


The Key Role of Mitochondrial Function in Health and Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondria, the Key Aerobic Microbe for Eukaryotic Cell Evolution
3. Mitochondrial Bioenergetics Are Complex and Intertwined
4. Mitochondria, the Main Producers of Reactive Oxygen Species (ROS)
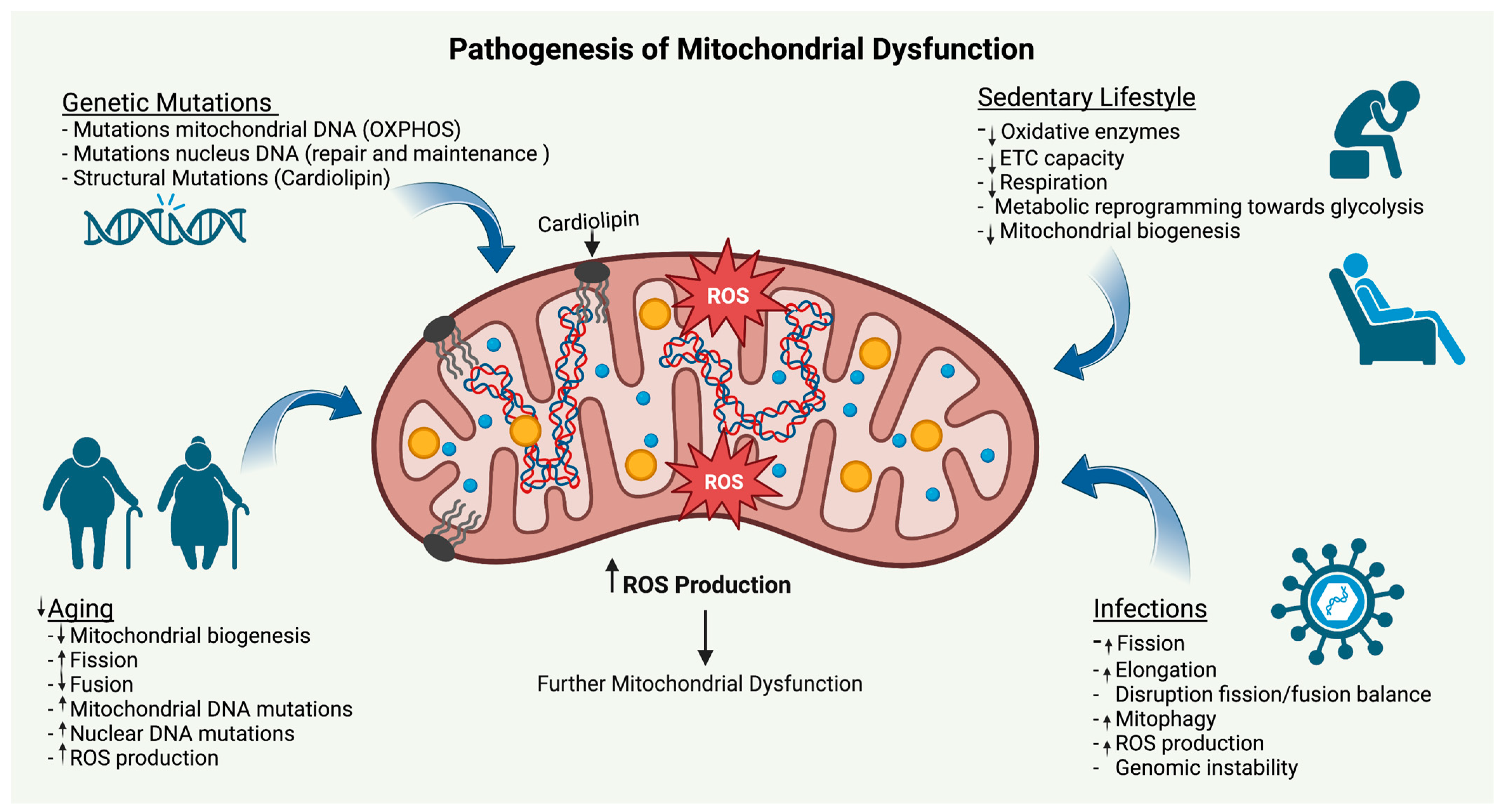
5. Etiologies of Mitochondrial Dysfunction
5.1. Genetic Mutations
5.2. Aging
5.3. Infections
5.4. Lack of Physical Activity
6. The Role of Mitochondrial Function in Multiple Diseases
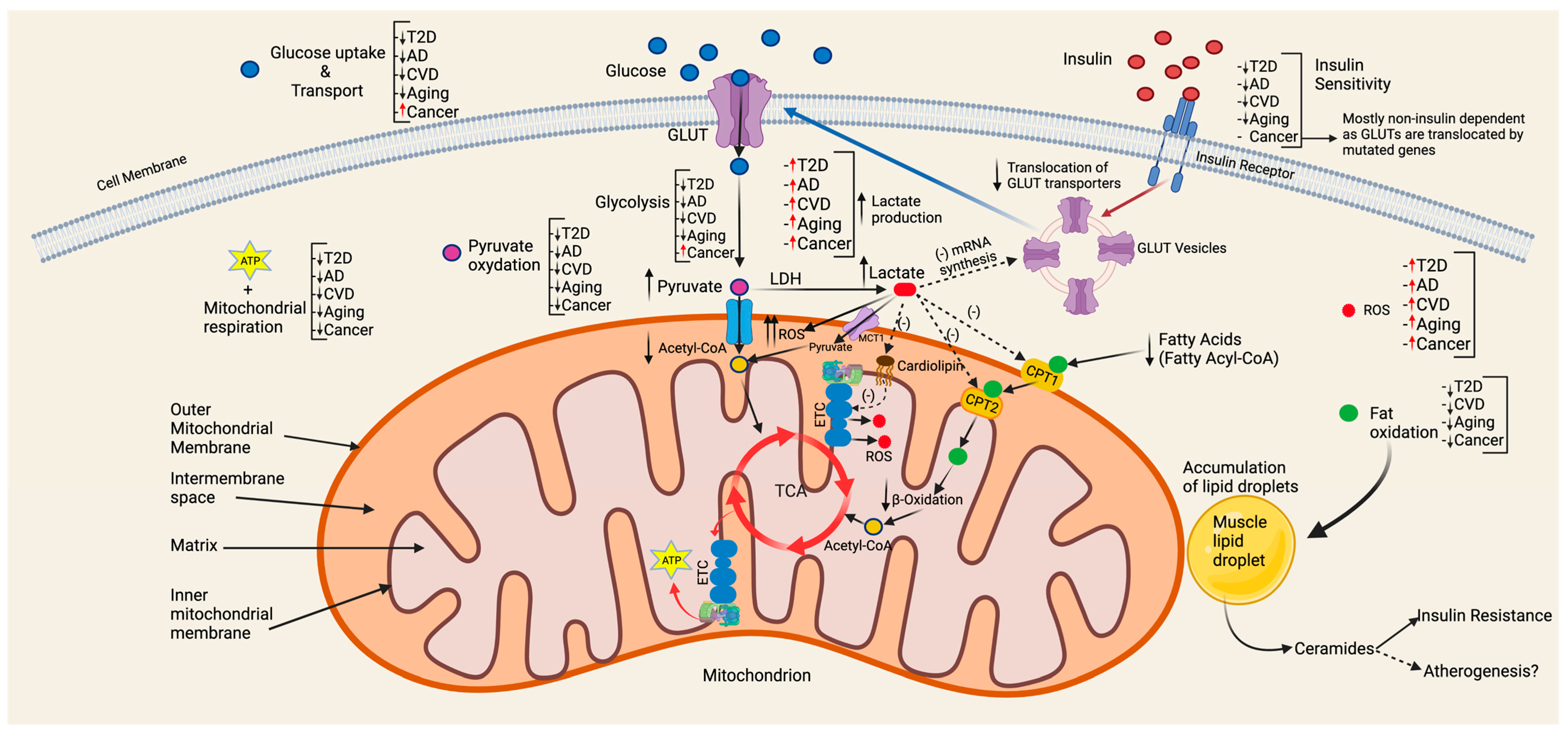
6.1. Type 2 Diabetes
6.2. Cardiovascular Disease
6.3. Mitochondrial Dysfunction at the Crossroads of the Connection between Type 2 Diabetes and Cardiovascular Disease
6.4. Alzheimer’s Disease, Is It the Brain’s Diabetes?
6.5. Cancer
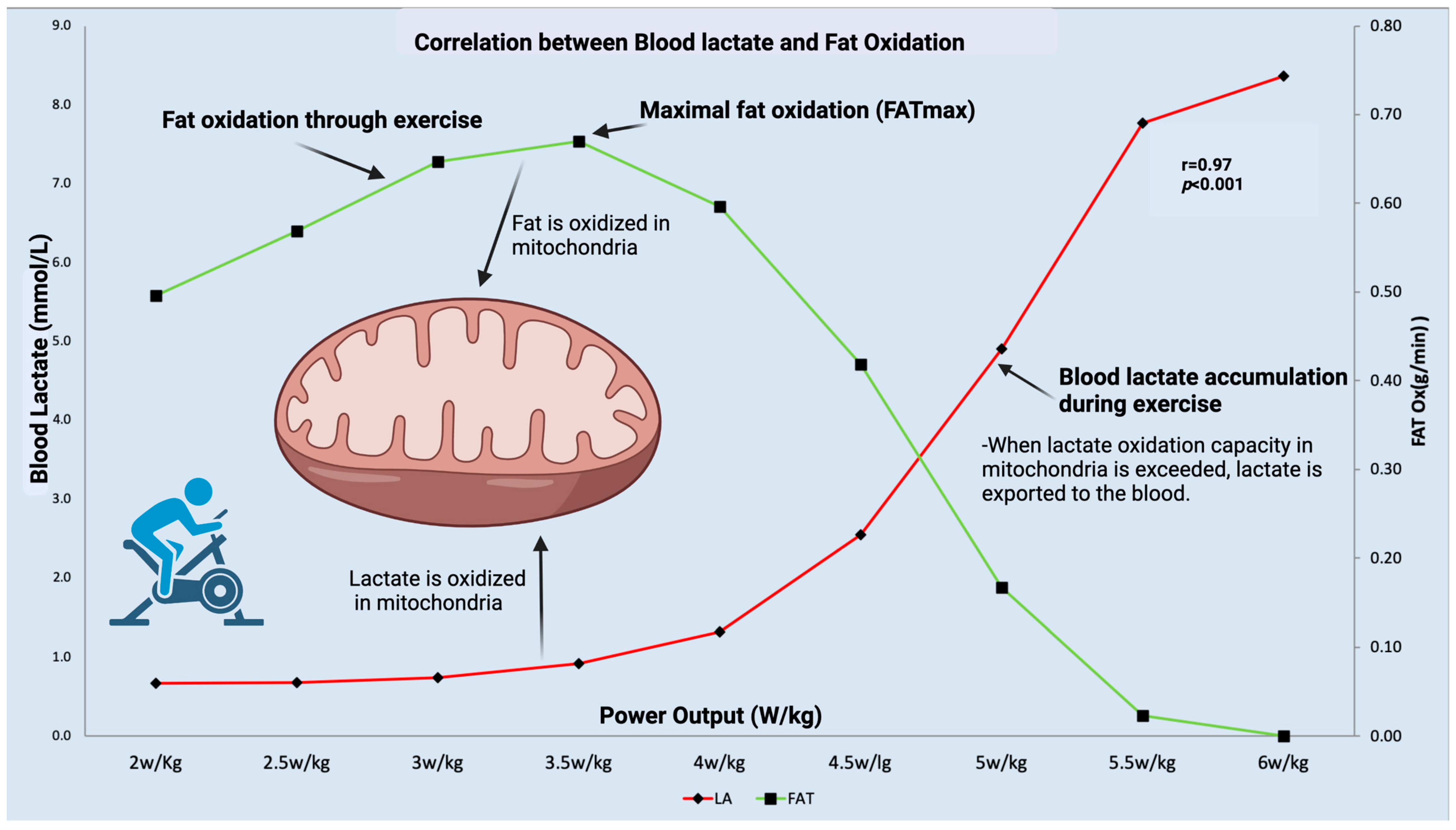
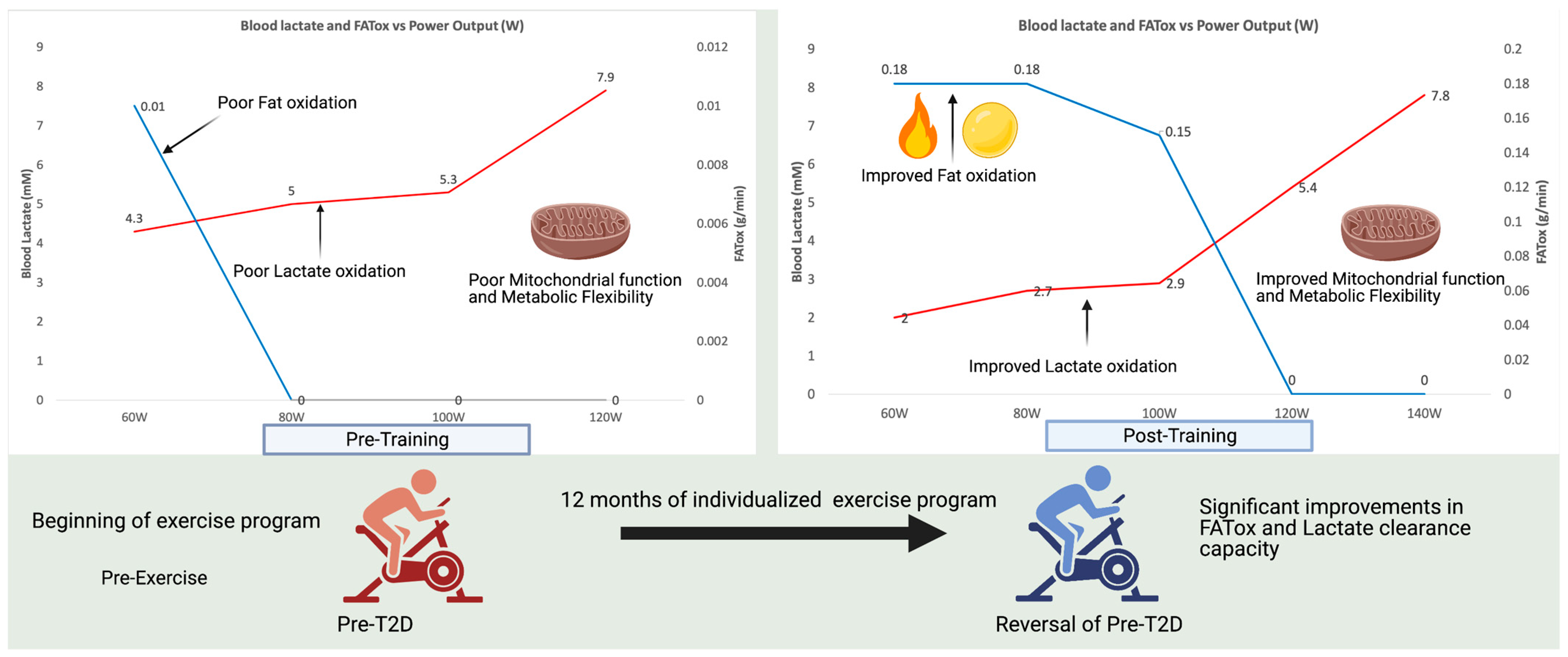
7. Exercise, the Only Known “Medicine” for Maintaining and Improving Mitochondrial Function
8. Assessment of Mitochondrial and Metabolic Function in the Clinical Setting
9. Summary and Future Directions
Funding
Conflicts of Interest
References
- Wallace, D.C. Bioenergetic origins of complexity and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1434–1455. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.A.; McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Schmidt, C.; Kassai, M.; Kew, K.A.; McClung, J.M.; Neufer, P.D.; Brophy, P. Intrinsic OXPHOS limitations underlie cellular bioenergetics in leukemia. eLife 2021, 10, e63104. [Google Scholar] [CrossRef]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaría, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar]
- Alam, M.M.; Lal, S.; FitzGerald, K.E.; Zhang, L. A holistic view of cancer bioenergetics: Mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin. Transl. Med. 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagan, L. On the origin of mitosing cells. J. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Macovschi, E. Biostructura; Romanian Academy Publishing: Bucharest, Romania, 1969; Volume 57. [Google Scholar]
- Drochioiu, G. Biological Systems: A Structural–Phenomenological Approach; na. 2008. [Google Scholar]
- Murariu, M.; Drochioiu, G. Biostructural theory of the living systems. Biosystems 2012, 109, 126–132. [Google Scholar] [CrossRef]
- Macovschi, E. The confirmation of the biostructural theory by the high–voltage electron–microscopy. Rev. Roum. Biochim. 1982, 19, 177–186. [Google Scholar]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, S.P.; Munn, E.A.; Brooks, G.A. Mitochondrial reticulum in limb skeletal muscle. Am. J. Physiol. 1986, 251, C395–C402. [Google Scholar] [CrossRef]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, D.R.; Howley, E.T. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med. Sci. Sport. Exerc. 2000, 32, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.W. Malonyl–CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.A. The tortuous path of lactate shuttle discovery: From cinders and boards to the lab and ICU. J. Sport. Health Sci. 2020, 9, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Hussien, R.; Brooks, G.A. Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: Evidence of a mitochondrial lactate oxidation complex. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1237–E1244. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hussien, R.; Cho, H.S.; Kaufer, D.; Brooks, G.A. Evidence for the mitochondrial lactate oxidation complex in rat neurons: Demonstration of an essential component of brain lactate shuttles. PLoS ONE 2008, 3, e2915. [Google Scholar] [CrossRef]
- Lombardi, A.M.; Fabris, R.; Bassetto, F.; Serra, R.; Leturque, A.; Federspil, G.; Girard, J.; Vettor, R. Hyperlactatemia reduces muscle glucose uptake and GLUT–4 mRNA while increasing (E1α)PDH gene expression in rat. Am. J. Physiol.–Endocrinol. Metab. 1999, 276, E922–E929. [Google Scholar] [CrossRef]
- Cai, T.Q.; Ren, N.; Jin, L.; Cheng, K.; Kash, S.; Chen, R.; Wright, S.D.; Taggart, A.K.; Waters, M.G. Role of GPR81 in lactate–mediated reduction of adipose lipolysis. Biochem. Biophys. Res. Commun. 2008, 377, 987–991. [Google Scholar] [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G–protein–coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [Green Version]
- San–Millan, I.; Sparagna, G.C.; Chapman, H.L.; Warkins, V.L.; Chatfield, K.C.; Shuff, S.R.; Martinez, J.L.; Brooks, G.A. Chronic Lactate Exposure Decreases Mitochondrial Function by Inhibition of Fatty Acid Uptake and Cardiolipin Alterations in Neonatal Rat Cardiomyocytes. Front. Nutr. 2022, 9, 809485. [Google Scholar] [CrossRef] [PubMed]
