

TNF-α-Mediated Endothelial Cell Apoptosis Is Rescued by Hydrogen Sulfide

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Highlights
- Hydrogen sulfide ameliorates TNF-α mediated endothelial dysfunction in human endothelial cells by regulating the intrinsic apoptosis pathway.
- Beneficial effects of hydrogen sulfide against TNF-α mediated apoptosis in endothelial cells are associated with S-sulfhydration of pro-caspase 3.
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
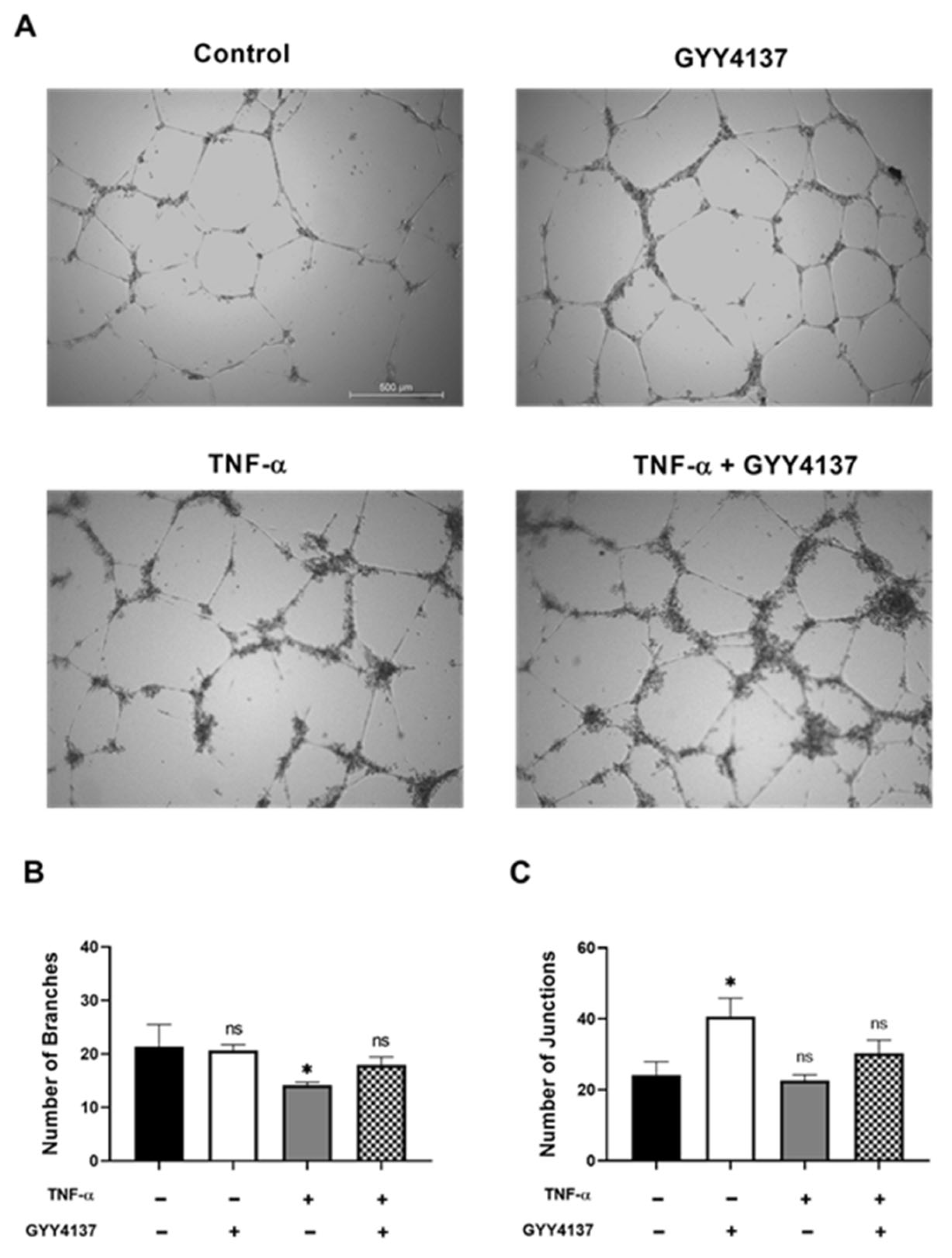
2.3. Tube Formation Assay
2.4. Measurement of Intracellular ROS Formation
2.5. Measurement of MitoSox Oxidation
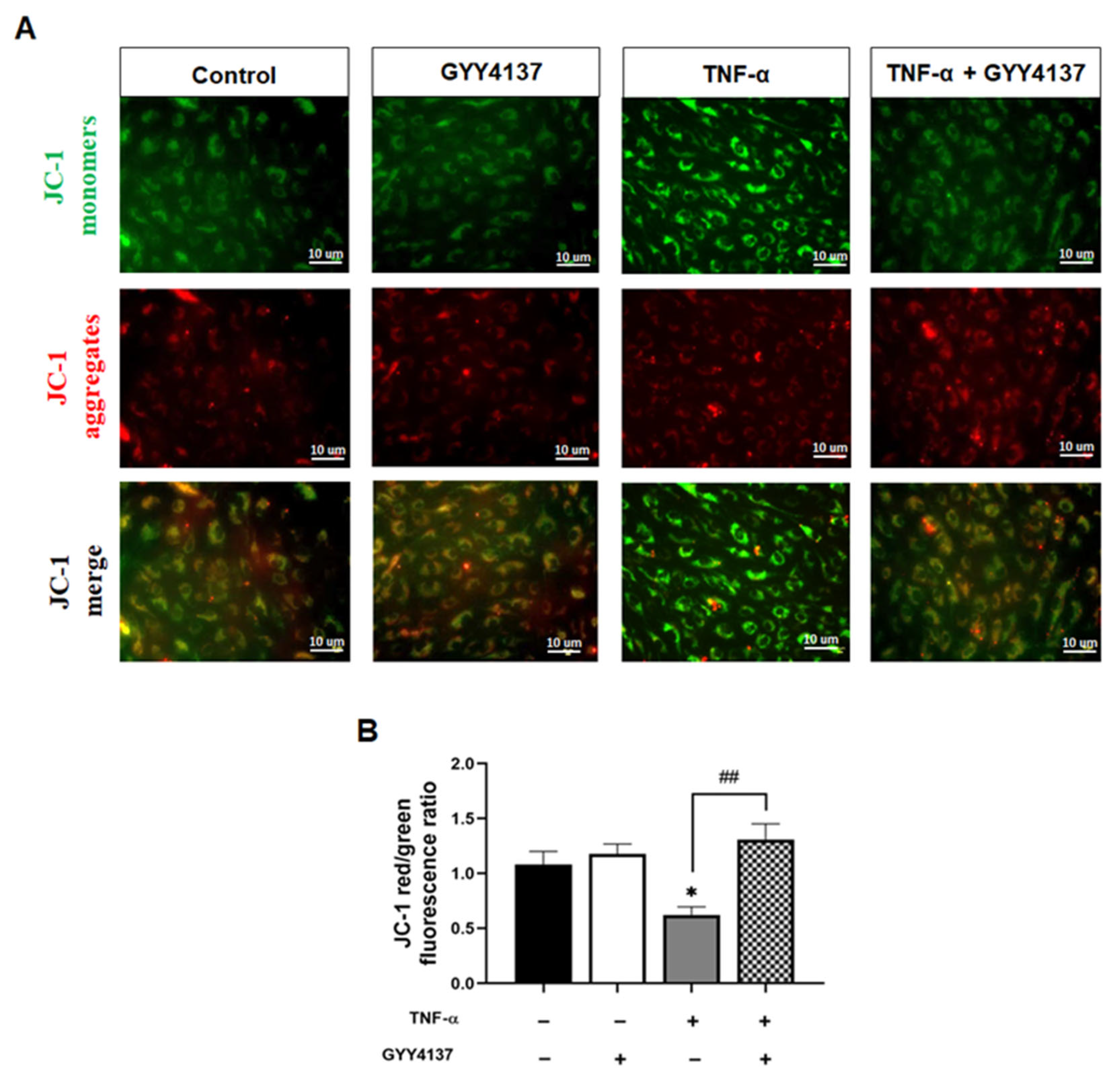
2.6. Assessment of Mitochondrial Membrane Potential (Δψm)
2.7. Detection of H2S
2.8. Interleukin-6 (IL-6) Levels
2.9. ICAM-1 (Intercellular Adhesion Molecule 1) Levels
2.10. Western Blotting
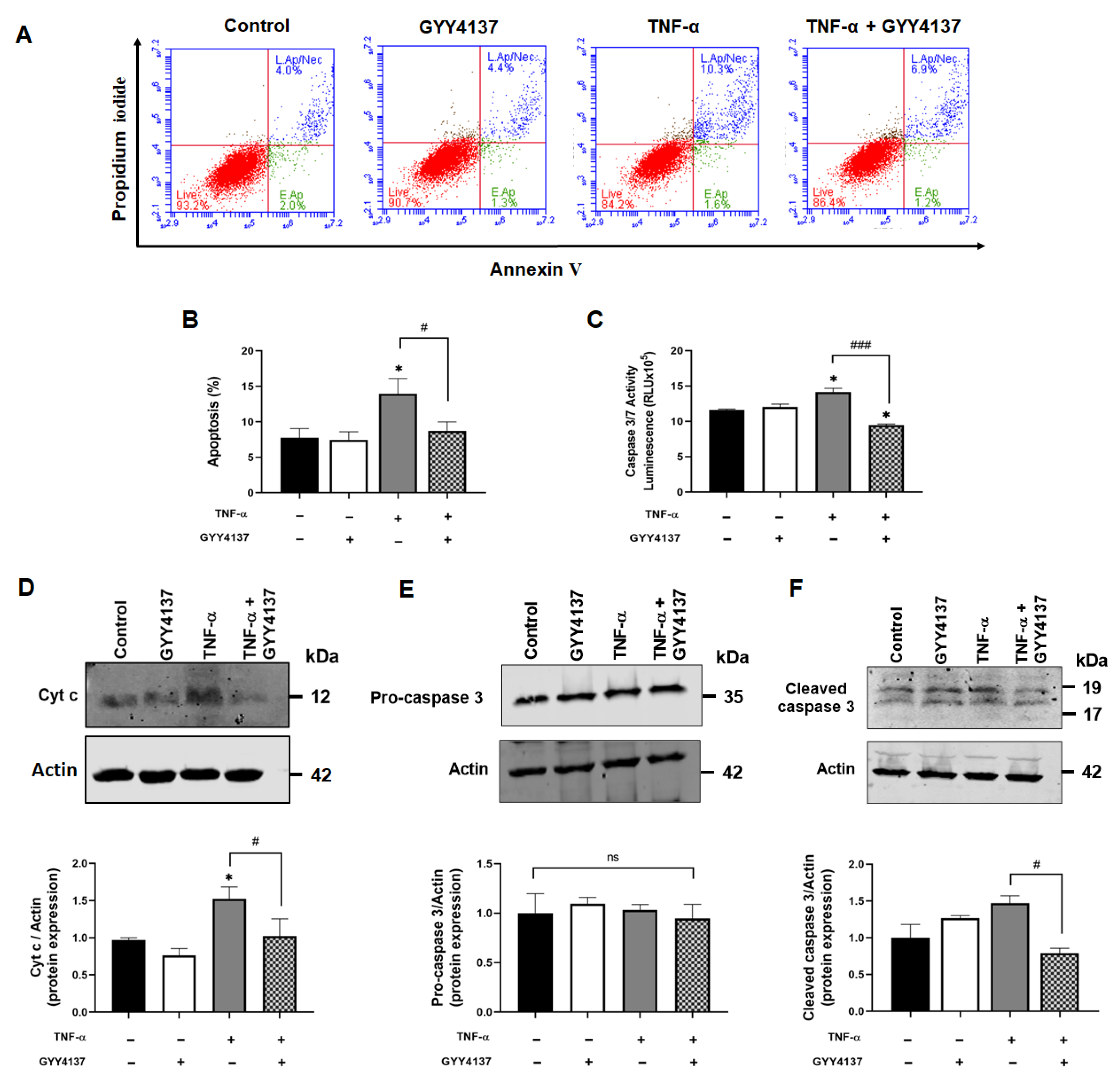
2.11. Flow Cytometry Analysis of Apoptosis
2.12. Caspase 3/7 Activity
2.13. Biotin Switch Assay
2.14. Mitochondrial Network Assay
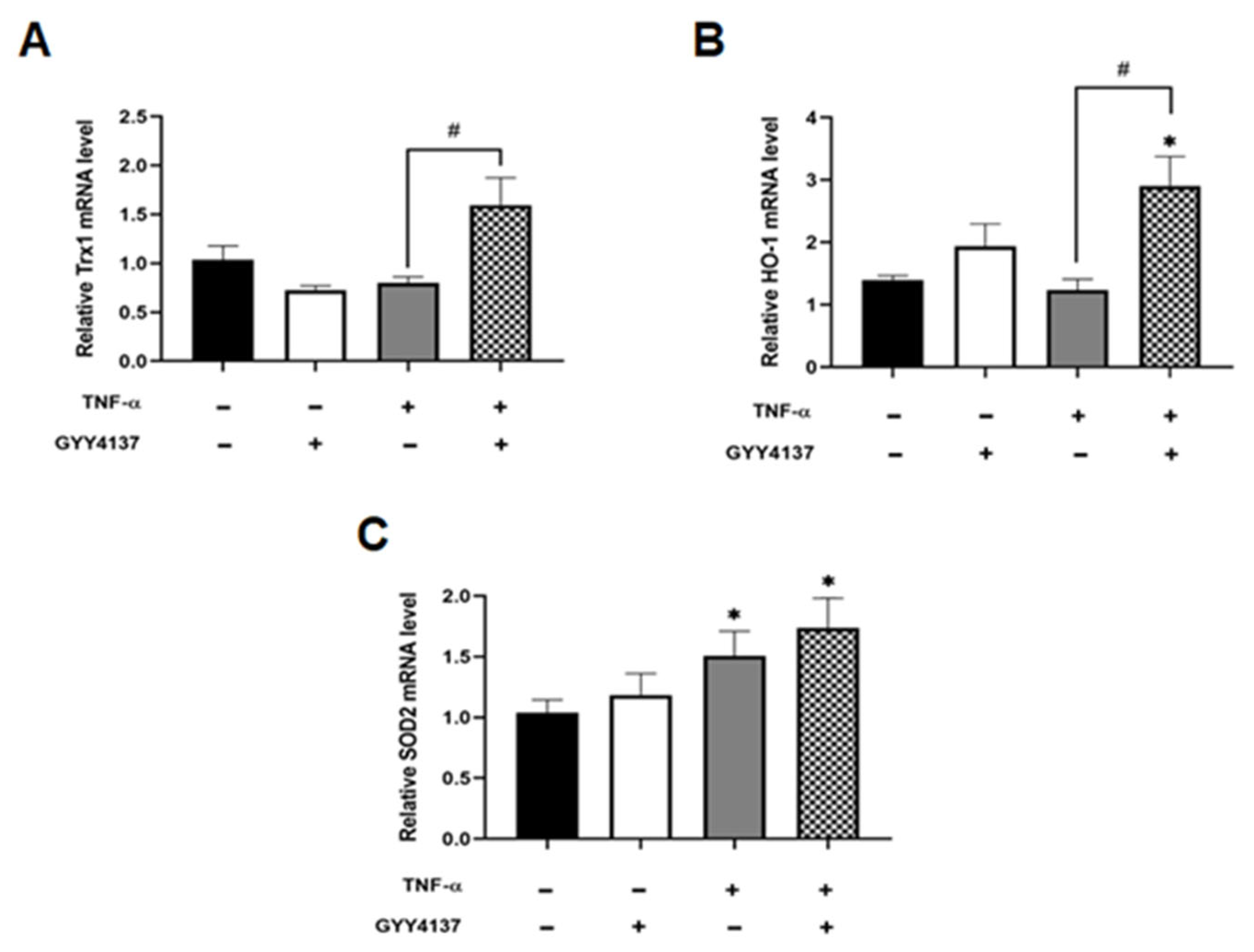
2.15. Real-Time PCR
2.16. Statistical Analysis
3. Results
3.1. Exogenous H2S Ameliorates Intrinsic Apoptotic Pathways in Endothelial Cells
3.2. Exogenous H2S Enhanced S-Sulfhydration of Caspase 3 in Endothelial Cells
3.3. Exogenous H2S Restores the Antioxidant Gene Response and Mitosox Oxidation in TNF-α Treated Endothelial Cells
3.4. Exogenous H2S Improves Mitochondrial Δψm in Endothelial Cells
