
Muscle Oxidative Stress Plays a Role in Hyperthyroidism-Linked Insulin Resistance

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Glucose Tolerance Test
2.3. Tissue Preparations
2.4. Isolation of Muscle Mitochondria
2.5. Oxidative Damage Assessment and In Vitro Susceptibility to Oxidative Stress
2.6. Total ROS Content
2.7. NADPH Oxidase (NOX) Activity Assay
2.8. H2O2 Mitochondrial Release
2.9. Antioxidant Enzymes Activities
2.10. Tissue and Mitochondrial Respiration
2.11. Response of Skeletal Muscle to Insulin and Immunoblotting Analyses
2.12. RNA Isolation, RT-PCR, and qPCR
2.13. Data Analysis
3. Results
3.1. Body Parameters
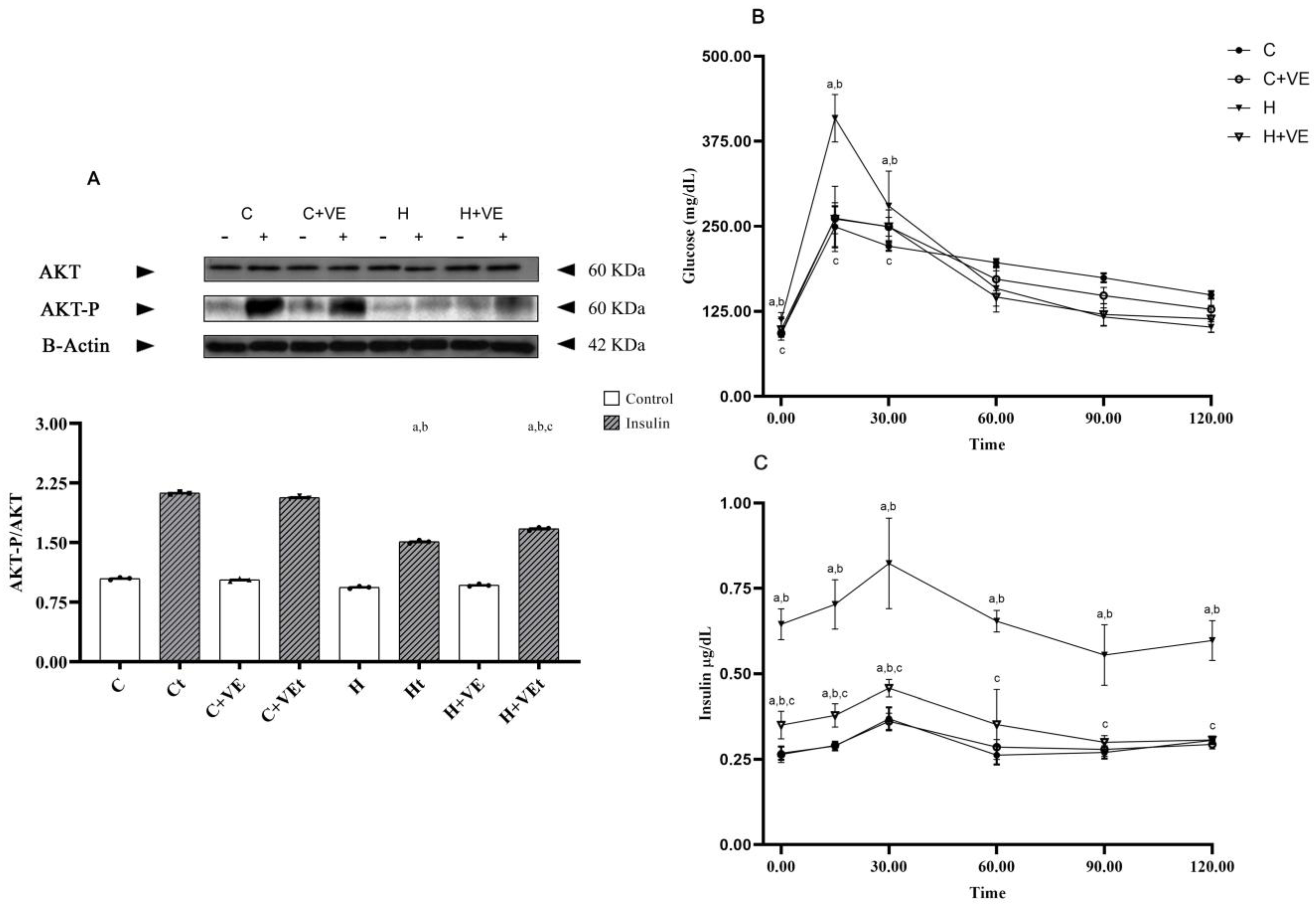
3.2. Glucose Tolerance Test and Insulin Levels
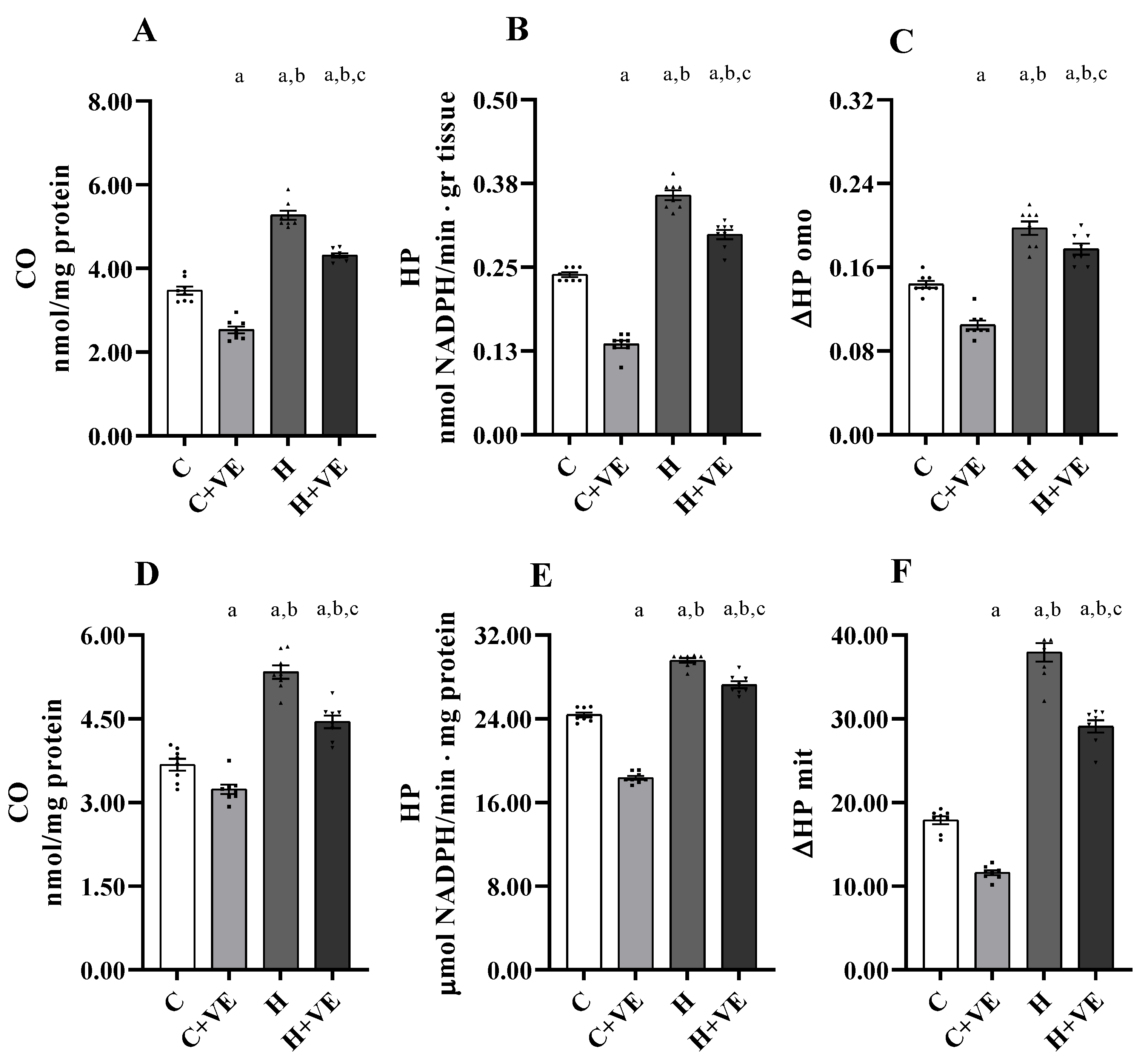
3.3. Oxidative Damage to Lipids and Proteins and Susceptibility to Oxidative Damage
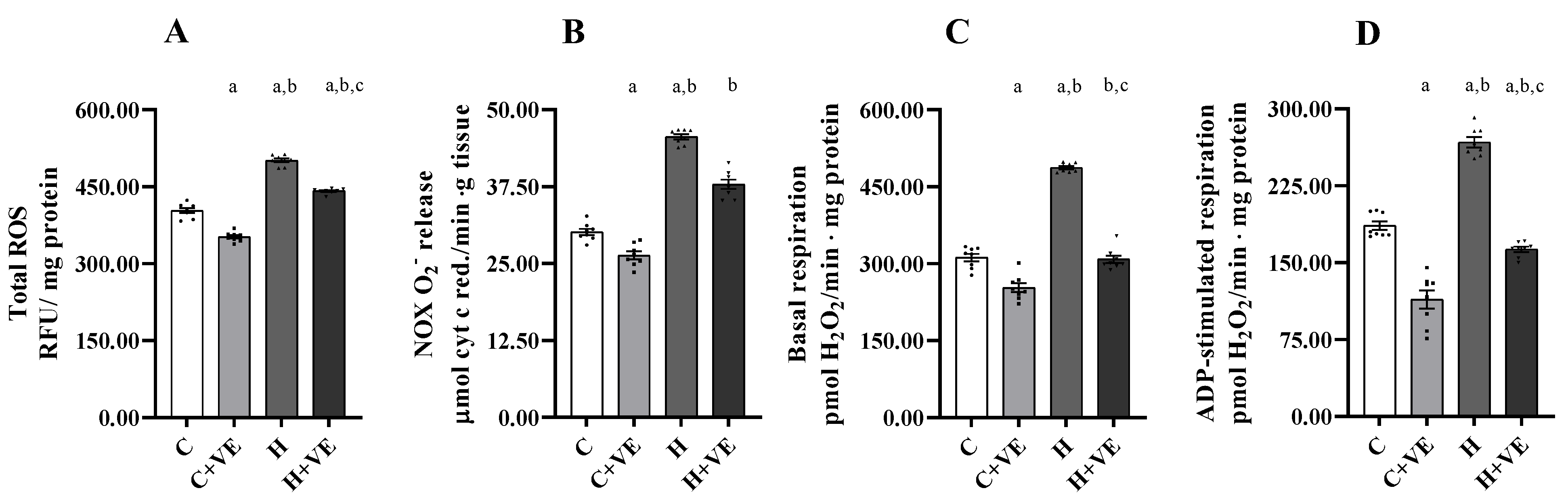
3.4. Muscle Total ROS Content, Mitochondrial H2O2 Release and NADPH Oxidase (NOX) Activity
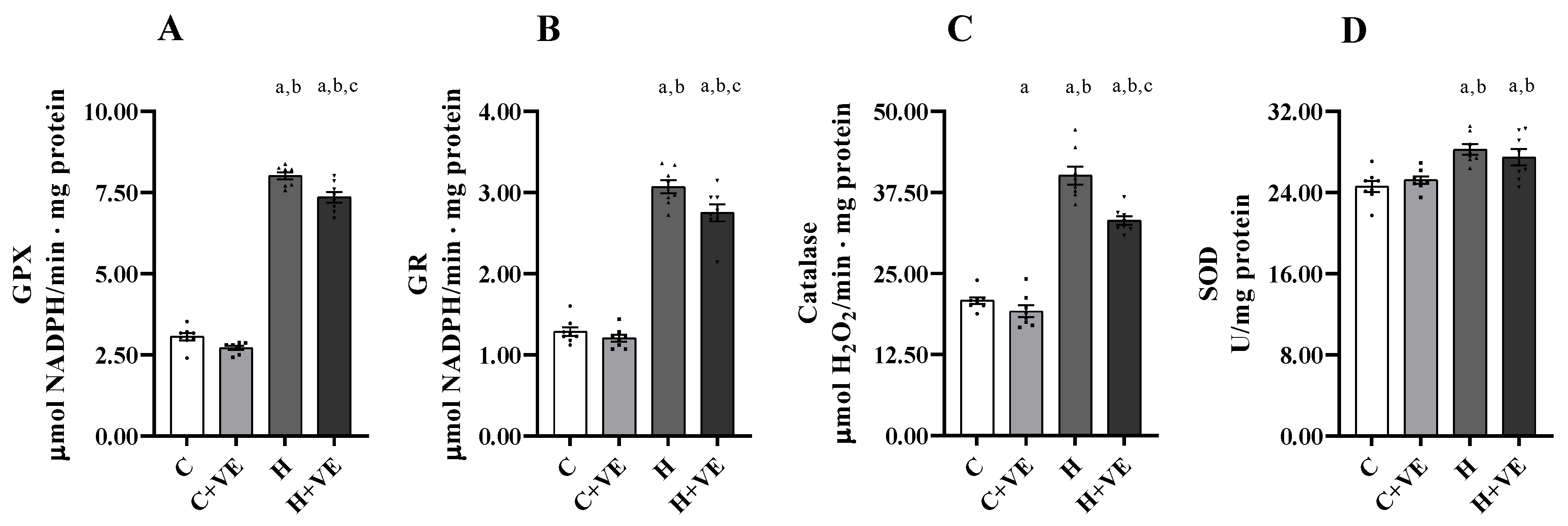
3.5. Antioxidant Enzyme Activity of Skeletal Muscle Homogenate
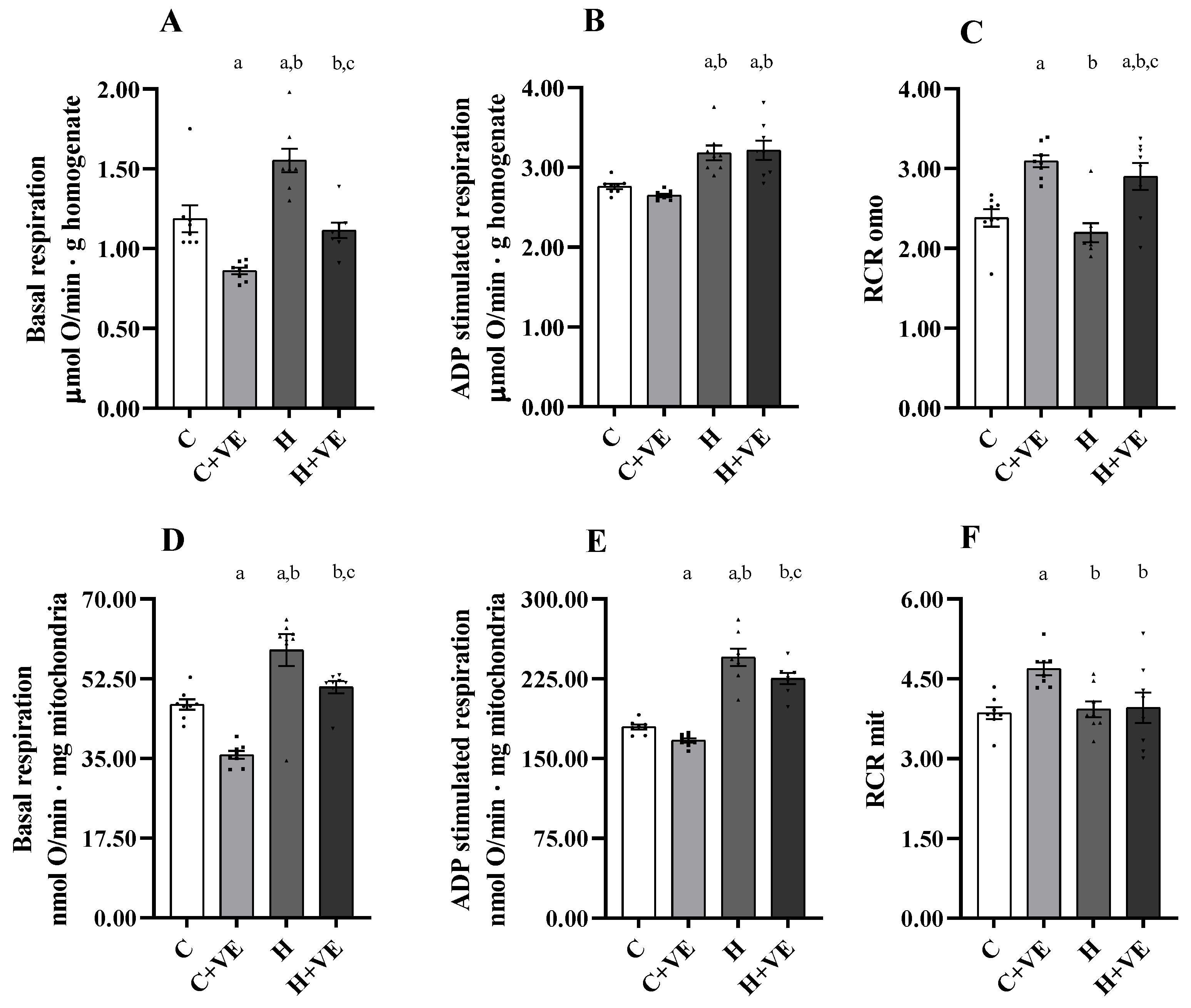
3.6. Oxygen Consumption of Skeletal Muscle Homogenate and Isolated Mitochondria
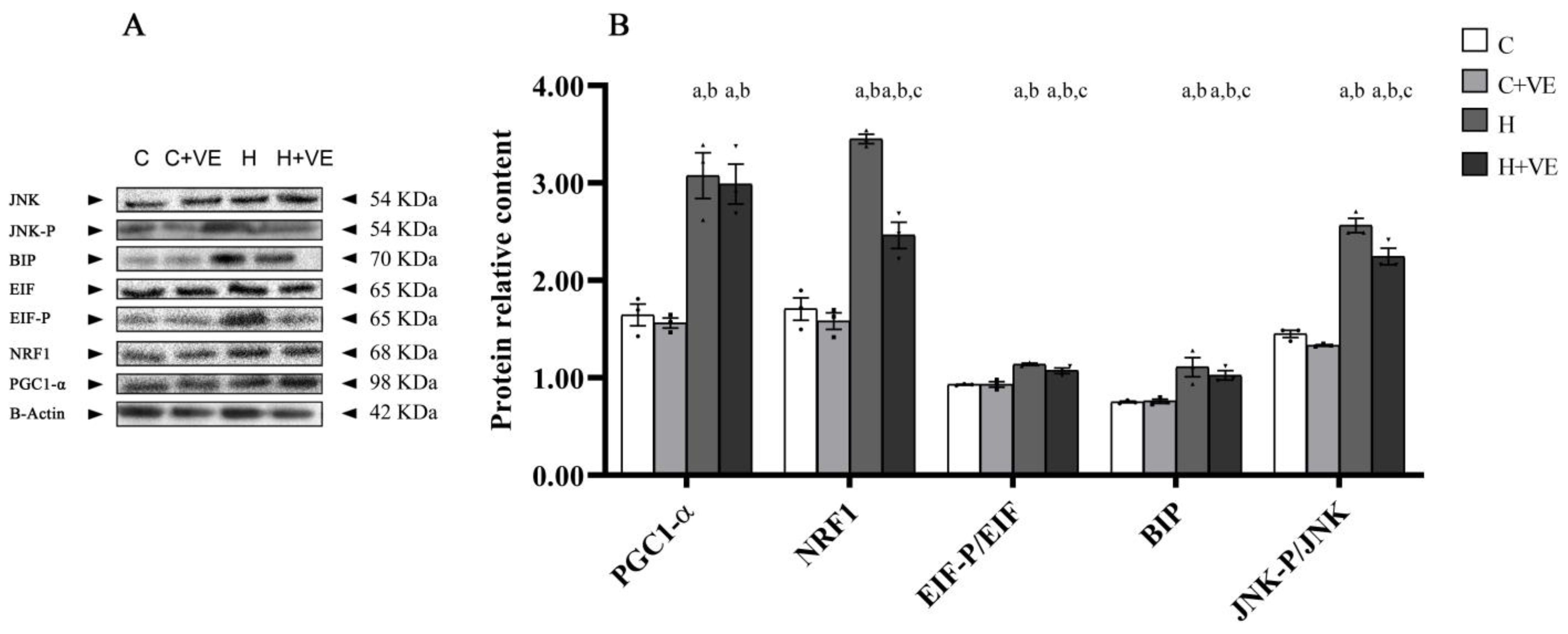
3.7. Factors Involved in Stress Response
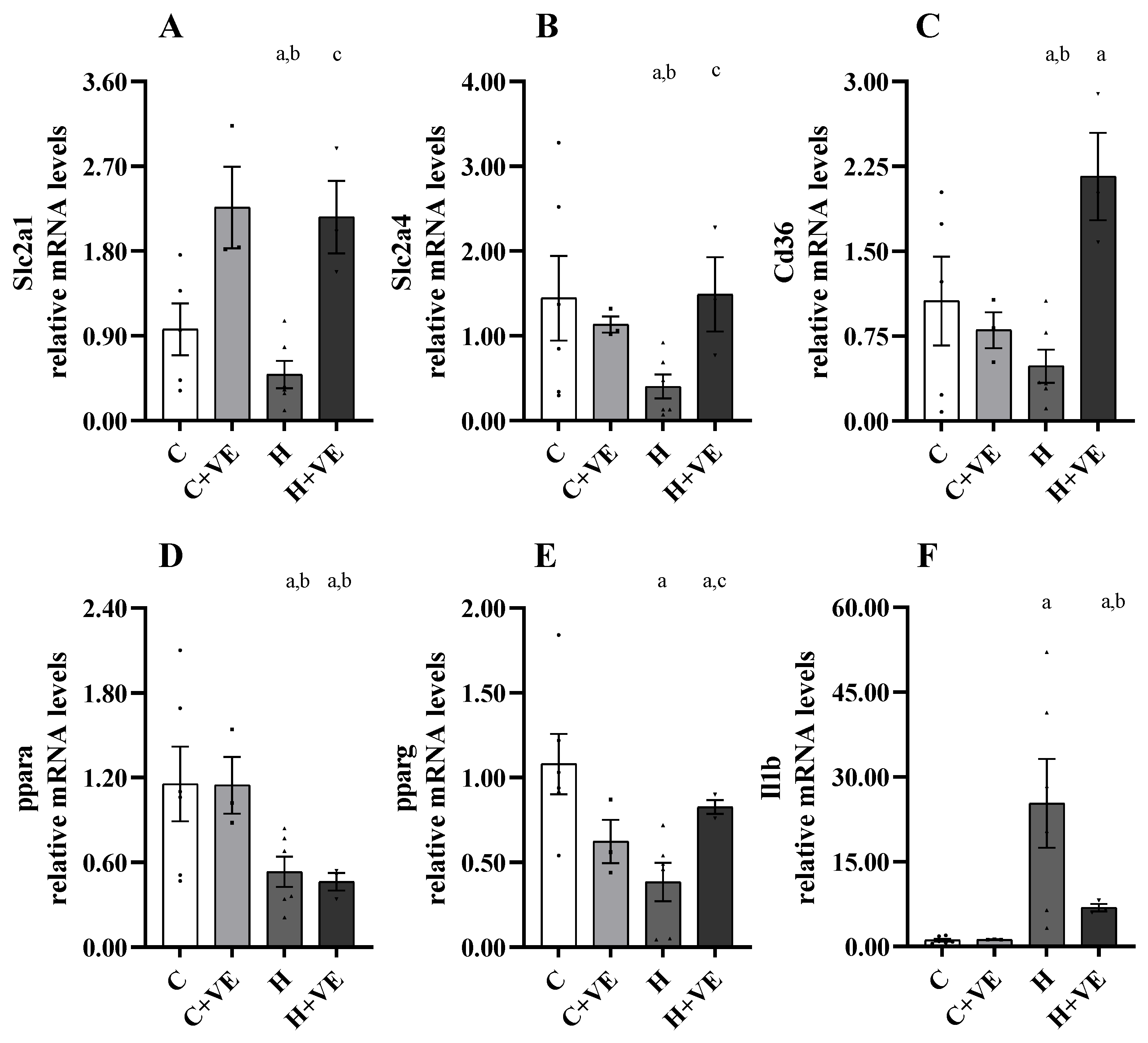
3.8. Gene Expression Analysis of Slc2a1, Slc2a4, Pparg, Ppara, Cd36 and Il1b
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Napolitano, G.; Fasciolo, G.; Venditti, P. The Ambiguous Aspects of Oxygen. Oxygen 2022, 2, 382–409. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Reed, T.T.; Victor, V.M.; Di Meo, S. Insulin resistance and diabetes in hyperthyroidism: A possible role for oxygen and nitrogen reactive species. Free Radic. Res. 2019, 53, 248–268. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.E.B.F.; Moreira, N.C.S.; Sakamoto-Hojo, E.T. Mechanisms underlying the pathophysiology of type 2 diabetes: From risk factors to oxidative stress, metabolic dysfunction, and hyperglycemia. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2022, 874–875, 503437. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Wassmann, S.; Czech, T.; Münzel, T.; Eisenhauer, M.; Böhm, M.; Nickenig, G. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chen, K.; Whaley-Connell, A.T.; Stump, C.S.; Ibdah, J.A.; Sowers, J.R. Skeletal muscle insulin resistance: Role of inflammatory cytokines and reactive oxygen species. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R673–R680. [Google Scholar] [CrossRef] [Green Version]
- Rohdenburg, G.L. Thyroid diabetes. Endocrinology 1920, 4, 63–70. [Google Scholar] [CrossRef]
- Foss, M.C.; Paccola, G.M.; Saad, M.J.; Pimenta, W.P.; Piccinato, C.E.; Iazigi, N. Peripheral glucose metabolism in human hyperthyroidism. J. Clin. Endocrinol. Metab. 1990, 70, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.P.; O’Boyle, E.; Haber, R.S. Thyroid hormone increases basal and insulin-stimulated glucose transport in skeletal muscle. The role of GLUT4 glucose transporter expression. Diabetes 1994, 43, 1185–1189. [Google Scholar] [CrossRef]
- Casla, A.; Rovira, A.; Wells, J.A.; Dohm, G.L. Increased glucose transporter (GLUT4) protein expression in hyperthyroidism. Biochem. Biophys. Res. Commun. 1990, 171, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Boutati, E.; Maratou, E.; Koukkou, E.; Panagiotakos, D.; Tountas, N.; Economopoulos, T.; Raptis, S.A. Insulin stimulated rates of glucose uptake in muscle in hyperthyroidism: The importance of blood flow. J. Clin. Endocrinol. Metab. 2008, 93, 2413–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, R.W.; White, A.E.; Metcalf, M.D.; Winters, B.R.; Diggs, B.S.; Zhu, X.; Marks, D.L. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism 2012, 61, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Dimitriadis, G.; Parry-Billings, M.; Bevan, S.; Leighton, B.; Krause, U.; Piva, T.; Tegos, K.; Challiss, R.A.; Wegener, G.; Newsholme, E.A. The effects of insulin on transport and metabolism of glucose in skeletal muscle from hyperthyroid and hypothyroid rats. Eur. J. Clin. Investig. 1997, 27, 475–483. [Google Scholar] [CrossRef]
