
Voltage-Gated Proton Channel Hv1 Regulates Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease Models


 , , , and
, , , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. HVCN1 Expression Analysis in GEO Datasets
2.2. Animal Care and Treatment Paradigms
2.3. Tissue Preparation
2.4. Primary Microglial Culture and Treatments
2.5. Reactive Oxygen Species and Nitric Oxide Quantification
2.6. N27 Cell Culture and Conditioned Media Experiments
2.7. Immunohistochemistry and Immunocytochemistry
2.8. Unbiased Stereology
2.9. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.10. Data Analysis
3. Results
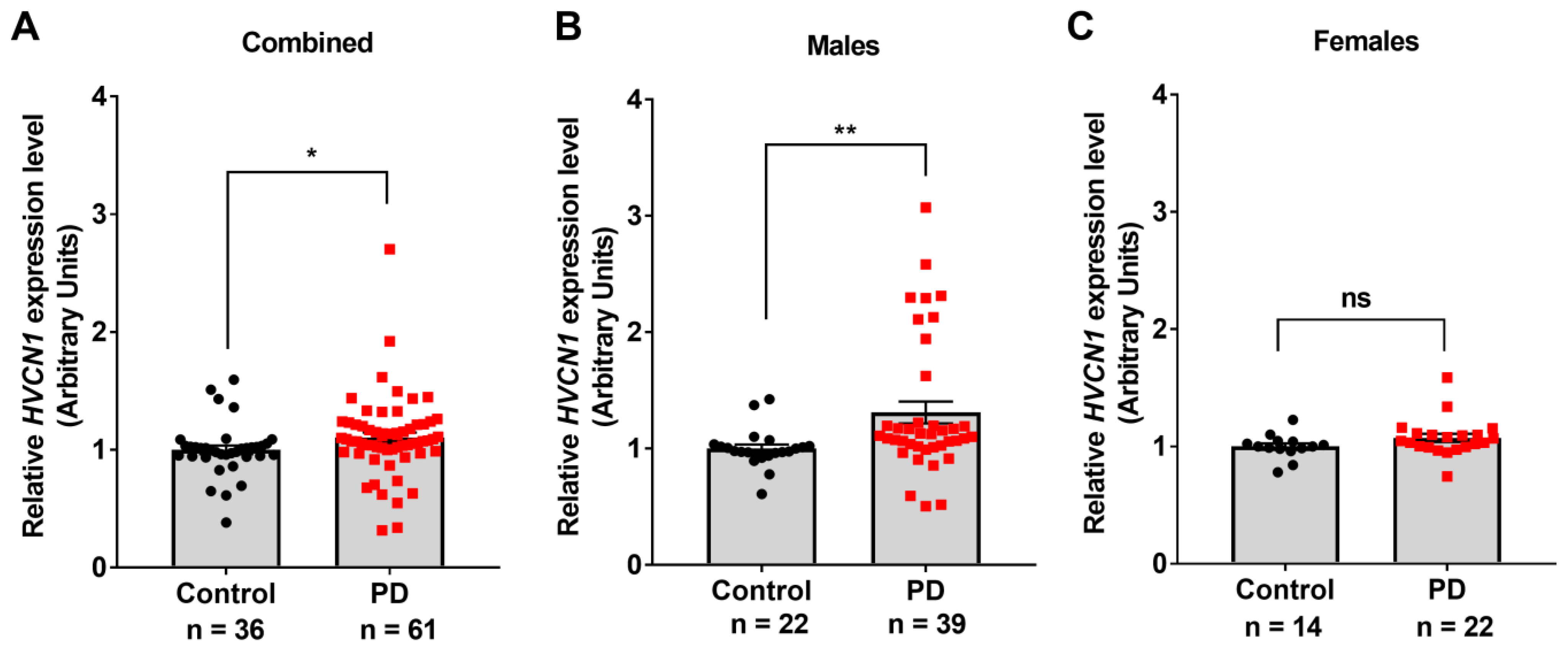
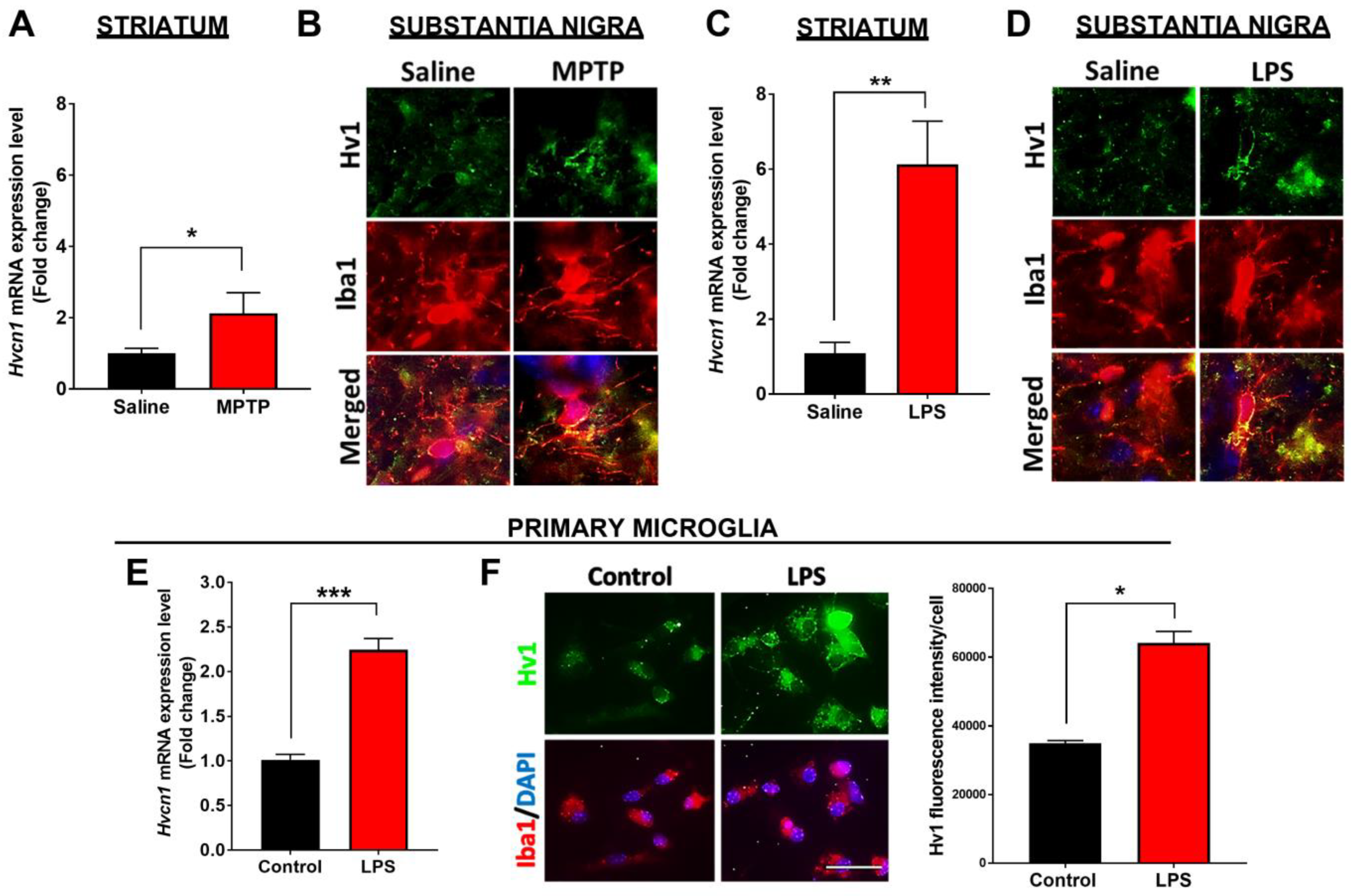
3.1. Hv1 Levels Are Increased in the Brains of PD Patients, Animal Models of PD, and Cultured Microglia following MPTP or LPS Treatment
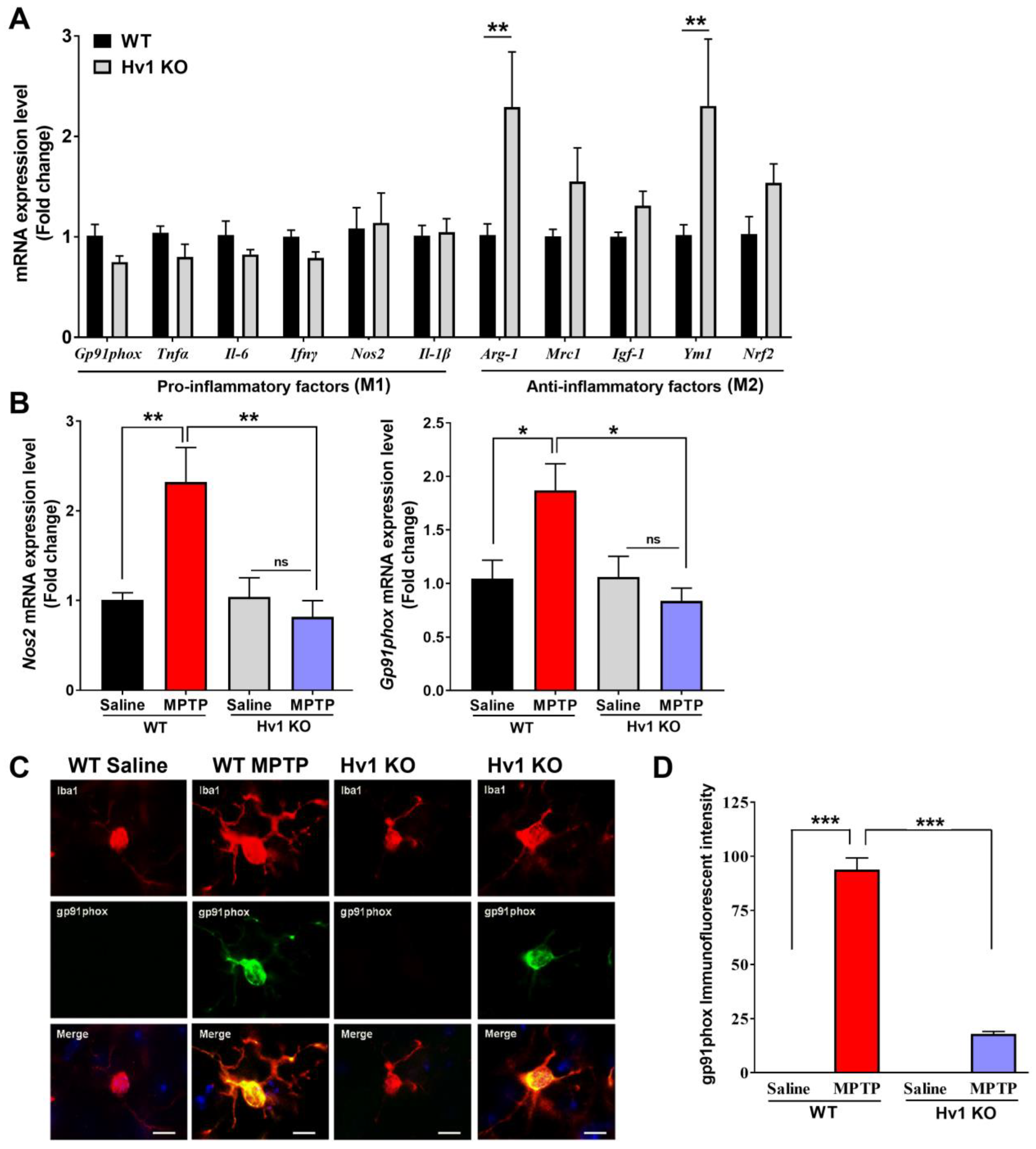
3.2. Hv1 KO Mice Have Lower Basal Inflammatory Status and Exhibit a Reduced MPTP-Mediated Inflammatory Response in the Mouse Nigrostriatal Pathway
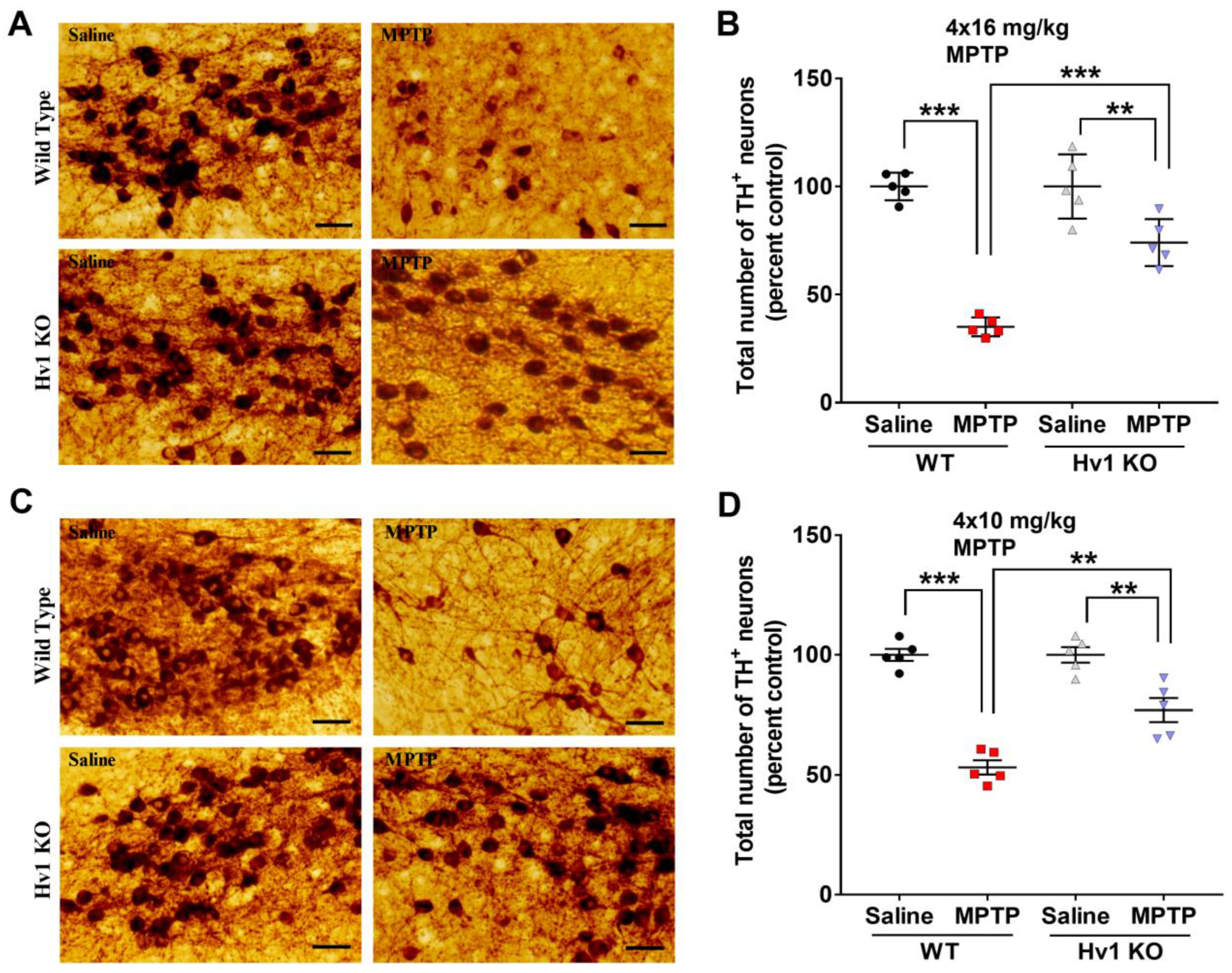
3.3. Hv1 KO Mice Are Protected from MPTP-Induced Dopaminergic Neurotoxicity
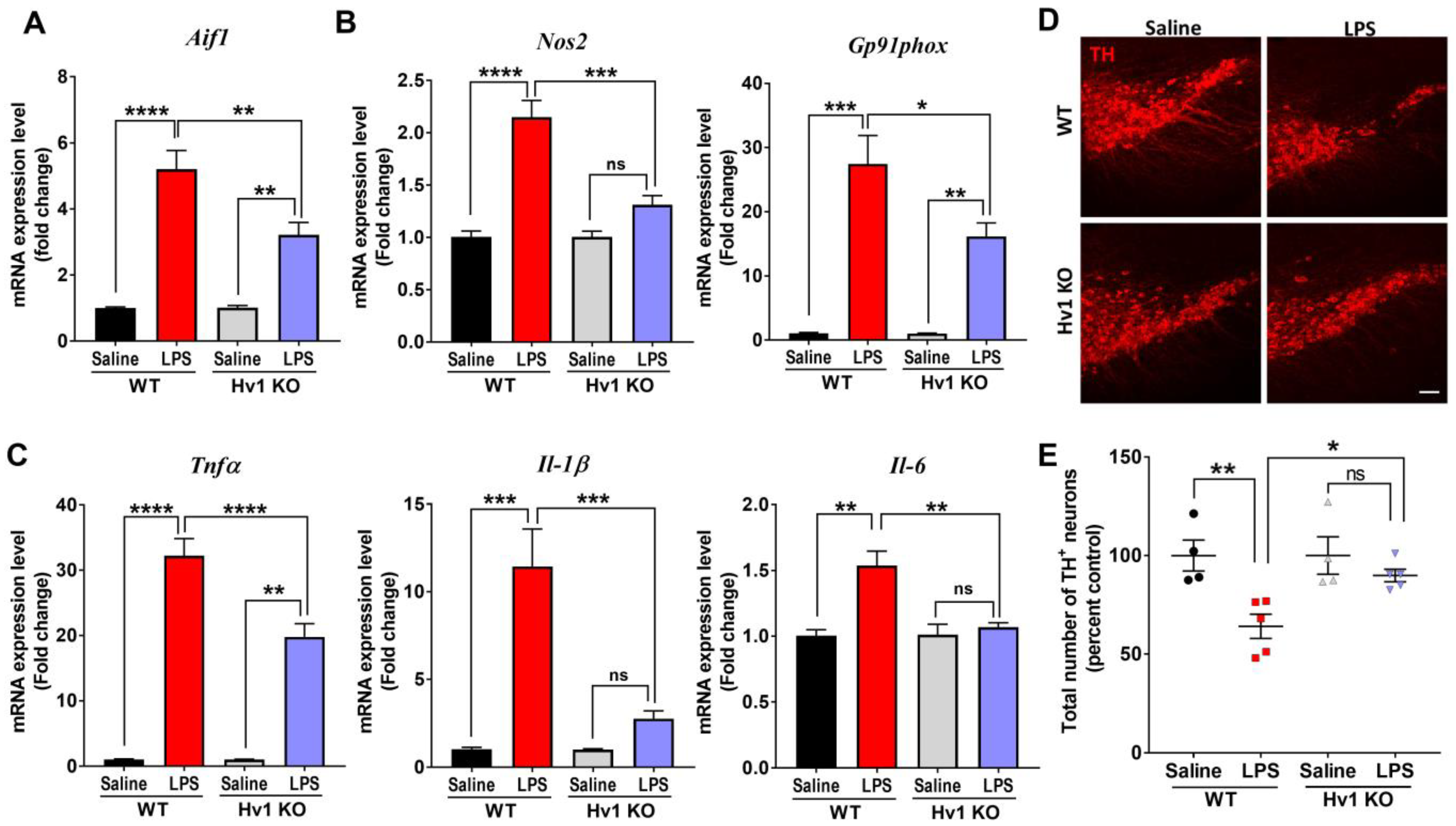
3.4. Hv1 KO Mice Are Protected from LPS-Induced Neuroinflammation and Dopaminergic Neurotoxicity
3.5. Hv1 KO Attenuates LPS-Induced Reactive Oxygen and Nitrogen Species Generation in Primary Mouse Microglia
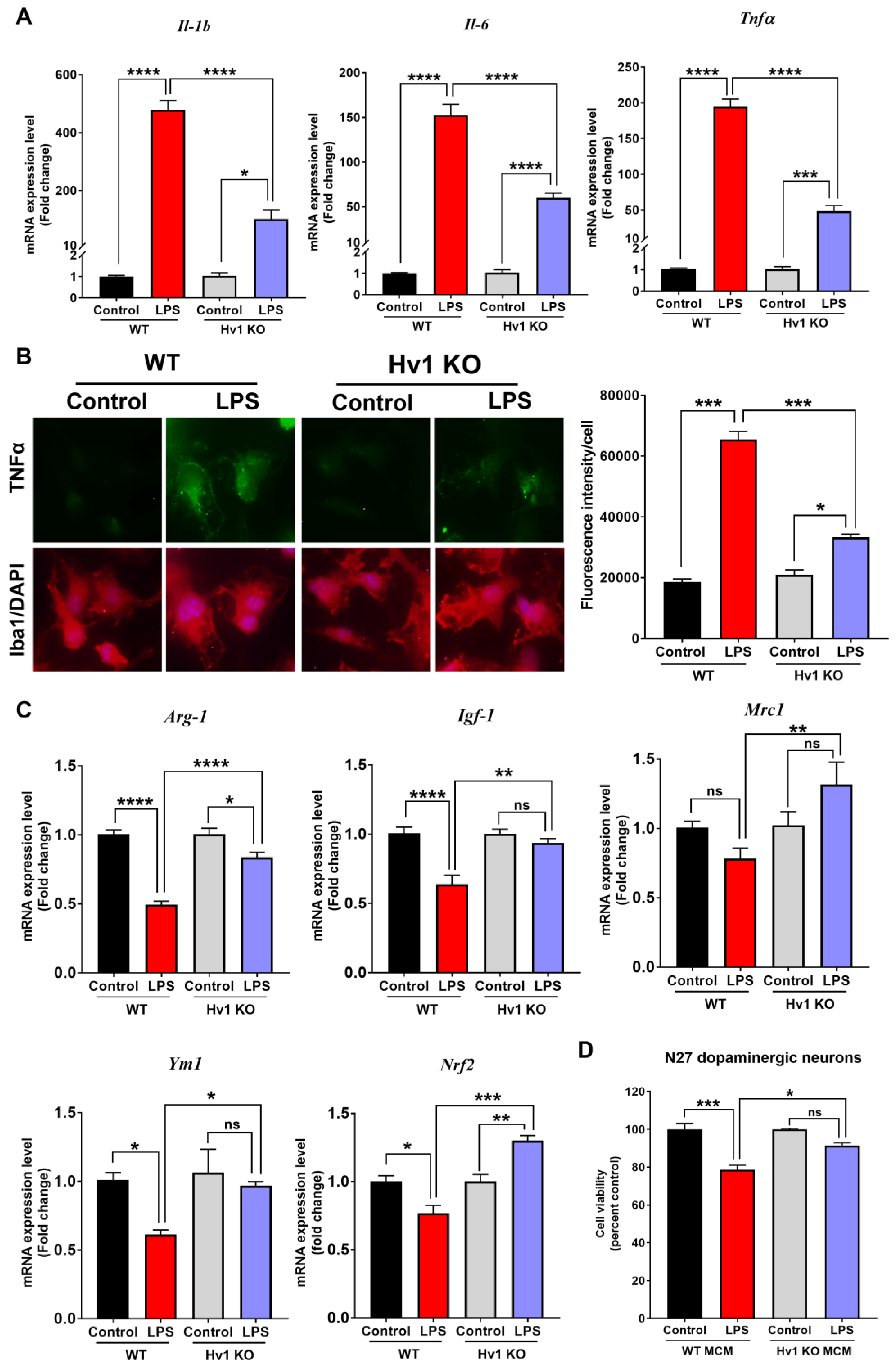
3.6. Hv1 KO Attenuates LPS-Induced Inflammatory Cytokine Production and Blocks the Reduction of Anti-Inflammatory Factors in Primary Mouse Microglia
3.7. Hv1 Deficiency in Microglia Protects Dopaminergic Neuron Viability from LPS-Mediated Microglial Inflammatory Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Seidl, S.E.; Potashkin, J.A. The promise of neuroprotective agents in Parkinson’s disease. Front. Neurol. 2011, 2, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: Role of tumor necrosis factor-alpha. J. Neurochem. 2006, 96, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Flood, P.M.; Hong, J.S. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. J. Neural Transm. 2010, 117, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2016, 155, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Wang, T.; Wei, S.-J.; Block, M.L.; Wilson, B.; Liu, B.; Hong, J.-S. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem. 2004, 279, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Hamill, C.E.; Caudle, W.M.; Richardson, J.R.; Yuan, H.; Pennell, K.D.; Greene, J.G.; Miller, G.W.; Traynelis, S.F. Exacerbation of dopaminergic terminal damage in a mouse model of Parkinson’s disease by the G-protein-coupled receptor protease-activated receptor 1. Mol. Pharmacol. 2007, 72, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Purisai, M.G.; McCormack, A.L.; Cumine, S.; Li, J.; Isla, M.Z.; Di Monte, D.A. Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol. Dis. 2007, 25, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.C.; Teismann, P.; Tieu, K.; Vila, M.; Jackson-Lewis, V.; Ischiropoulos, H.; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 6145–6150. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Qin, L.; Liu, Y.; Hong, J.-S.; Crews, F.T. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 2013, 61, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Pallares, J.; Parga, J.A.; Muñoz, A.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007, 103, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sugama, S.; Chirichigno, J.W.; Gregorio, J.; Lorenzl, S.; Shin, D.H.; Browne, S.E.; Shimizu, Y.; Joh, T.H.; Beal, M.F.; et al. Minocycline enhances MPTP toxicity to dopaminergic neurons. J. Neurosci. Res. 2003, 74, 278–285. [Google Scholar] [CrossRef]
- Diguet, E.; Fernagut, P.-O.; Wei, X.; Du, Y.; Rouland, R.; Gross, C.; Bezard, E.; Tison, F. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur. J. Neurosci. 2004, 19, 3266–3276. [Google Scholar] [CrossRef]
- Gordon, P.H.; Moore, D.H.; Miller, R.G.; Florence, J.M.; Verheijde, J.L.; Doorish, C.; Hilton, J.F.; Spitalny, G.M.; MacArthur, R.B.; Mitsumoto, H.; et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomised trial. Lancet Neurol. 2007, 6, 1045–1053. [Google Scholar] [CrossRef]
- Ninds Net-Pd Investigators. A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin. Neuropharmacol. 2008, 31, 141–150. [Google Scholar] [CrossRef] [Green Version]