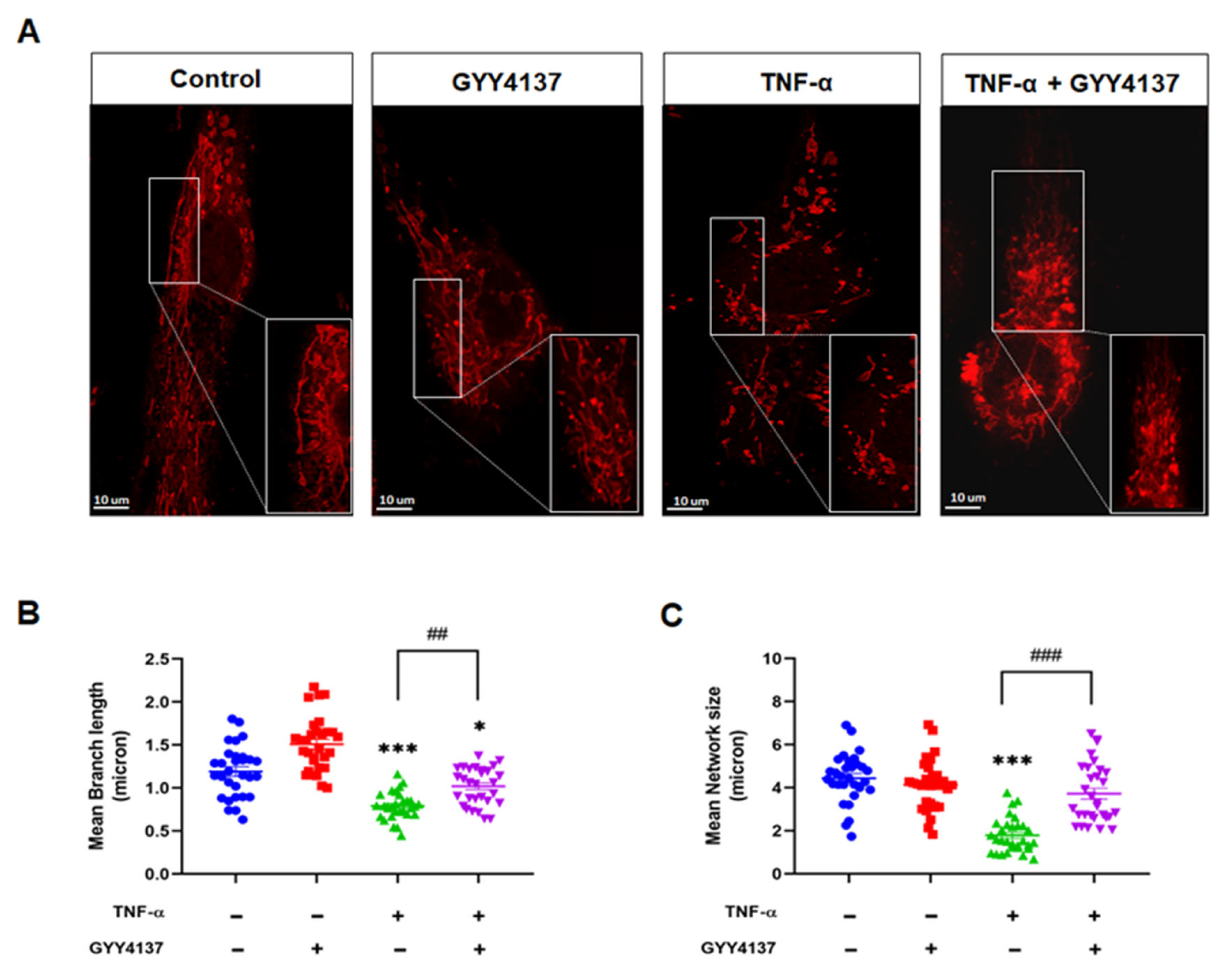
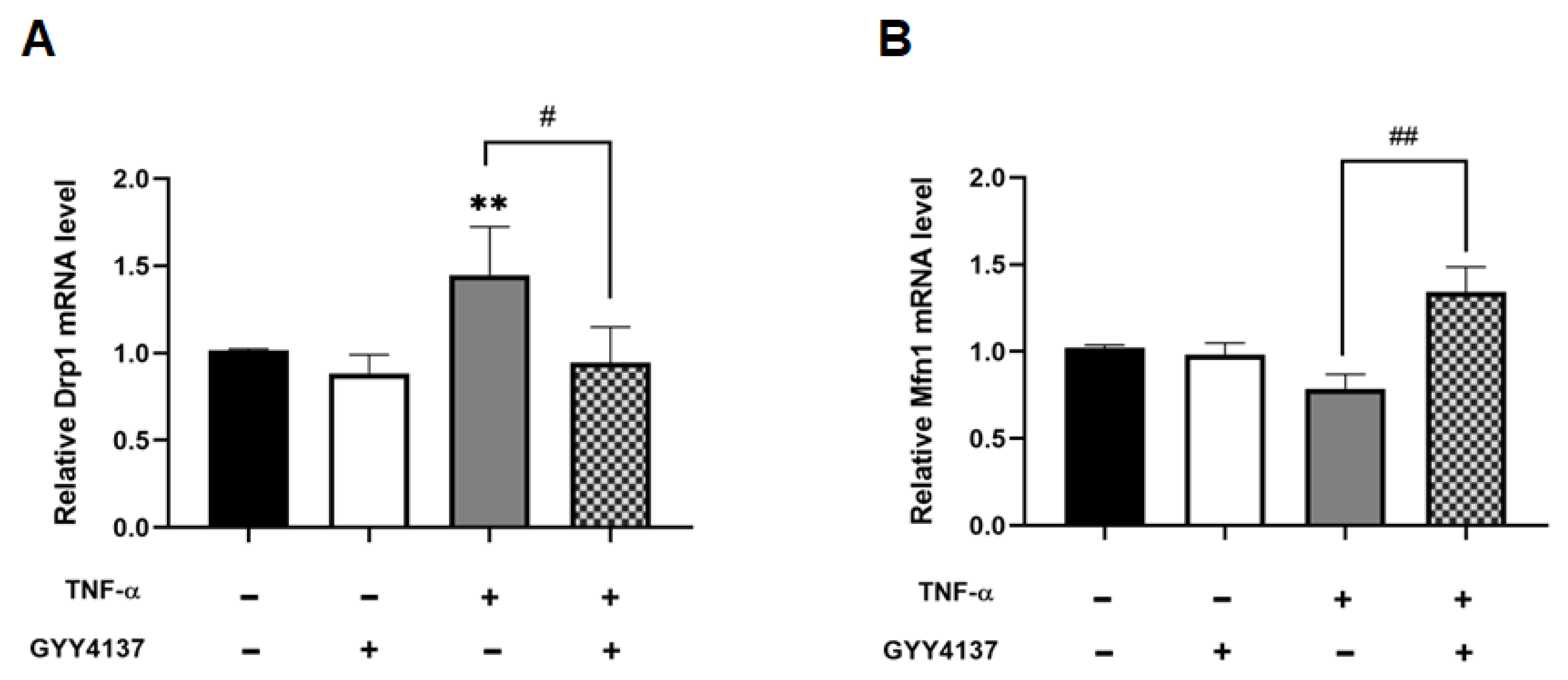
3.5. Exogenous H2S Improved Mitochondrial Morphology in TNF-α-Treated Endothelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Steyers, C.; Miller, F.; Steyers, C.M.; Miller, F.J. Endothelial Dysfunction in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in the metabolic disorders. Inflamm. Res. 2018, 67, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFα in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, C.; Schinzari, F.; Mores, N.; Mettimano, M.; Melina, D.; Zoli, A.; Ferraccioli, G. Intravascular tumor necrosis factor α blockade reverses endothelial dysfunction in rheumatoid arthritis. Clin. Pharmacol. Ther. 2006, 80, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Biniecka, M.; Kennedy, A.; Ng, C.T.; Chang, T.C.; Balogh, E.; Fox, E.; Veale, D.J.; Fearon, U.; O’Sullivan, J.N. Successful tumour necrosis factor (TNF) blocking therapy suppresses oxidative stress and hypoxia-induced mitochondrial mutagenesis in inflammatory arthritis. Arthritis Res. Ther. 2011, 13, R121. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.-Y.; Wang, M.; Li, Y.-N.; Jiang, H.-H.; Sun, X.-J.; Chen, Z.-W. Vascular Protection of Hydrogen Sulfide on Cerebral Ischemia/Reperfusion Injury in Rats. Front. Neurol. 2018, 9, 779. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cai, X.; Zhang, Q.; Li, X.; Li, S.; Ma, J.; Zhu, W.; Liu, X.; Wei, M.; Tu, W.; et al. Hydrogen sulfide restores sevoflurane postconditioning mediated cardioprotection in diabetic rats: Role of SIRT1/Nrf2 signaling-modulated mitochondrial dysfunction and oxidative stress. J. Cell. Physiol. 2020, 7, 5052–5068. [Google Scholar] [CrossRef]