- Venditti, P.; Napolitano, G.; Barone, D.; Di Meo, S. Effect of training and vitamin E administration on rat liver oxidative metabolism. Free Radic. Res. 2014, 48, 322–332. [Google Scholar] [CrossRef]
- Venditti, P.; De Leo, T.; Di Meo, S. Vitamin E administration attenuates the tri-iodothyronine-induced modification of heart electrical activity in the rat. J. Exp. Biol. 1997, 200, 909–914. [Google Scholar] [CrossRef]
- Lang, J.K.; Gohil, K.; Packer, L. Simultaneous determination of tocopherols, ubiquinols, and ubiquinones in blood, plasma, tissue homogenates, and subcellular fractions. Anal. Biochem. 1986, 157, 106–116. [Google Scholar] [CrossRef]
- Heath, R.L.; Tappel, A.L. A new sensitive assay for the measurement of hydroperoxides. Anal. Biochem. 1976, 76, 184–191. [Google Scholar] [CrossRef]
- Mesquita, C.S.; Oliveira, R.; Bento, F.; Geraldo, D.; Rodrigues, J.V.; Marcos, J.C. Simplified 2,4-dinitrophenylhydrazine spectrophotometric assay for quantification of carbonyls in oxidized proteins. Anal. Biochem. 2014, 458, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Fasciolo, G.; Napolitano, G.; Aprile, M.; Cataldi, S.; Costa, V.; Ciccodicola, A.; Di Meo, S.; Venditti, P. Hepatic Insulin Resistance in Hyperthyroid Rat Liver: Vitamin E Supplementation Highlights a Possible Role of ROS. Antioxidants 2022, 11, 1295. [Google Scholar] [CrossRef] [PubMed]
- Driver, A.S.; Kodavanti, P.R.; Mundy, W.R. Age-related changes in reactive oxygen species production in rat brain homogenates. Neurotoxicol. Teratol. 2000, 22, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Lehrer, R.I. NAD(P)H oxidase activity in human neutrophils stimulated by phorbol myristate acetate. J. Clin. Investig. 