- San–Millan, I.; Brooks, G.A. Assessment of Metabolic Flexibility by Means of Measuring Blood Lactate, Fat, and Carbohydrate Oxidation Responses to Exercise in Professional Endurance Athletes and Less–Fit Individuals. Sport. Med. 2018, 48, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.A.; Benedict, F.G. A Biometric Study of Human Basal Metabolism. Proc. Natl. Acad. Sci. USA 1918, 4, 370–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.A.B.; Benedict, F.G. Biometric Standards for Energy Requirements in Human Nutrition. Sci. Mon. 1919, 8, 385–402. [Google Scholar]
- Benedict, F.G. Vital Energetics: A Study in Comparative Basal Metabolism. J. Am. Med. Assoc. 1939, 112, 2089. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141. [Google Scholar] [CrossRef]
- Kelley, D.E.; Simoneau, J.A. Impaired free fatty acid utilization by skeletal muscle in non–insulin–dependent diabetes mellitus. J. Clin. Investig. 1994, 94, 2349–2356. [Google Scholar] [CrossRef]
- Kelley, D.E.; Mokan, M.; Simoneau, J.A.; Mandarino, L.J. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J. Clin. Investig. 1993, 92, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; Mandarino, L.J. Fuel selection in human skeletal muscle in insulin resistance: A reexamination. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The Effect of Insulin on the Disposal of Intravenous Glucose: Results from Indirect Calorimetry and Hepatic and Femoral Venous Catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gong, G.; Wang, X.; Wei–LaPierre, L.; Cheng, H.; Dirksen, R.; Sheu, S.S. Mitochondrial Flash: Integrative Reactive Oxygen Species and pH Signals in Cell and Organelle Biology. Antioxid. Redox Signal. 2016, 25, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Canali, R.; Rettori, D.; Kaplowitz, N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol. Pharmacol. 2003, 64, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The redox code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Schaar, C.E.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Cooper, J.F.; Senchuk, M.; Hekimi, S.; Van Raamsdonk, J.M. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015, 11, e1004972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, S.; Schmeisser, K.; Weimer, S.; Groth, M.; Priebe, S.; Fazius, E.; Kuhlow, D.; Pick, D.; Einax, J.W.; Guthke, R.; et al. Mitochondrial hormesis links low–dose arsenite exposure to lifespan extension. Aging Cell 2013, 12, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristow, M. Unraveling the Truth About Antioxidants: Mitohormesis explains ROS–induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Blake, R.; Trounce, I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Et. Biophys. Acta (BBA)–Gen. Subj. 2014, 1840, 1404–1412. [Google Scholar] [CrossRef]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; de Souza–Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free. Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2003, 53, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxidative Med. Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Gross, M.; Lee, D.-H.; Holvoet, P.; Himes, J.H.; Shikany, J.M.; Jacobs, D.R. Oxidative stress and insulin resistance: The coronary artery risk development in young adults study. Diabetes Care 2009, 32, 1302–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2015, 16, 193–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.; Liu, F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal. 2011, 23, 1528–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, L.G.; Judge, S.; Cruz, A.; Pourang, D.; Mathews, C.E.; Stacpoole, P.W. Increased superoxide accumulation in pyruvate dehydrogenase complex deficient fibroblasts. Mol. Genet. Metab. 2011, 104, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Tauffenberger, A.; Fiumelli, H.; Almustafa, S.; Magistretti, P.J. Lactate and pyruvate promote oxidative stress resistance through hormetic ROS signaling. Cell Death Dis. 2019, 10, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babbar, N.; Casero, R.A., Jr. Tumor necrosis factor–alpha increases reactive oxygen species by inducing spermine oxidase in human lung epithelial cells: A potential mechanism for inflammation–induced carcinogenesis. Cancer Res. 2006, 66, 11125–11130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Sanfeliu, C.; Sebastià, J.; Kim, S.U. Methylmercury neurotoxicity in cultures of human neurons, astrocytes, neuroblastoma cells. Neurotoxicology 2001, 22, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Liu, J.; Ames, B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: Relevance to aging. J. Biol. Chem. 2001, 276, 48410–48416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Rusecka, J.; Kaliszewska, M.; Bartnik, E.; Tońska, K. Nuclear genes involved in mitochondrial diseases caused by instability of mitochondrial DNA. J. Appl. Genet. 2018, 59, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J. Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [Green Version]