- LaPorta, V.N.; Nikitakis, N.G.; Sindler, A.J.; Reynolds, M.A. Minocycline-associated intra-oral soft-tissue pigmentation: Clinicopathologic correlations and review. J. Clin. Periodontol. 2005, 32, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Cascio, A.; Di Liberto, C.; D’Angelo, M.; Iaria, C.; Scarlata, F.; Titone, L.; Campisi, G. No findings of dental defects in children treated with minocycline. Antimicrob. Agents Chemother. 2004, 48, 2739–2741. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.; Harrison, A. Minocycline-induced lupus: A case series. N. Z. Med. J. 2003, 116, U384. [Google Scholar]
- Shetty, A.K. Tetracyclines in pediatrics revisited. Clin. Pediatr. 2002, 41, 203–209. [Google Scholar] [CrossRef]
- Smith, K.; Leyden, J.J. Safety of doxycycline and minocycline: A systematic review. Clin. Ther. 2005, 27, 1329–1342. [Google Scholar] [CrossRef]
- Tazzeo, T.; Worek, F.; Janssen, L. The NADPH oxidase inhibitor diphenyleneiodonium is also a potent inhibitor of cholinesterases and the internal Ca(2+) pump. Br. J. Pharmacol. 2009, 158, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, C.; DeCoursey, T.E. Voltage-gated proton channels in microglia. Prog. Neurobiol. 2001, 64, 277–305. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J. Voltage-gated proton channel HV1 in microglia. Neuroscientist 2014, 20, 599–609. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mony, L.; Stroebel, D.; Isacoff, E.Y. Dimer interaction in the Hv1 proton channel. Proc. Natl. Acad. Sci. USA 2020, 117, 20898–20907. [Google Scholar] [CrossRef]
- He, J.; Ritzel, R.M.; Wu, J. Functions and Mechanisms of the Voltage-Gated Proton Channel Hv1 in Brain and Spinal Cord Injury. Front. Cell. Neurosci. 2021, 15, 662971. [Google Scholar] [CrossRef]
- Li, Y.; Ritzel, R.M.; He, J.; Cao, T.; Sabirzhanov, B.; Li, H.; Liu, S.; Wu, L.J.; Wu, J. The voltage-gated proton channel Hv1 plays a detrimental role in contusion spinal cord injury via extracellular acidosis-mediated neuroinflammation. Brain Behav. Immun. 2021, 91, 267–283. [Google Scholar] [CrossRef]
- Wu, L.J.; Wu, G.; Sharif, M.R.A.; Baker, A.; Jia, Y.; Fahey, F.H.; Luo, H.R.; Feener, E.P.; Clapham, D.E. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 2012, 15, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tian, D.; Murugan, M.; Eyo, U.B.; Dreyfus, C.F.; Wang, W.; Wu, L.-J. Microglial Hv1 proton channel promotes cuprizone-induced demyelination through oxidative damage. J. Neurochem. 2015, 135, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, I.S.; Ruchti, E.; Kaczmarek, J.S.; Clapham, D.E. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc. Natl. Acad. Sci. USA 2009, 106, 7642–7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnick, T.G.; Papapetropoulos, S.; Mash, D.C.; Ffrench-Mullen, J.; Shehadeh, L.; de Andrade, M.; Henley, J.; A Rocca, W.; Ahlskog, J.E.; Maraganore, D.M. A genomic pathway approach to a complex disease: Axon guidance and Parkinson disease. PLoS Genet. 2007, 3, e98. [Google Scholar] [CrossRef]
- Duke, D.C.; Moran, L.B.; Kalaitzakis, M.E.; Deprez, M.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Transcriptome analysis reveals link between proteasomal and mitochondrial pathways in Parkinson’s disease. Neurogenetics 2006, 7, 139–148. [Google Scholar] [CrossRef]
- Moran, L.B.; Duke, D.; Deprez, M.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics 2006, 7, 1–11. [Google Scholar] [CrossRef]
- Dijkstra, A.A.; Ingrassia, A.; de Menezes, R.X.; van Kesteren, R.E.; Rozemuller, A.J.; Heutink, P.; van de Berg, W.D. Evidence for Immune Response, Axonal Dysfunction and Reduced Endocytosis in the Substantia Nigra in Early Stage Parkinson’s Disease. PLoS ONE 2015, 10, e0128651. [Google Scholar] [CrossRef] [Green Version]
- Corradini, B.R.; Iamashita, P.; Tampellini, E.; Farfel, J.M.; Grinberg, L.T.; Moreira-Filho, C.A. Complex network-driven view of genomic mechanisms underlying Parkinson’s disease: Analyses in dorsal motor vagal nucleus, locus coeruleus, and substantia nigra. Biomed. Res. Int. 2014, 2014, 543673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Park. Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Serdar, C.C.; Cihan, M.; Yücel, D.; A Serdar, M. Sample size, power and effect size revisited: Simplified and practical approaches in pre-clinical, clinical and laboratory studies. Biochem. Med. 2021, 31, 010502. [Google Scholar] [CrossRef] [PubMed]
- Beier, E.E.; Neal, M.; Alam, G.; Edler, M.; Wu, L.-J.; Richardson, J.R. Alternative microglial activation is associated with cessation of progressive dopamine neuron loss in mice systemically administered lipopolysaccharide. Neurobiol. Dis. 2017, 108, 115–127. [Google Scholar] [CrossRef]