1980, 66, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Minkenberg, I.; Ferber, E. Lucigenin-dependent chemiluminescence as a new assay for NAD(P)H-oxidase activity in particulate fractions of human polymorphonuclear leukocytes. J. Immunol. Methods 1984, 71, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Hyslop, P.A.; Sklar, L.A. A quantitative fluorimetric assay for the determination of oxidant production by polymorphonuclear leukocytes: Its use in the simultaneous fluorimetric assay of cellular activation processes. Anal. Biochem. 1984, 141, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Günzler, W.A. Assays of glutathione peroxidase. Methods Enzymol. 1984, 105, 114–121. [Google Scholar]
- Carlberg, I.; Mannervik, B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar]
- Flohé, L.; Otting, F. Superoxide dismutase assays. Methods Enzymol. 1984, 105, 93–104. [Google Scholar]
- Amouzou, C.; Breuker, C.; Fabre, O.; Bourret, A.; Lambert, K.; Birot, O.; Fédou, C.; Dupuy, A.M.; Cristol, J.P.; Sutra, T.; et al. Skeletal Muscle Insulin Resistance and Absence of Inflammation Characterize Insulin-Resistant Grade I Obese Women. PLoS ONE 2016, 11, e0154119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditti, P.; Napolitano, G.; Fasciolo, G.; Di Meo, S. Thyroid state affects H2O2 removal by rat heart mitochondria. Arch. Biochem. Biophys. 2019, 662, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Fasciolo, G.; Di Meo, S.; Venditti, P. Vitamin E Supplementation and Mitochondria in Experimental and Functional Hyperthyroidism: A Mini-Review. Nutrients 2019, 11, 2900. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Weng, S.W.; Chang, Y.H.; Su, J.; Chang, M.; Tsai, C.J.; Shen, F.C.; Chuang, J.H.; Lin, T.K.; Liou, W.; et al. The causal role of mitochondrial dynamics in regulating insulin resistance in diabetes: Link through mitochondrial reactive oxygen species. Oxid. Med. Cell Longev. 2018, 2018, 7514383. [Google Scholar] [CrossRef]