- Chatfield, K.C.; Sparagna, G.C.; Specht, K.S.; Whitcomb, L.A.; Omar, A.K.; Miyamoto, S.D.; Wolfe, L.M.; Chicco, A.J. Long—Chain fatty acid oxidation and respiratory complex I deficiencies distinguish Barth Syndrome from idiopathic pediatric cardiomyopathy. J. Inherit. Metab. Dis. 2022, 45, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury–Ecob, R.; Clayton, N.; Martin, R.P.; Tsai–Goodman, B.; Garratt, V.; Ashworth, M. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, C.H.; Benage, L.G.; Specht, K.S.; Li Puma, L.C.; Mulligan, C.M.; Heuberger, A.L.; Prenni, J.E.; Claypool, S.M.; Chatfield, K.C.; Sparagna, G.C.; et al. Tafazzin deficiency impairs CoA–dependent oxidative metabolism in cardiac mitochondria. J. Biol. Chem. 2020, 295, 12485–12497. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Kagan, J.; Sidransky, D.; Srivastava, S. Proteomic analysis of cancer–cell mitochondria. Nat. Rev. Cancer 2003, 3, 789–795. [Google Scholar] [CrossRef]
- Alonso, A.; Martin, P.; Albarran, C.; Aguilera, B.; Garcia, O.; Guzman, A.; Oliva, H.; Sancho, M. Detection of somatic mutations in the mitochondrial DNA control region of colorectal and gastric tumors by heteroduplex and single—Strand conformation analysis. Electrophoresis 1997, 18, 682–685. [Google Scholar] [CrossRef]
- Anderson, L.; Seilhamer, J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis 1997, 18, 533–537. [Google Scholar] [CrossRef]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [CrossRef]
- Lee, H.-C.; Huang, K.-H.; Yeh, T.-S.; Chi, C.-W. Somatic alterations in mitochondrial DNA and mitochondrial dysfunction in gastric cancer progression. World J. Gastroenterol. 2014, 20, 3950. [Google Scholar] [CrossRef]
- Warburg, O.; Minami, S. Versuche an Überlebendem Carcinom–gewebe. Klin. Wochenschr. 1923, 2, 776–777. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen–Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez–Freire, M.; De Cabo, R.; Bernier, M.; Sollott, S.J.; Fabbri, E.; Navas, P.; Ferrucci, L. Reconsidering the role of mitochondria in aging. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2015, 70, 1334–1342. [Google Scholar] [CrossRef] [Green Version]
- Hebert, S.L.; Marquet–de Rougé, P.; Lanza, I.R.; McCrady–Spitzer, S.K.; Levine, J.A.; Middha, S.; Carter, R.E.; Klaus, K.A.; Therneau, T.M.; Highsmith, E.W. Mitochondrial aging and physical decline: Insights from three generations of women. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2015, 70, 1409–1417. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.N.; Pauly, M.; Tryon, L.D.; Hood, D.A. Effect of contractile activity on PGC–1alpha transcription in young and aged skeletal muscle. J. Appl. Physiol. 2018, 124, 1605–1615. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuchner, J.; Kuznetsov, A.; Obexer, P.; Ausserlechner, M. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2013, 32, 4748–4757. [Google Scholar] [CrossRef] [Green Version]
- Rossin, F.; D’eletto, M.; Falasca, L.; Sepe, S.; Cocco, S.; Fimia, G.; Campanella, M.; Mastroberardino, P.; Farrace, M.; Piacentini, M. Transglutaminase 2 ablation leads to mitophagy impairment associated with a metabolic shift towards aerobic glycolysis. Cell Death Differ. 2015, 22, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Arnheim, N.; Cortopassi, G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat. Res./DNAging 1992, 275, 157–167. [Google Scholar] [CrossRef]
- Raule, N.; Sevini, F.; Li, S.; Barbieri, A.; Tallaro, F.; Lomartire, L.; Vianello, D.; Montesanto, A.; Moilanen, J.S.; Bezrukov, V.; et al. The co–occurrence of mtDNA mutations on different oxidative phosphorylation subunits, not detected by haplogroup analysis, affects human longevity and is population specific. Aging Cell 2014, 13, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Sevini, F.; Giuliani, C.; Vianello, D.; Giampieri, E.; Santoro, A.; Biondi, F.; Garagnani, P.; Passarino, G.; Luiselli, D.; Capri, M. mtDNA mutations in human aging and longevity: Controversies and new perspectives opened by high–throughput technologies. Exp. Gerontol. 2014, 56, 234–244. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal. Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive oxygen species and mitochondrial dynamics: The yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Monlun, M.; Hyernard, C.; Blanco, P.; Lartigue, L.; Faustin, B. Mitochondria as Molecular Platforms Integrating Multiple Innate Immune Signalings. J. Mol. Biol. 2017, 429, 1–13. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.A.; Serger, E.; Campos, S.; Divakaruni, A.S.; Kim, C.; Smith, K.; Trejo, M.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV alters neuronal mitochondrial fission/fusion in the brain during HIV–associated neurocognitive disorders. Neurobiol. Dis. 2016, 86, 154–169. [Google Scholar] [CrossRef] [Green Version]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.-I.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1–F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatti, P.; Ilamathi, H.S.; Todkar, K.; Germain, M. Mitochondria Targeted Viral Replication and Survival Strategies—Prospective on SARS-CoV-2. Front. Pharmacol. 2020, 11, 578599. [Google Scholar] [CrossRef] [PubMed]
- Ganji, R.; Reddy, P.H. Impact of COVID–19 on Mitochondrial–Based Immunity in Aging and Age–Related Diseases. Front. Aging Neurosci. 2021, 12, 614650. [Google Scholar] [CrossRef] [PubMed]
- de Boer, E.; Petrache, I.; Goldstein, N.M.; Olin, J.T.; Keith, R.C.; Modena, B.; Mohning, M.P.; Yunt, Z.X.; San–Millán, I.; Swigris, J.J. Decreased Fatty Oxidation and Altered Lactate Production During Exercise in Post–Acute COVID–19 Patients. Am. J. Respir. Crit. Care Med. 2021, 205, 126–129. [Google Scholar] [CrossRef]
- Guntur, V.P.; Nemkov, T.; de Boer, E.; Mohning, M.P.; Baraghoshi, D.; Cendali, F.I.; San–Millán, I.; Petrache, I.; D’Alessandro, A. Signatures of Mitochondrial Dysfunction and Impaired Fatty Acid Metabolism in Plasma of Patients with Post–Acute Sequelae of COVID–19 (PASC). Metabolites 2022, 12, 1026. [Google Scholar] [CrossRef]
- Singer, M. The role of mitochondrial dysfunction in sepsis–induced multi–organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef]
- Rahmel, T.; Marko, B.; Nowak, H.; Bergmann, L.; Thon, P.; Rump, K.; Kreimendahl, S.; Rassow, J.; Peters, J.; Singer, M.; et al. Mitochondrial dysfunction in sepsis is associated with diminished intramitochondrial TFAM despite its increased cellular expression. Sci. Rep. 2020, 10, 21029. [Google Scholar] [CrossRef]
- Gornik, I.; Vujaklija, A.; Lukić, E.; Madžarac, G.; Gašparović, V. Hyperglycemia in sepsis is a risk factor for development of type II diabetes. J. Crit. Care 2010, 25, 263–269. [Google Scholar] [CrossRef]
- Mankowski, R.T.; Yende, S.; Angus, D.C. Long–term impact of sepsis on cardiovascular health. Intensive Care Med. 2019, 45, 78–81. [Google Scholar] [CrossRef]
- Ahmed, H.M.; Blaha, M.J.; Nasir, K.; Rivera, J.J.; Blumenthal, R.S. Effects of physical activity on cardiovascular disease. Am. J. Cardiol. 2012, 109, 288–295. [Google Scholar] [CrossRef]
- Durstine, J.L.; Gordon, B.; Wang, Z.; Luo, X. Chronic disease and the link to physical activity. J. Sport. Health Sci. 2013, 2, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Powell, K.E.; Thompson, P.D.; Caspersen, C.J.; Kendrick, J.S. Physical activity and the incidence of coronary heart disease. Annu. Rev. Public. Health 1987, 8, 253–287. [Google Scholar] [CrossRef]
- Kohl, H., 3rd. Physical activity and cardiovascular disease: Evidence for a dose response. Med. Sci. Sport. Exerc. 2001, 33, S472–S483, discussion S493. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.; Schwarzschild, M.; Hernan, M.; Ascherio, A. Physical activity and the risk of Parkinson disease. Neurology 2005, 64, 664–669. [Google Scholar] [CrossRef]
- Berlin, J.A.; Colditz, G.A. A meta–analysis of physical activity in the prevention of coronary heart disease. Am. J. Epidemiol. 1990, 132, 612–628. [Google Scholar] [CrossRef]
- Colberg, S.R.; Sigal, R.J.; Yardley, J.E.; Riddell, M.C.; Dunstan, D.W.; Dempsey, P.C.; Horton, E.S.; Castorino, K.; Tate, D.F. Physical activity/exercise and diabetes: A position statement of the American Diabetes Association. Diabetes Care 2016, 39, 2065–2079. [Google Scholar] [CrossRef] [Green Version]
- LaMonte, M.J.; Blair, S.N.; Church, T.S. Physical activity and diabetes prevention. J. Appl. Physiol. 2005, 99, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Grill, S.; Yahiaoui–Doktor, M.; Dukatz, R.; Lammert, J.; Ullrich, M.; Engel, C.; Pfeifer, K.; Basrai, M.; Siniatchkin, M.; Schmidt, T. Smoking and physical inactivity increase cancer prevalence in BRCA–1 and BRCA–2 mutation carriers: Results from a retrospective observational analysis. Arch. Gynecol. Obstet. 2017, 296, 1135–1144. [Google Scholar] [CrossRef]