- Sanchez-Aranguren, L.C.; Ahmad, S.; Dias, I.H.K.; Alzahrani, F.A.; Rezai, H.; Wang, K.; Ahmed, A. Bioenergetic effects of hydrogen sulfide suppress soluble Flt-1 and soluble endoglin in cystathionine gamma-lyase compromised endothelial cells. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, G.; Murphy, B.; Dey, A.; Dwivedi, S.K.D.; Zhang, Y.; Roy, R.V.; Chakraborty, P.; Bhattacharya, R.; Mukherjee, P. Cystathionine beta synthase regulates mitochondrial dynamics and function in endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 9372. [Google Scholar] [CrossRef]
- Sen, T.; Saha, P.; Jiang, T.; Sen, N. Sulfhydration of AKT triggers Tau-phosphorylation by activating glycogen synthase kinase 3β in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 4418–4427. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Oláh, G.; Módis, K.; Hellmich, M.R.; Szabo, C. H(2)S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem. Pharmacol. 2017, 136, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.D.; Sanchez-Aranguren, L.; Marwah, M.; Wang, K.; Spickett, C.M.; Griffiths, H.R.; Dias, I.H.K. Exploring mitochondrial hydrogen sulfide signalling for therapeutic interventions in vascular diseases. Adv. Redox Res. 2022, 4, 100030. [Google Scholar] [CrossRef]