- Lark, D.S.; Fisher-Wellman, K.H.; Neufer, P.D. High-fat load: Mechanism(s) of insulin resistance in skeletal muscle. Int. J. Obes. 2012, 2 (Suppl. S2), S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; De Rosa, R.; Portero-Otin, M.; Pamplona, R.; Di Meo, S. Cold-induced hyperthyroidism produces oxidative damage in rat tissues and increases susceptibility to oxidants. Int. J. Biochem. Cell Biol. 2004, 36, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox. Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Sakellariou, G.K.; Jackson, M.J.; Vasilaki, A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014, 48, 12–29. [Google Scholar] [CrossRef]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Duarte, S.; Montoya-Pérez, R.; Márquez-Gamiño, S.; Vera-Delgado, K.S.; Caudillo-Cisneros, C.; Sotelo-Barroso, F.; Sánchez-Briones, L.A.; Sánchez-Duarte, E. Apocynin Attenuates Diabetes-Induced Skeletal Muscle Dysfunction by Mitigating ROS Generation and Boosting Antioxidant Defenses in Fast-Twitch and Slow-Twitch Muscles. Life 2022, 12, 674. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Salbitani, G.; Venditti, P. Chlorella sorokiniana Dietary Supplementation Increases Antioxidant Capacities and Reduces Ros Release in Mitochondria of Hyperthyroid Rat Liver. Antioxidants 2020. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Meo, S. Thyroid hormone-induced oxidative stress. Cell Mol.. Life Sci. 2006, 63, 414–434. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, J.M.; Iwen, K.A. Coordination of mitochondrial biogenesis by thyroid hormone. Mol. Cell Endocrinol. 2011, 342, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giammanco, M.; Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. Int. J. Mol. Sci. 2020, 21, 4140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gauthier, K.; Ho, J.P.; Lim, A.; Zhu, X.G.; Han, C.R.; Sinha, R.A.; Cheng, S.Y.; Yen, P.M. Thyroid Hormone Receptor α Regulates Autophagy, Mitochondrial Biogenesis, and Fatty Acid Use in Skeletal Muscle. Endocrinology 2021, 162, bqab112. [Google Scholar] [CrossRef]