- McTiernan, A.; Friedenreich, C.M.; Katzmarzyk, P.T.; Powell, K.E.; Macko, R.; Buchner, D.; Pescatello, L.S.; Bloodgood, B.; Tennant, B.; Vaux–Bjerke, A. Physical activity in cancer prevention and survival: A systematic review. Med. Sci. Sport. Exerc. 2019, 51, 1252. [Google Scholar] [CrossRef]
- Friedenreich, C.M.; Ryder–Burbidge, C.; McNeil, J. Physical activity, obesity and sedentary behavior in cancer etiology: Epidemiologic evidence and biologic mechanisms. Mol. Oncol. 2021, 15, 790–800. [Google Scholar] [CrossRef]
- Rolland, Y.; van Kan, G.A.; Vellas, B. Physical activity and Alzheimer’s disease: From prevention to therapeutic perspectives. J. Am. Med. Dir. Assoc. 2008, 9, 390–405. [Google Scholar] [CrossRef]
- Santos–Lozano, A.; Pareja–Galeano, H.; Sanchis–Gomar, F.; Quindós–Rubial, M.; Fiuza–Luces, C.; Cristi–Montero, C.; Emanuele, E.; Garatachea, N.; Lucia, A. Physical activity and Alzheimer disease: A protective association. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2016; Volume 91, pp. 999–1020. [Google Scholar]
- Stephen, R.; Hongisto, K.; Solomon, A.; Lönnroos, E. Physical activity and Alzheimer’s disease: A systematic review. J. Gerontol. Ser. A 2017, 72, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Blair, S.N. Physical inactivity: The biggest public health problem of the 21st century. Br. J. Sport. Med. 2009, 43, 1–2. [Google Scholar]
- Houston, M.E.; Bentzen, H.; Larsen, H. Interrelationships between skeletal muscle adaptations and performance as studied by detraining and retraining. Acta Physiol. Scand. 1979, 105, 163–170. [Google Scholar] [CrossRef]
- Coyle, E.F.; Martin, W.H., 3rd; Bloomfield, S.A.; Lowry, O.H.; Holloszy, J.O. Effects of detraining on responses to submaximal exercise. J. Appl. Physiol. 1985, 59, 853–859. [Google Scholar] [CrossRef]
- Fritzen, A.M.; Thøgersen, F.B.; Thybo, K.; Vissing, C.R.; Krag, T.O.; Ruiz–Ruiz, C.; Risom, L.; Wibrand, F.; Høeg, L.D.; Kiens, B.; et al. Adaptations in Mitochondrial Enzymatic Activity Occurs Independent of Genomic Dosage in Response to Aerobic Exercise Training and Deconditioning in Human Skeletal Muscle. Cells 2019, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Houmard, J.A.; Hortobágyi, T.; Johns, R.A.; Bruno, N.J.; Nute, C.C.; Shinebarger, M.H.; Welborn, J.W. Effect of short–term training cessation on performance measures in distance runners. Int. J. Sport. Med. 1992, 13, 572–576. [Google Scholar] [CrossRef]
- Bergouignan, A.; Rudwill, F.; Simon, C.; Blanc, S. Physical inactivity as the culprit of metabolic inflexibility: Evidence from bed–rest studies. J. Appl. Physiol. 2011, 111, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Rudwill, F.; O’Gorman, D.; Lefai, E.; Chery, I.; Zahariev, A.; Normand, S.; Pagano, A.F.; Chopard, A.; Damiot, A.; Laurens, C.; et al. Metabolic Inflexibility Is an Early Marker of Bed–Rest–Induced Glucose Intolerance Even When Fat Mass Is Stable. J. Clin. Endocrinol. Metab. 2018, 103, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Alibegovic, A.C.; Sonne, M.P.; Højbjerre, L.; Bork–Jensen, J.; Jacobsen, S.; Nilsson, E.; Færch, K.; Hiscock, N.; Mortensen, B.; Friedrichsen, M. Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am. J. Physiol.–Endocrinol. Metab. 2010, 299, E752–E763. [Google Scholar] [CrossRef] [Green Version]
- Bergouignan, A.; Schoeller, D.A.; Normand, S.; Gauquelin–Koch, G.; Laville, M.; Shriver, T.; Desage, M.; Maho, Y.L.; Ohshima, H.; Gharib, C. Effect of physical inactivity on the oxidation of saturated and monounsaturated dietary fatty acids: Results of a randomized trial. PLoS Clin. Trials 2006, 1, e27. [Google Scholar] [CrossRef] [PubMed]
- Lutwak, L.; Whedon, G.D. The effect of physical conditioning on glucose tolerance. Clin. Res. 1959, 7, 143. [Google Scholar]
- Dolkas, C.; Greenleaf, J. Insulin and glucose responses during bed rest with isotonic and isometric exercise. J. Appl. Physiol. 1977, 43, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, N.M.; McMackin, C.J.; Huang, A.L.; Shenouda, S.M.; Widlansky, M.E.; Schulz, E.; Gokce, N.; Ruderman, N.B.; Keaney, J.F., Jr.; Vita, J.A. Physical inactivity rapidly induces insulin resistance and microvascular dysfunction in healthy volunteers. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2650–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikines, K.J.; Richter, E.A.; Dela, F.; Galbo, H. Seven days of bed rest decrease insulin action on glucose uptake in leg and whole body. J. Appl. Physiol. 1991, 70, 1245–1254. [Google Scholar] [CrossRef]
