, 39, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.; Piredda, R.; Calabrese, F.M.; Simone, D.; Lang, M.; Calabrese, C.; Petruzzella, V.; Tommaseo-Ponzetta, M.; Gasparre, G.; Attimonelli, M. HmtDB, a Genomic Resource for Mitochondrion-Based Human Variability Studies. Nucleic Acids Res. 2012, 40, D1150–D1159. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to Accessing Data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice-Improving Genome-Wide Variant Effect Prediction Using Deep Learning-Derived Splice Scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the Pathogenicity of Novel Variants in Mitochondrial TRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef]
- Kelsen, J.R.; Dawany, N.; Moran, C.J.; Petersen, B.-S.; Sarmady, M.; Sasson, A.; Pauly-Hubbard, H.; Martinez, A.; Maurer, K.; Soong, J.; et al. Exome Sequencing Analysis Reveals Variants in Primary Immunodeficiency Genes in Patients with Very Early Onset Inflammatory Bowel Disease. Gastroenterology 2015, 149, 1415–1424. [Google Scholar] [CrossRef]
- NC_012920.1:M.15682A>G and Familial Cancer of Breast-ClinVar–NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/RCV000133436/ (accessed on 31 October 2023).
- Reddy, T.V.; Govatati, S.; Deenadayal, M.; Sisinthy, S.; Bhanoori, M. Impact of Mitochondrial DNA Copy Number and Displacement Loop Alterations on Polycystic Ovary Syndrome Risk in South Indian Women. Mitochondrion 2019, 44, 35–40. [Google Scholar] [CrossRef]
- Shen, X.; Du, A. The Non-Syndromic Clinical Spectrums of MtDNA 3243A>G Mutation. Neurosciences 2021, 26, 128–133. [Google Scholar] [CrossRef]
- Chin, J.; Marotta, R.; Chiotis, M.; Allan, E.H.; Collins, S.J. Detection Rates and Phenotypic Spectrum of m.3243A>G in the MT-TL1 Gene: A Molecular Diagnostic Laboratory Perspective. Mitochondrion 2014, 17, 34–41. [Google Scholar] [CrossRef]
- Miiller-Hocker, J.; Jacob, U.; Seibel, P. Hashimoto Thyreoiditis Is Associated with Defects of Cytochrome-c Oxidase in Oxyphil Askanazy Cells and with the Common Deletion (4977) of Mitochondrial DNA. Ultrastruct. Pathol. 1998, 22, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Bergman, O.; Ben-Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) Deficits in Schizophrenia: Possible Interactions with Cellular Processes. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Uyanikoglu, H.; Sabuncu, T.; Dursun, H.; Sezen, H.; Aksoy, N. Circulating Levels of Apoptotic Markers and Oxidative Stress Parameters in Women with Polycystic Ovary Syndrome: A Case-Controlled Descriptive Study. Biomarkers 2017, 22, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhu, M.; Xu, W. Roles of Oxidative Stress in Polycystic Ovary Syndrome and Cancers. Oxid. Med. Cell. Longev. 2016, 2016, 8589318. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Bruno, C.; Vergani, E.; d’Abate, C.; Giacchi, E.; Silvestrini, A. Oxidative Stress and Low-Grade Inflammation in Polycystic Ovary Syndrome: Controversies and New Insights. Int. J. Mol. Sci. 2021, 22, 1667. [Google Scholar] [CrossRef]