- Feng, S.; Chen, S.; Yu, W.; Zhang, D.; Zhang, C.; Tang, C.; Du, J.; Jin, H. H(2)S inhibits pulmonary arterial endothelial cell inflammation in rats with monocrotaline-induced pulmonary hypertension. Lab. Investig. 2017, 97, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behera, J.; Kelly, K.E.; Tyagi, N. Hydrogen sulfide prevents ethanol-induced ZO-1 CpG promoter hypermethylation-dependent vascular permeability via miR-218/DNMT3a axis. J. Cell. Physiol. 2021, 236, 6852–6867. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.J.; Jiang, Z.S.; Qiu, J.; Zhou, S.H.; Liu, Q.M. Protective effects of hydrogen sulfide against angiotensin II-induced endoplasmic reticulum stress in HUVECs. Mol. Med. Rep. 2017, 15, 2213–2222. [Google Scholar] [CrossRef] [Green Version]
- Kamat, P.K.; Kalani, A.; Tyagi, S.C.; Tyagi, N. Hydrogen Sulfide Epigenetically Attenuates Homocysteine-Induced Mitochondrial Toxicity Mediated Through NMDA Receptor in Mouse Brain Endothelial (bEnd3) Cells. J. Cell. Physiol. 2015, 230, 378–394. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.L.; Yan, L.; Chen, Y.H.; Zeng, G.H.; Zhou, Y.; Chen, H.P.; Peng, W.J.; He, M.; Huang, Q.R. A role for diallyl trisulfide in mitochondrial antioxidative stress contributes to its protective effects against vascular endothelial impairment. Eur. J. Pharmacol. 2014, 725, 23–31. [Google Scholar] [CrossRef]
- Cheung, S.H.; Lau, J.Y.W. Hydrogen sulfide mediates athero-protection against oxidative stress via S-sulfhydration. PLoS ONE 2018, 13, e0194176. [Google Scholar]