- Stuart, C.A.; Shangraw, R.E.; Prince, M.J.; Peters, E.J.; Wolfe, R.R. Bed–rest–induced insulin resistance occurs primarily in muscle. Metabolism 1988, 37, 802–806. [Google Scholar] [CrossRef]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and Trends in Diabetes Among Adults in the United States, 1988–2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Li, X.; Wan, G.; Sun, L.; Zhu, X.; Che, F.; Yang, Z. Type 2 diabetes epidemic in East Asia: A 35–year systematic trend analysis. Oncotarget 2017, 9, 6718–6727. [Google Scholar] [CrossRef] [Green Version]
- Tamayo, T.; Rosenbauer, J.; Wild, S.H.; Spijkerman, A.M.; Baan, C.; Forouhi, N.G.; Herder, C.; Rathmann, W. Diabetes in Europe: An update. Diabetes Res. Clin. Pr. 2014, 103, 206–217. [Google Scholar] [CrossRef]
- Arnold Domínguez, Y.; González Hernández, O.; Martínez Vázquez, N.; Formental Hidalgo, B.I.; de Lourdes Arnold Alfonso, M.; González Calero, T.M.; Conesa González, A.I. Incidencia de la diabetes mellitus en Cuba, según tipo, en menores de 18 años de edad. Rev. Cuba. Endocrinol. 2017, 28, 1–19. [Google Scholar]
- Sulaiman, N.; Mahmoud, I.; Hussein, A.; Elbadawi, S.; Abusnana, S.; Zimmet, P.; Shaw, J. Diabetes risk score in the United Arab Emirates: A screening tool for the early detection of type 2 diabetes mellitus. BMJ Open. Diabetes Res. Care 2018, 6, e000489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangwung, P.; Petersen, K.F.; Shulman, G.I.; Knowles, J.W. Mitochondrial Dysfunction, Insulin Resistance, and Potential Genetic Implications. Endocrinology 2020, 161, bqaa017. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe–Marsh, N. Mitochondrial (dys) function and insulin resistance: From pathophysiological molecular mechanisms to the impact of diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Heilbronn, L.K. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol. Metab. 2008, 19, 324–330. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez–Franquesa, A.; Patti, M.-E. Insulin Resistance and Mitochondrial Dysfunction. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 465–520. [Google Scholar] [CrossRef]
- Chomentowski, P.; Coen, P.M.; Radikova, Z.; Goodpaster, B.H.; Toledo, F.G. Skeletal muscle mitochondria in insulin resistance: Differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J. Clin. Endocrinol. Metab. 2011, 96, 494–503. [Google Scholar] [CrossRef]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Asmann, Y.W.; Stump, C.S.; Short, K.R.; Coenen–Schimke, J.M.; Guo, Z.; Bigelow, M.L.; Nair, K.S. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 2006, 55, 3309–3319. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsøe, R.; Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [Green Version]
- Abdul–Ghani, M.A.; DeFronzo, R.A. Mitochondrial dysfunction, insulin resistance, and type 2 diabetes mellitus. Curr. Diabetes Rep. 2008, 8, 173–178. [Google Scholar] [CrossRef]
- Schrauwen–Hinderling, V.; Kooi, M.; Hesselink, M.; Jeneson, J.; Backes, W.; Van Echteld, C.; Van Engelshoven, J.; Mensink, M.; Schrauwen, P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI–matched control subjects. Diabetologia 2007, 50, 113–120. [Google Scholar] [CrossRef]
- Blaak, E.E.; van Aggel–Leijssen, D.P.; Wagenmakers, A.J.; Saris, W.H.; van Baak, M.A. Impaired oxidation of plasma–derived fatty acids in type 2 diabetic subjects during moderate–intensity exercise. Diabetes 2000, 49, 2102–2107. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D. Banting lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E. Skeletal muscle fat oxidation: Timing and flexibility are everything. J. Clin. Invest. 2005, 115, 1699–1702. [Google Scholar] [CrossRef] [Green Version]
- Amati, F.; Dubé, J.J.; Alvarez–Carnero, E.; Edreira, M.M.; Chomentowski, P.; Coen, P.M.; Switzer, G.E.; Bickel, P.E.; Stefanovic–Racic, M.; Toledo, F.G.S.; et al. Skeletal Muscle Triglycerides, Diacylglycerols, and Ceramides in Insulin Resistance. Another Parad. Endur.–Trained Athl. 2011, 60, 2588–2597. [Google Scholar] [CrossRef] [Green Version]