- Nuñez-Calonge, R.; Cortés, S.; Gutierrez Gonzalez, L.M.; Kireev, R.; Vara, E.; Ortega, L.; Caballero, P.; Rancan, L.; Tresguerres, J. Oxidative Stress in Follicular Fluid of Young Women with Low Response Compared with Fertile Oocyte Donors. Reprod. Biomed. Online 2016, 32, 446–456. [Google Scholar] [CrossRef]
- Yalcinkaya, E.; Cakiroglu, Y.; Doger, E.; Budak, O.; Cekmen, M.; Caliskan, E. Effect of Follicular Fluid NO, MDA and GSH Levels on in Vitro Fertilization Outcomes. J. Turk. Ger. Gynecol. Assoc. 2013, 14, 136–141. [Google Scholar] [CrossRef]
- Zimmermann, F.A.; Neureiter, D.; Feichtinger, R.G.; Trost, A.; Sperl, W.; Kofler, B.; Mayr, J.A. Deficiency of Respiratory Chain Complex I in Hashimoto Thyroiditis. Mitochondrion 2016, 26, 1–6. [Google Scholar] [CrossRef]
- Kochman, J.; Jakubczyk, K.; Bargiel, P.; Janda-Milczarek, K. The Influence of Oxidative Stress on Thyroid Diseases. Antioxidants 2021, 10, 1442. [Google Scholar] [CrossRef]
- Rostami, R.; Aghasi, M.R.; Mohammadi, A.; Nourooz-Zadeh, J. Enhanced Oxidative Stress in Hashimoto’s Thyroiditis: Inter-Relationships to Biomarkers of Thyroid Function. Clin. Biochem. 2013, 46, 308–312. [Google Scholar] [CrossRef]
- Yehuda-Shnaidman, E.; Kalderon, B.; Bar-Tana, J. Thyroid Hormone, Thyromimetics, and Metabolic Efficiency. Endocr. Rev. 2014, 35, 35–58. [Google Scholar] [CrossRef] [PubMed]
- Ates, I.; Arikan, M.F.; Altay, M.; Yilmaz, F.M.; Yilmaz, N.; Berker, D.; Guler, S. The Effect of Oxidative Stress on the Progression of Hashimoto’s Thyroiditis. Arch. Physiol. Biochem. 2018, 124, 351–356. [Google Scholar] [CrossRef]
- Wang, Q.; Moley, K.H. Maternal Diabetes and Oocyte Quality. Mitochondrion 2010, 10, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ratchford, A.M.; Chi, M.M.-Y.; Schoeller, E.; Frolova, A.; Schedl, T.; Moley, K.H. Maternal Diabetes Causes Mitochondrial Dysfunction and Meiotic Defects in Murine Oocytes. Mol. Endocrinol. 2009, 23, 1603–1612. [Google Scholar] [CrossRef]
- Jimenez, P.T.; Frolova, A.I.; Chi, M.M.; Grindler, N.M.; Willcockson, A.R.; Reynolds, K.A.; Zhao, Q.; Moley, K.H. DHEA-Mediated Inhibition of the Pentose Phosphate Pathway Alters Oocyte Lipid Metabolism in Mice. Endocrinology 2013, 154, 4835–4844. [Google Scholar] [CrossRef]
- Sarkar, M.; Terrault, N.; Chan, W.; Cedars, M.I.; Huddleston, H.G.; Duwaerts, C.C.; Balitzer, D.; Gill, R.M. Polycystic Ovary Syndrome (PCOS) Is Associated with NASH Severity and Advanced Fibrosis. Liver Int. 2020, 40, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Vassilatou, E. Nonalcoholic Fatty Liver Disease and Polycystic Ovary Syndrome. World J. Gastroenterol. 2014, 20, 8351. [Google Scholar] [CrossRef]