- Campbell, M.T.; Dagher, P.; Hile, K.L.; Zhang, H.; Meldrum, D.R.; Rink, R.C.; Meldrum, K. Tumor necrosis factor-alpha induces intrinsic apoptotic signaling during renal obstruction through truncated bid activation. J. Urol. 2008, 180, 2694–2700. [Google Scholar] [CrossRef] [Green Version]
- Oberst, A.; Green, D.R. It cuts both ways: Reconciling the dual roles of caspase 8 in cell death and survival. Nat. Rev. Mol. Cell Biol. 2011, 12, 757–763. [Google Scholar] [CrossRef]
- Braunstein, I.; Engelman, R.; Yitzhaki, O.; Ziv, T.; Galardon, E.; Benhar, M. Opposing effects of polysulfides and thioredoxin on apoptosis through caspase persulfidation. J. Biol. Chem. 2020, 295, 3590–3600. [Google Scholar] [CrossRef]
- Chazotte, B. Labeling mitochondria with JC-1. Cold Spring Harb. Protoc. 2011, 2011, 065490. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of endothelial dysfunction in cardiovascular diseases: The link between inflammation and hydrogen sulfide. Frontiers Media S.A. 2020, 10, 1568. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Aranguren, L.C.; Rezai, H.; Ahmad, S.; Alzahrani, F.A.; Sparatore, A.; Wang, K.; Ahmed, A. MZe786 Rescues Cardiac Mitochondrial Activity in High sFlt-1 and Low HO-1 Environment. Antioxidants 2020, 9, 598. [Google Scholar] [CrossRef]
- Wu, D.; Hu, Q.; Liu, X.; Pan, L.; Xiong, Q.; Zhu, Y.Z. Hydrogen sulfide protects against apoptosis under oxidative stress through SIRT1 pathway in H9c2 cardiomyocytes. Nitric Oxide 2015, 46, 204–212. [Google Scholar] [CrossRef]