- National Research Council Committee. The National Academies Collection: Reports funded by National Institutes of Health. In Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Hossain, M.M.; DiCicco-Bloom, E.; Richardson, J.R. Hippocampal ER stress and learning deficits following repeated pyrethroid exposure. Toxicol. Sci. 2015, 143, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Alam, G.; Edler, M.; Burchfield, S.; Richardson, J.R. Single low doses of MPTP decrease tyrosine hydroxylase expression in the absence of overt neuron loss. Neurotoxicology 2017, 60, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.M.; Sonsalla, P.K.; Richardson, J.R. Coordinated role of voltage-gated sodium channels and the Na+/H+ exchanger in sustaining microglial activation during inflammation. Toxicol. Appl. Pharmacol. 2013, 273, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Hogan, C.E.; Neal, M.L.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. A simple magnetic separation method for high-yield isolation of pure primary microglia. J. Neurosci. Methods 2011, 194, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Malovic, E.; Plante, B.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Rapid and Refined CD11b Magnetic Isolation of Primary Microglia with Enhanced Purity and Versatility. J. Vis. Exp. 2017, 122, e55364. [Google Scholar]
- Neal, M.L.; Boyle, A.M.; Budge, K.M.; Safadi, F.F.; Richardson, J.R. The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J. Neuroinflamm. 2018, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Baquet, Z.C.; Williams, D.; Brody, J.; Smeyne, R. A comparison of model-based (2D) and design-based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Neuroscience 2009, 161, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Bradner, J.M.; Suragh, T.A.; Wilson, W.W.; Lazo, C.R.; Stout, K.A.; Kim, H.M.; Wang, M.Z.; Walker, D.I.; Pennell, K.D.; Richardson, J.R.; et al. Exposure to the polybrominated diphenyl ether mixture DE-71 damages the nigrostriatal dopamine system: Role of dopamine handling in neurotoxicity. Exp. Neurol. 2013, 241, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, M.L.; Fleming, S.M.; Budge, K.M.; Boyle, A.M.; Kim, C.; Alam, G.; Beier, E.E.; Wu, L.J.; Richardson, J.R. Pharmacological inhibition of CSF1R by GW2580 reduces microglial proliferation and is protective against neuroinflammation and dopaminergic neurodegeneration. FASEB J. 2020, 34, 1679–1694. [Google Scholar] [CrossRef] [Green Version]