- Perreault, L.; Newsom, S.A.; Strauss, A.; Kerege, A.; Kahn, D.E.; Harrison, K.A.; Snell–Bergeon, J.K.; Nemkov, T.; D’Alessandro, A.; Jackman, M.R.; et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 2018, 3, e96805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, B.C.; Hunerdosse, D.M.; Kerege, A.; Playdon, M.C.; Perreault, L. Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia 2012, 55, 1140–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, B.C.; Goodpaster, B.H. Exercise and Muscle Lipid Content, Composition, and Localization: Influence on Muscle Insulin Sensitivity. Diabetes 2020, 69, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Zierath, J.; He, L.; Guma, A.; Wahlström, E.O.; Klip, A.; Wallberg–Henriksson, H. Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia 1996, 39, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.M.; Kim, J.K.; Yakar, S.; Dupont, J.; Hernandez–Sanchez, C.; Castle, A.L.; Filmore, J.; Shulman, G.I.; Le Roith, D. Functional inactivation of the IGF–I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes. Dev. 2001, 15, 1926–1934. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Ivanova, E.A.; Sobenin, I.A.; Yet, S.F.; Orekhov, A.N. The Role of Mitochondria in Cardiovascular Diseases. Biology 2020, 9, 137. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free. Radic. Biol. Med. 2005, 38, 1278–1295. [Google Scholar] [CrossRef]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid beta–oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, D.H.; Wang, Z.V. Glucose metabolism in cardiac hypertrophy and heart failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casademont, J.; Miró, Ò. Electron transport chain defects in heart failure. Heart Fail. Rev. 2002, 7, 131–139. [Google Scholar] [CrossRef]
- Aubert, G.; Vega, R.B.; Kelly, D.P. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim. Biophys. Acta (BBA)–Mol. Cell Res. 2013, 1833, 840–847. [Google Scholar] [CrossRef] [Green Version]
- Corral–Debrinski, M.; Shoffner, J.; Lott, M.; Wallace, D. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat. Res./DNAging 1992, 275, 169–180. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Khasanova, Z.B.; Postnov, A.Y.; Yarygina, E.I.; Orekhov, A.N.; Sobenin, I.A. Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis. Oxid. Med. Cell Longev. 2017, 2017, 6934394. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher–risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arter. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef] [Green Version]
- Yetkin–Arik, B.; Vogels, I.M.C.; Neyazi, N.; van Duinen, V.; Houtkooper, R.H.; van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. Endothelial tip cells in vitro are less glycolytic and have a more flexible response to metabolic stress than non–tip cells. Sci. Rep. 2019, 9, 10414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yetkin–Arik, B.; Vogels, I.M.C.; Nowak–Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Luo, Y.-X.; Chen, H.-Z.; Liu, D.-P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; De Saedeleer, C.J.; Préat, V.; Thissen, J.P.; Feron, O.; Sonveaux, P. Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis 2012, 15, 581–592. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, T.; Guo, H.; Cui, H.; Li, P.; Feng, D.; Hu, E.; Huang, Q.; Yang, A.; Zhou, J.; et al. Lactate potentiates angiogenesis and neurogenesis in experimental intracerebral hemorrhage. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Beckert, S.; Farrahi, F.; Aslam, R.S.; Scheuenstuhl, H.; Konigsrainer, A.; Hussain, M.Z.; Hunt, T.K. Lactate stimulates endothelial cell migration. Wound Repair. Regen. 2006, 14, 321–324. [Google Scholar] [CrossRef]
- San–Millan, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is Lactate an Oncometabolite? Evidence Supporting a Role for Lactate in the Regulation of Transcriptional Activity of Cancer–Related Genes in MCF7 Breast Cancer Cells. Front. Oncol. 2019, 9, 1536. [Google Scholar] [CrossRef]
- Peng, H.; Wang, X.; Du, J.; Cui, Q.; Huang, Y.; Jin, H. Metabolic Reprogramming of Vascular Endothelial Cells: Basic Research and Clinical Applications. Front. Cell Dev. Biol. 2021, 9, 626047. [Google Scholar] [CrossRef]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol.–Heart Circ. Physiol. 2009, 297, H1876–H1881. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.-F.; Rabinovitch, P.S.; Ungvari, Z.; Sinclair, D.; North, B. Mitochondria and Cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I. Vascular Cell Senescence. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jendrach, M.; Pohl, S.; Vöth, M.; Kowald, A.; Hammerstein, P.; Bereiter–Hahn, J. Morpho–dynamic changes of mitochondria during ageing of human endothelial cells. Mech. Ageing Dev. 2005, 126, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkman, D.L.; Robinson, A.T.; Rossman, M.J.; Seals, D.R.; Edwards, D.G. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2080–H2100. [Google Scholar] [CrossRef] [PubMed]
- Kozieł, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen–Dürr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef]