- Gao, S.; Long, F.; Jiang, Z.; Shi, J.; Ma, D.; Yang, Y.; Bai, J.; Han, T.-L. The Complex Metabolic Interactions of Liver Tissue and Hepatic Exosome in PCOS Mice at Young and Middle Age. Front. Physiol. 2022, 13, 990987. [Google Scholar] [CrossRef]
- Arneth, B.M. The Impact of Krebs Cycle Intermediates on the Endocrine System and Immune System: A Comparison. Endocrines 2023, 4, 179–193. [Google Scholar] [CrossRef]
- Mendez, D.A.; Ortiz, R.M. Thyroid Hormones and the Potential for Regulating Glucose Metabolism in Cardiomyocytes during Insulin Resistance and T2DM. Physiol. Rep. 2021, 9, e14858. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.H.; Verkaart, S.; Visch, H.J.; van Emst-de Vries, S.; Nijtmans, L.G.J.; Smeitink, J.A.M.; Willems, P.H.G.M. Human NADH:Ubiquinone Oxidoreductase Deficiency: Radical Changes in Mitochondrial Morphology? Am. J. Physiol. Cell Physiol. 2007, 293, C22–C29. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, B.; Duleba, A.J.; Spaczynski, R.Z.; Pawelczyk, L. Metformin Therapy Decreases Hyperandrogenism and Hyperinsulinemia in Women with Polycystic Ovary Syndrome. Fertil. Steril. 2000, 73, 1149–1154. [Google Scholar] [CrossRef]
- Hirsch, A.; Hahn, D.; Kempná, P.; Hofer, G.; Nuoffer, J.-M.; Mullis, P.E.; Flück, C.E. Metformin Inhibits Human Androgen Production by Regulating Steroidogenic Enzymes HSD3B2 and CYP17A1 and Complex I Activity of the Respiratory Chain. Endocrinology 2012, 153, 4354–4366. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Giacco, A.; Goglia, F.; Silvestri, E. Bioenergetic Aspects of Mitochondrial Actions of Thyroid Hormones. Cells 2022, 11, 997. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Di Segni, C.; Raimondo, S.; Olivieri, G.; Silvestrini, A.; Meucci, E.; Currò, D. Thyroid Hormones, Oxidative Stress, and Inflammation. Mediat. Inflamm. 2016, 2016, 6757154. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fromme, T. Uncoupling Protein 1 Does Not Produce Heat without Activation. Int. J. Mol. Sci. 2022, 23, 2406. [Google Scholar] [CrossRef] [PubMed]
- Shorakae, S.; Jona, E.; de Courten, B.; Lambert, G.W.; Lambert, E.A.; Phillips, S.E.; Clarke, I.J.; Teede, H.J.; Henry, B.A. Brown Adipose Tissue Thermogenesis in Polycystic Ovary Syndrome. Clin. Endocrinol. 2018, 90, 425–432. [Google Scholar] [CrossRef]
- Yuan, X.; Hu, T.; Zhao, H.; Huang, Y.; Ye, R.; Lin, J.; Zhang, C.; Zhang, H.; Wei, G.; Zhou, H.; et al. Brown Adipose Tissue Transplantation Ameliorates Polycystic Ovary Syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 2708–2713. [Google Scholar] [CrossRef]
- Jarczak, J.; Grochowalski, Ł.; Marciniak, B.; Lach, J.; Słomka, M.; Sobalska-Kwapis, M.; Lorkiewicz, W.; Pułaski, Ł.; Strapagiel, D. Mitochondrial DNA Variability of the Polish Population. Eur. J. Hum. Genet. 2019, 27, 1304–1314. [Google Scholar] [CrossRef]