- West, M.J.; Slomianka, L.; Gundersen, H.J. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 1991, 231, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7 (Suppl. S1), S11–S19. [Google Scholar] [CrossRef] [Green Version]
- Risiglione, P.; Leggio, L.; Cubisino, S.A.; Reina, S.; Paternò, G.; Marchetti, B.; Magrì, A.; Iraci, N.; Messina, A. High-Resolution Respirometry Reveals MPP(+) Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7809. [Google Scholar] [CrossRef]
- Heikkila, R.E.; Hess, A.; Duvoisin, R.C. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science 1984, 224, 1451–1453. [Google Scholar] [CrossRef]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonnekes, J.; Post, B.; Tetrud, J.W.; Langston, J.W.; Bloem, B.R. MPTP-induced parkinsonism: An historical case series. Lancet Neurol. 2018, 17, 300–301. [Google Scholar] [CrossRef] [Green Version]
- Noelker, C.; Morel, L.; Lescot, T.; Osterloh, A.; Alvarez-Fischer, D.; Breloer, M.; Henze, C.; Depboylu, C.; Skrzydelski, D.; Michel, P.P.; et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci. Rep. 2013, 3, 1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, J.P.; Miller, D.B.; Reinhard, J.F. Characterization of the origins of astrocyte response to injury using the dopaminergic neurotoxicant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 1990, 521, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Park. Dis 2011, 1, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.-C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.S.; Li, C.-Y.; Qin, C.; Murugan, M.; Wu, L.-J.; Liu, J.-L. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J. Neurochem. 2016, 139, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.R.; Hossain, M.M. Microglial ion channels as potential targets for neuroprotection in Parkinson’s disease. Neural Plast. 2013, 2013, 587418. [Google Scholar] [CrossRef] [Green Version]
- Duda, J.; Pötschke, C.; Liss, B. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 156–178. [Google Scholar] [CrossRef] [Green Version]
- Oltra, J.; Uribe, C.; Campabadal, A.; Inguanzo, A.; Monté-Rubio, G.C.; Martí, M.J.; Compta, Y.; Valldeoriola, F.; Junque, C.; Segura, B. Sex Differences in Brain and Cognition in de novo Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 791532. [Google Scholar] [CrossRef]
- Lynch, M.A. Exploring Sex-Related Differences in Microglia May Be a Game-Changer in Precision Medicine. Front. Aging Neurosci. 2022, 14, 868448. [Google Scholar] [CrossRef]