- Cachia, O.; Benna, J.E.; Pedruzzi, E.; Descomps, B.; Gougerot-Pocidalo, M.A.; Leger, C.L. alpha-tocopherol inhibits the respiratory burst in human monocytes. Attenuation of p47(phox) membrane translocation and phosphorylation. J. Biol Chem. 1998, 273, 32801–32805. [Google Scholar] [CrossRef] [Green Version]
- Calvisi, D.F.; Ladu, S.; Hironaka, K.; Factor, V.M.; Thorgeirsson, S.S. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer. J. Hepatol. 2004, 41, 815–822. [Google Scholar] [CrossRef]
- Chow, C.K. Vitamin E regulation of mitochondrial superoxide generation. Biol. Signals Recept. 2001. [Google Scholar] [CrossRef]
- Venditti, P.; Balestrieri, M.; Di Meo, S.; De Leo, T. Effect of thyroid state on lipid peroxidation, antioxidant defences, and susceptibility to oxidative stress in rat tissues. J. Endocrinol. 1997, 155, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Leutner, S.; Eckert, A.; Müller, W.E. ROS generation, lipid peroxidation and antioxidant enzyme activities in the aging brain. J. Neural. Transm. 2001, 108, 955–967. [Google Scholar] [CrossRef]
- Zamora, M.; Villena, J. Targeting mitochondrial biogenesis to treat insulin resistance. Curr. Pharm. Des. 2014, 20, 5527–5557. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditti, P.; Costagliola, I.R.; Di Meo, S. H2O2 production and response to stress conditions by mitochondrial fractions from rat liver. J. Bioenerg. Biomembr. 2002, 34, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.; Vidal-Puig, A.; Brand, M.D. Stimulation of mitochondrial proton conductance by hydroxynonenal requires a high membrane potential. Biosci. Rep. 2008, 28, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox. Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [Green Version]
- Gordaliza-Alaguero, I.; Cantó, C.; Zorzano, A. Metabolic implications of organelle–mitochondria communication. EMBO Rep. 2019, 20, e47928. [Google Scholar] [CrossRef]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-associated ER membranes—The origin site of autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef]