- Dong, J.; Wong, L.-J.; Mims, M.P. Mitochondrial Inheritance and Cancer. Transl. Res. 2018, 202, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Gomez-Duran, A. Oldies but Goldies MtDNA Population Variants and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 682. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Parameter | PCOS N = 20 | HT N = 20 | PCOS + HT N = 20 | Control N = 24 | Kruskal–Wallis |
| Median (IQR) | Median (IQR) | Median (IQR) | Median (IQR) | padj-Value | |
| BMI (kg/m2) | 23.52 (7.75) | 22.11 (3.99) | 26.77 (10.16) | 21.63 (2.85) | ns. |
| Age (years) | 24 (8) | 33.5 (5.75) | 25 (3.25) | 30 (8) | 7.9 × 10−4 |
| FSH (mIU/mL) | 5.19 (1.93) | 5.19 (2.12) | 4.43 (1.02) | 5.55 (1.07) | ns. |
| LH (mIU/mL) | 8.66 (5.52) | 5.3 (2.79) | 6.59 (4.15) | 4.77 (0.85) | 1.9 × 10−3 |
| LH/FSH | 1.7 (0.97) | 1.01 (0.79) | 1.33 (1.03) | 0.87 (0.23) | 5.3 × 10−4 |
| E2 (pg/mL) | 36.5 (18) | 39 (37.75) | 41.5 (12) | 32.5 (20.25) | ns. |
| T (ng/mL) | 0.55 (0.16) | 0.32 (0.12) | 0.53 (0.17) | 0.32 (0.12) | 8.9 × 10−9 |
| A (ng/mL) | 4.46 (1.63) | 1.94 (0.59) | 3.77 (1.95) | 1.96 (0.23) | 6.6 × 10−11 |
| PRG (ng/mL) | 0.3 (0.2) | 9.75 (9.12) | 0.27 (0.44) | 11 (4.38) | 1.9 × 10−9 |
| 17-OH-PRG (ng/mL) | 2.65 (0.97) | 1.27 (0.85) | 1.42 (1.16) | 1.23 (0.45) | 5.1 × 10−3 |
| DHEAS (µmol/l) | 9.24 (3.52) | 6.28 (4.3) | 9.51 (5.45) | 6.88 (2.98) | 1.5 × 10−2 |
| TSH (µIU/mL) | 1.31 (0.77) | 1.31 (1.15) | 1.66 (1.07) | 1.57 (0.8) | ns. |
| fT4 (pmol/mL) | 12.43 (1.46) | 13.23 (1.84) | 13.74 (1.9) | 12.5 (0.64) | 1.7 × 10−2 |
| TPO-Ab (IU/mL) | 0.3 (0.2) | 276.5 (601) | 122 (219.25) | 0.69 (0.64) | 6.6 × 10−11 |
| Tg-Ab (IU/mL) | 1.75 (1.79) | 41.5 (58.25) | 34.5 (35.5) | 1 (0.51) | 6.6 × 10−11 |
| Haplogroup | PCOS % in Group N = 20 | HT % in Group N = 20 | PCOS + HT % in Group N = 20 | Control % in Group N = 24 |
|---|---|---|---|---|
| R0 | 35 | 50 | 35 | 33 |
| U | 40 | 30 | 30 | 29 |
| JT | 10 | 10 | 20 | 25 |
| other | 15 | 10 | 15 | 13 |
| Fisher’s Exact Test | Mann–Whitney U Test | |||||||
|---|---|---|---|---|---|---|---|---|
| PCOS + HT vs. PCOS | ||||||||
| Gene | Variant | Estimate | p-Value | padj-Value | Gene | Variant | p-Value | padj-Value |
| MT-CO1 | 6698a | 0 | 1.29 × 10−5 | 0.0049 | MT-ATP6 | 8860G | 4.72 × 10−6 | 0.0002 |
| MT-CO3 | 9499t | 0 | 4.51 × 10−5 | 0.0085 | MT-CO1 | 6698a | 2.56 × 10−5 | 0.0004 |
| D-loop | 438c | 0 | 0.0004 | 0.0548 | MT-CO3 | 9499t | 6.67 × 10−5 | 0.0008 |
| MT-ND6 | 14628M | 0.05 | 0.0012 | 0.1176 | MT-ND2 | 4611a | 0.0002 | 0.0020 |
| MT-ND5 | 13237a | 0.08 | 0.0022 | 0.1659 | D-loop | 438c | 0.0004 | 0.0027 |
| D-loop | 302M | 0.06 | 0.0033 | 0.1800 | MT-ND6 | 14628M | 0.0005 | 0.0027 |