- Lopez-Cerdan, A.; Andreu, Z.; Hidalgo, M.R.; Grillo-Risco, R.; Català-Senent, J.F.; Soler-Sáez, I.; Neva-Alejo, A.; Gordillo, F.; de la Iglesia-Vayá, M.; García-García, F. Unveiling sex-based differences in Parkinson’s disease: A comprehensive meta-analysis of transcriptomic studies. Biol. Sex Differ. 2022, 13, 68. [Google Scholar] [CrossRef]
- Siani, F.; Greco, R.; Levandis, G.; Ghezzi, C.; Daviddi, F.; Demartini, C.; Vegeto, E.; Fuzzati-Armentero, M.-T.; Blandini, F. Influence of Estrogen Modulation on Glia Activation in a Murine Model of Parkinson’s Disease. Front. Neurosci. 2017, 11, 306. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Chakrabarti, N.; Dutta, S.S.; Ray, S.; Bhattacharya, P.; Sinha, P.; Bhattacharyya, A. Gender-specific brain regional variation of neurons, endogenous estrogen, neuroinflammation and glial cells during rotenone-induced mouse model of Parkinson’s disease. Neuroscience 2015, 292, 46–70. [Google Scholar] [CrossRef] [PubMed]
- Pattarini, R.; Smeyne, R.J.; Morgan, J.I. Temporal mRNA profiles of inflammatory mediators in the murine 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrimidine model of Parkinson’s disease. Neuroscience 2007, 145, 654–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattarini, R.; Rong, Y.; Qu, C.; Morgan, J. Distinct mechanisms of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrimidine resistance revealed by transcriptome mapping in mouse striatum. Neuroscience 2008, 155, 1174–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson-Lewis, V.; Jakowec, M.; E Burke, R.; Przedborski, S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration 1995, 4, 257–269. [Google Scholar] [CrossRef]
- Jiang, L.; Wu, X.; Wang, S.; Chen, S.-H.; Zhou, H.; Wilson, B.; Jin, C.-Y.; Lu, R.-B.; Xie, K.; Wang, Q.; et al. Clozapine metabolites protect dopaminergic neurons through inhibition of microglial NADPH oxidase. J. Neuroinflamm. 2016, 13, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capasso, M.; Bhamrah, M.K.; Henley, T.; Boyd, R.S.; Langlais, C.; Cain, K.; Dinsdale, D.; Pulford, K.; Khan, M.; Musset, B.; et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat. Immunol. 2010, 11, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Okochi, Y.; Sasaki, M.; Iwasaki, H.; Okamura, Y. Voltage-gated proton channel is expressed on phagosomes. Biochem. Biophys. Res. Commun. 