- Wallace, J.L.; Nagy, P.; Feener, T.D.; Allain, T.; Ditrói, T.; Vaughan, D.J.; Muscara, M.N.; de Nucci, G.; Buret, A.G. A proof-of-concept, Phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br. J. Pharm. 2020, 177, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W.F.; et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine γ-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E-/- mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-X.; Huang, X.-Y.; Wang, P.; Lin, W.-T.; Xu, W.-X.; Zeng, M. Effects and mechanism of arachidonic acid against TNF-α induced apoptosis of endothelial cells. Clin. Hemorheol. Microcirc. 2021, 77, 259–265. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-α-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-XL. Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, Y.; Hirashima, S.; Ishihara, N.; Kusukawa, J.; Nakamura, K.I.; Ohta, K. Uncoupled mitochondria quickly shorten along their long axis to form indented spheroids, instead of rings, in a fission-independent manner. Sci. Rep. 2018, 8, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-F.; Zhao, B.; Tang, X.-Y.; Li, W.; Zhu, L.-L.; Tang, C.-S.; Du, J.-B.; Jin, H.-F. Hydrogen sulfide regulates vascular endoplasmic reticulum stress in apolipoprotein E knockout mice. Chin. Med. J. 2011, 124, 3460–3467. [Google Scholar]