| MT-ND2 | 5251t | 0.06 | 0.0033 | 0.1800 | MT-RNR1 | 750G | 0.0012 | 0.0052 |
| MT-TG | 10053a | 0 | 0.0083 | 0.3152 | MT-ND5 | 13237a | 0.0013 | 0.0052 |
| MT-CYB | 15682a | 0 | 0.0083 | 0.3152 | MT-ND2 | 5251t | 0.0014 | 0.0052 |
| MT-CYB | 15787t | 0.07 | 0.0084 | 0.3152 | D-loop | 302M | 0.0015 | 0.0052 |
| D-loop | 309.1C | 0.14 | 0.0095 | 0.3266 | MT-CYB | 15787t | 0.0028 | 0.0089 |
| MT-ND1 | 3571c | 0.18 | 0.0407 | 1 | MT-ND2 | 4769G | 0.0041 | 0.0120 |
| MT-ND2 | 4611a | 0.18 | 0.0407 | 1 | MT-RNR1 | 1438G | 0.0070 | 0.0187 |
| MT-ND6 | 14429c | 0 | 0.0471 | 1 | D-loop | 16380c | 0.0143 | 0.0345 |
| MT-ATP6 | 8695a | 0 | 0.0471 | 1 | MT-ND1 | 3571c | 0.0148 | 0.0345 |
| PCOS + HT vs. HT | ||||||||
| MT-ND5 | 13237a | 0.12 | 0.0138 | 1 | MT-ND5 | 13237a | 0.0059 | 0.1655 |
| D-loop | 309c | 0.0350 | 0.4904 | |||||
| PCOS + HT vs. C | ||||||||
| MT-CO1 | 6698a | 0 | 2.67 × 10−6 | 0.0010 | MT-CO1 | 6698a | 1.28 × 10−5 | 0.0004 |
| MT-CO3 | 9499t | 0 | 0.0003 | 0.0641 | MT-ND2 | 4769G | 8.77 × 10−5 | 0.0014 |
| MT-ND5 | 13237a | 0.07 | 0.0005 | 0.0641 | MT-ND5 | 13237a | 0.0004 | 0.0036 |
| MT-ND2 | 5251t | 0.05 | 0.0008 | 0.0734 | MT-ND2 | 5251t | 0.0006 | 0.0036 |
| MT-ND4 | 11038a | 6.7 | 0.0052 | 0.3980 | MT-CO3 | 9499t | 0.0007 | 0.0036 |
| D-loop | 438c | 0 | 0.0111 | 0.7020 | MT-ND4 | 11038del | 0.0007 | 0.0036 |
| MT-ND4 | 11038del | 0.19 | 0.0138 | 0.7533 | MT-ND2 | 4611a | 0.0020 | 0.0088 |
| MT-ND4 | 11038a | 0.0103 | 0.0398 | |||||
| MT-ND1 | 3571c | 0.0261 | 0.0817 | |||||
| MT-TS1 | 7452a | 0.0263 | 0.0817 | |||||
| MT-ND4 | 11431c | 0.0452 | 0.1274 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeber-Lubecka, N.; Kulecka, M.; Suchta, K.; Dąbrowska, M.; Ciebiera, M.; Hennig, E.E. Association of Mitochondrial Variants with the Joint Occurrence of Polycystic Ovary Syndrome and Hashimoto’s Thyroiditis. Antioxidants 2023, 12, 1983. https://doi.org/10.3390/antiox12111983
Zeber-Lubecka N, Kulecka M, Suchta K, Dąbrowska M, Ciebiera M, Hennig EE. Association of Mitochondrial Variants with the Joint Occurrence of Polycystic Ovary Syndrome and Hashimoto’s Thyroiditis. Antioxidants. 2023; 12(11):1983. https://doi.org/10.3390/antiox12111983
Chicago/Turabian StyleZeber-Lubecka, Natalia, Maria Kulecka, Katarzyna Suchta, Michalina Dąbrowska, Michał Ciebiera, and Ewa E. Hennig. 2023. "Association of Mitochondrial Variants with the Joint Occurrence of Polycystic Ovary Syndrome and Hashimoto’s Thyroiditis" Antioxidants 12, no. 11: 1983. https://doi.org/10.3390/antiox12111983
APA StyleZeber-Lubecka, N., Kulecka, M., Suchta, K., Dąbrowska, M., Ciebiera, M., & Hennig, E. E. (2023). Association of Mitochondrial Variants with the Joint Occurrence of Polycystic Ovary Syndrome and Hashimoto’s Thyroiditis. Antioxidants, 12(11), 1983. https://doi.org/10.3390/antiox12111983