2009, 382, 274–279. [Google Scholar] [CrossRef]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, A.; Gonzalez Leon, C. In pursuit of an inhibitory drug for the proton channel. Proc. Natl. Acad. Sci. USA 2014, 111, 9673–9674. [Google Scholar] [CrossRef] [Green Version]
- Capasso, M.; DeCoursey, T.E.; Dyer, M.J. pH regulation and beyond: Unanticipated functions for the voltage-gated proton channel, HVCN1. Trends Cell Biol. 2011, 21, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.A.; Valente, A.J. Nuclear factor kappa B activation by NADPH oxidases. Mech. Ageing Dev. 2004, 125, 799–810. [Google Scholar] [CrossRef]
- Bassani, T.B.; Vital, M.A.; Rauh, L.K. Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq. Neuropsiquiatr. 2015, 73, 616–623. [Google Scholar] [CrossRef]
- Flood, P.; Arbabzada, N.; Sharma, M. Inflammation: Role in Parkinson’s Disease and Target for Therapy. In Challenges in Parkinson’s Disease; INTECH: London, UK, 2016. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neal, M.L.; Beier, E.E.; Hossain, M.M.; Boyle, A.; Zheng, J.; Kim, C.; Mhatre-Winters, I.; Wu, L.-J.; Richardson, J.R. Voltage-Gated Proton Channel Hv1 Regulates Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease Models. Antioxidants 2023, 12, 582. https://doi.org/10.3390/antiox12030582
Neal ML, Beier EE, Hossain MM, Boyle A, Zheng J, Kim C, Mhatre-Winters I, Wu L-J, Richardson JR. Voltage-Gated Proton Channel Hv1 Regulates Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease Models. Antioxidants. 2023; 12(3):582. https://doi.org/10.3390/antiox12030582
Chicago/Turabian StyleNeal, Matthew L., Eric E. Beier, Muhammad M. Hossain, Alexa Boyle, Jiaying Zheng, Chunki Kim, Isha Mhatre-Winters, Long-Jun Wu, and Jason R. Richardson. 2023. "Voltage-Gated Proton Channel Hv1 Regulates Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease Models" Antioxidants 12, no. 3: 582. https://doi.org/10.3390/antiox12030582
APA StyleNeal, M. L., Beier, E. E., Hossain, M. M., Boyle, A., Zheng, J., Kim, C., Mhatre-Winters, I., Wu, L. -J., & Richardson, J. R. (2023). Voltage-Gated Proton Channel Hv1 Regulates Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease Models. Antioxidants, 12(3), 582. https://doi.org/10.3390/antiox12030582