- Zhuge, W.; Zhuge, Q.; Wang, W.; Lu, X.; You, R.; Liu, L.; Yu, H.; Wang, J.; Wang, X.; Ye, Y.; et al. Hydrogen sulphide ameliorates dopamine-induced astrocytic inflammation and neurodegeneration in minimal hepatic encephalopathy. J. Cell Mol. Med. 2020, 24, 13634–13647. [Google Scholar] [CrossRef]
- Fang, B.; Boross, P.I.; Tozser, J.; Weber, I.T. Structural and kinetic analysis of caspase-3 reveals role for s5 binding site in substrate recognition. J. Mol. Biol. 2006, 360, 654–666. [Google Scholar] [CrossRef]
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannick, J.B.; Hausladen, A.; Liu, L.; Hess, D.T.; Zeng, M.; Miao, Q.X.; Kane, L.S.; Gow, A.J.; Stamler, J.S. Fas-induced caspase denitrosylation. Science 1999, 284, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Berk, B.C. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: Key role for glutaredoxin in the death pathway. Circ. Res. 2007, 100, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.H.; Li, G.L.; Wang, B.A.; Qin, Y.; Bai, S.R.; Rong, J.; Deng, T.; Li, Q. Diallyl trisufide protects against oxygen glucose deprivation -induced apoptosis by scavenging free radicals via the PI3K/Akt -mediated Nrf2/HO-1 signaling pathway in B35 neural cells. Brain Res. 2015, 1614, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via s-sulfhydration of keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Pan, J.; Carroll, K.S. Persulfide Reactivity in the Detection of Protein S-Sulfhydration. ACS Chem. Biol. 2013, 8, 1110–1116. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Liu, K.; He, J.; Tian, C.; Yu, X.; Yang, J. Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid. Redox Signal. 2020, 33, 1061–1076. [Google Scholar] [CrossRef]