- Zhang, P.; Konja, D.; Zhang, Y.; Wang, Y. Communications between mitochondria and endoplasmic reticulum in the regulation of metabolic homeostasis. Cells 2021, 10, 2195. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Marañón, A.M.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melber, A.; Haynes, C. UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Venditti, P.; Bari, A.; Di Stefano, L.; Cardone, A.; Della Ragione, F.; D’Esposito, M.; Di Meo, S. Involvement of PGC-1, NRF-1, and NRF-2 in metabolic response by rat liver to hormonal and environmental signals. Mol. Cell Endocrinol. 2009, 305, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2016, 6, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Gao, Y.; Zhang, Y. Inhibition of JNK suppresses autophagy and attenuates insulin resistance in a rat model of nonalcoholic fatty liver disease. Mol. Med. Rep. 2017, 15, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The molecular Basis for Oxidative Stress-Induced Insulin Resistance. Antioxid. Redox. Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef]
- Birnbaum, M.J. Turning down insulin signalling. J. Clin. Investig. 2001, 108, 655–659. [Google Scholar] [CrossRef]
- Bloch-Damti, A.; Bashan, N. Proposed Mechanism for the Induction of Insulin Resistance by Oxidative Stress. Antioxid. Redox. Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef]
- Shen, H.M.; Liu, Z.G. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2006, 40, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, S.; Costa, V.; Ciccodicola, A.; Aprile, M. PPARγ and Diabetes: Beyond the Genome and Towards Personalized Medicine. Curr. Diab. Rep. 2021, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Gallo, M.A.; Letizia, F.; Aprile, M.; Casamassimi, A.; Ciccodicola, A. PPARG: Gene Expression Regulation and Next-Generation Sequencing for Unsolved Issues. PPAR Res. 2010, 2010, 409168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthier, A.; Johanns, M.; Zummo, F.P.; Lefebvre, P.; Staels, B. PPARs in liver physiology. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166097. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sense Primer (5′–3′) | Antisense Primer (5′–3′) | |
|---|---|---|
| 1 Slc2a1 | GCGGGCTGTGCTGTGCTC | CCACAGCAACAGCAGCAG |
| 2 Slc2a4 | ACCAGACCCGCCCTTTGC | CTGAAGGGAGCCAAGCAC |
| 3 Cd36 | TTACTGGAGCCGTTATTGGTG | CCTTGATCTTGCTGCTATTCT |
| 4 Ppara | CCACTTGAAGCAGATGACCT | CATTGCCAGGGGACTCATCT |
| 5 Pparg | GTCGGATCCACAAAAAGAGTA | TTTGTCTGTTGTCTTTCCTGT |
| 6 Il1b | AGGCTGACAGACCCCAAAAG | AAGCTCCACGGGCAAGACAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fasciolo, G.; Napolitano, G.; Aprile, M.; Cataldi, S.; Costa, V.; Muscari Tomajoli, M.T.; Lombardi, A.; Di Meo, S.; Venditti, P. Muscle Oxidative Stress Plays a Role in Hyperthyroidism-Linked Insulin Resistance. Antioxidants 2023, 12, 592. https://doi.org/10.3390/antiox12030592
Fasciolo G, Napolitano G, Aprile M, Cataldi S, Costa V, Muscari Tomajoli MT, Lombardi A, Di Meo S, Venditti P. Muscle Oxidative Stress Plays a Role in Hyperthyroidism-Linked Insulin Resistance. Antioxidants. 2023; 12(3):592. https://doi.org/10.3390/antiox12030592
Chicago/Turabian StyleFasciolo, Gianluca, Gaetana Napolitano, Marianna Aprile, Simona Cataldi, Valerio Costa, Maria Teresa Muscari Tomajoli, Assunta Lombardi, Sergio Di Meo, and Paola Venditti. 2023. "Muscle Oxidative Stress Plays a Role in Hyperthyroidism-Linked Insulin Resistance" Antioxidants 12, no. 3: 592. https://doi.org/10.3390/antiox12030592
APA StyleFasciolo, G., Napolitano, G., Aprile, M., Cataldi, S., Costa, V., Muscari Tomajoli, M. T., Lombardi, A., Di Meo, S., & Venditti, P. (2023). Muscle Oxidative Stress Plays a Role in Hyperthyroidism-Linked Insulin Resistance. Antioxidants, 12(3), 592. https://doi.org/10.3390/antiox12030592