- Bibli, S.-I.; Hu, J.; Looso, M.; Weigert, A.; Ratiu, C.; Wittig, J.; Drekolia, M.K.; Tombor, L.; Randriamboavonjy, V.; Leisegang, M.S.; et al. Mapping the Endothelial Cell <i>S</i>-Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation 2021, 143, 935–948. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz Sanchez, L.; Sanchez-Aranguren, L.; Wang, K.; Spickett, C.M.; Griffiths, H.R.; Dias, I.H.K. TNF-α-Mediated Endothelial Cell Apoptosis Is Rescued by Hydrogen Sulfide. Antioxidants 2023, 12, 734. https://doi.org/10.3390/antiox12030734
Diaz Sanchez L, Sanchez-Aranguren L, Wang K, Spickett CM, Griffiths HR, Dias IHK. TNF-α-Mediated Endothelial Cell Apoptosis Is Rescued by Hydrogen Sulfide. Antioxidants. 2023; 12(3):734. https://doi.org/10.3390/antiox12030734
Chicago/Turabian StyleDiaz Sanchez, Lorena, Lissette Sanchez-Aranguren, Keqing Wang, Corinne M. Spickett, Helen R. Griffiths, and Irundika H. K. Dias. 2023. "TNF-α-Mediated Endothelial Cell Apoptosis Is Rescued by Hydrogen Sulfide" Antioxidants 12, no. 3: 734. https://doi.org/10.3390/antiox12030734
APA StyleDiaz Sanchez, L., Sanchez-Aranguren, L., Wang, K., Spickett, C. M., Griffiths, H. R., & Dias, I. H. K. (2023). TNF-α-Mediated Endothelial Cell Apoptosis Is Rescued by Hydrogen Sulfide. Antioxidants, 12(3), 734. https://doi.org/10.3390/antiox12030734