- Davidson, S.M.; Duchen, M.R. Endothelial mitochondria: Contributing to vascular function and disease. Circ. Res. 2007, 100, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Martín–Timón, I.; Sevillano–Collantes, C.; Segura–Galindo, A.; del Cañizo–Gómez, F.J. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J. Diabetes 2014, 5, 444. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance–trained athletes. J. Clin. Endocrinol. Metab. 2001, 86, 5755–5761. [Google Scholar] [CrossRef]
- Krssak, M.; Petersen, K.F.; Bergeron, R.; Price, T.; Laurent, D.; Rothman, D.L.; Roden, M.; Shulman, G.I. Intramuscular glycogen and intramyocellular lipid utilization during prolonged exercise and recovery in man: A 13C and 1H nuclear magnetic resonance spectroscopy study. J. Clin. Endocrinol. Metab. 2000, 85, 748–754. [Google Scholar]
- Van Loon, L.J.; Koopman, R.; Stegen, J.H.; Wagenmakers, A.J.; Keizer, H.A.; Saris, W.H. Intramyocellular lipids form an important substrate source during moderate intensity exercise in endurance—Trained males in a fasted state. J. Physiol. 2003, 553, 611–625. [Google Scholar] [CrossRef] [Green Version]
- Watt, M.J.; Heigenhauser, G.J.; Dyck, D.J.; Spriet, L.L. Intramuscular triacylglycerol, glycogen and acetyl group metabolism during 4 h of moderate exercise in man. J. Physiol. 2002, 541, 969–978. [Google Scholar] [CrossRef]
- Bergman, B.C.; Perreault, L.; Strauss, A.; Bacon, S.; Kerege, A.; Harrison, K.; Brozinick, J.T.; Hunerdosse, D.M.; Playdon, M.C.; Holmes, W.; et al. Intramuscular triglyceride synthesis: Importance in muscle lipid partitioning in humans. Am. J. Physiol.–Endocrinol. Metab. 2018, 314, E152–E164. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin–resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Maza, M.P.; Rodriguez, J.; Hirsch, S.; Leiva, L.; Barrera, G.; Bunout, D. Skeletal muscle ceramide species in men with abdominal obesity. J. Nutr. Health Aging 2015, 19, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, E.; Blachnio–Zabielska, A. The role of ceramides in insulin resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef] [Green Version]
- Tan–Chen, S.; Guitton, J.; Bourron, O.; Le Stunff, H.; Hajduch, E. Sphingolipid Metabolism and Signaling in Skeletal Muscle: From Physiology to Physiopathology. Front. Endocrinol. 2020, 11, 491. [Google Scholar] [CrossRef]
- Chavez, J.A.; Summers, S.A. A ceramide–centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Coen, P.; Dube, J.J.; Amati, F.; Stefanovic–Racic, M.; Ferrell, R.E.; Toledo, F.G.; Goodpaster, B.H. Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes 2010, 59, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef]
- Colombini, M. Ceramide channels and mitochondrial outer membrane permeability. J. Bioenerg. Biomembr. 2017, 49, 57–64. [Google Scholar] [CrossRef]
- Gudz, T.I.; Tserng, K.-Y.; Hoppel, C.L. Direct inhibition of mitochondrial respiratory chain complex III by cell–permeable ceramide. J. Biol. Chem. 1997, 272, 24154–24158. [Google Scholar] [CrossRef] [Green Version]
- Law, B.A.; Liao, X.; Moore, K.S.; Southard, A.; Roddy, P.; Ji, R.; Szulc, Z.; Bielawska, A.; Schulze, P.C.; Cowart, L.A. Lipotoxic very–long–chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. 2018, 32, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGurk, K.A.; Keavney, B.D.; Nicolaou, A. Circulating ceramides as biomarkers of cardiovascular disease: Evidence from phenotypic and genomic studies. Atherosclerosis 2021, 327, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Petrocelli, J.J.; McKenzie, A.I.; Mahmassani, Z.S.; Reidy, P.T.; Stoddard, G.J.; Poss, A.M.; Holland, W.L.; Summers, S.A.; Drummond, M.J. Ceramide Biomarkers Predictive of Cardiovascular Disease Risk Increase in Healthy Older Adults After Bed Rest. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Vasile, V.C.; Meeusen, J.W.; Medina Inojosa, J.R.; Donato, L.J.; Scott, C.G.; Hyun, M.S.; Vinciguerra, M.; Rodeheffer, R.R.; Lopez–Jimenez, F.; Jaffe, A.S. Ceramide scores predict cardiovascular risk in the community. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1558–1569. [Google Scholar] [CrossRef]
- Lopez, X.; Goldfine, A.B.; Holland, W.L.; Gordillo, R.; Scherer, P.E. Plasma ceramides are elevated in female children and adolescents with type 2 diabetes. J. Pediatr. Endocrinol. Metab. 2013, 26, 995–998. [Google Scholar] [CrossRef]
- Boon, J.; Hoy, A.J.; Stark, R.; Brown, R.D.; Meex, R.C.; Henstridge, D.C.; Schenk, S.; Meikle, P.J.; Horowitz, J.F.; Kingwell, B.A. Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes 2013, 62, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Kirwan, J.P. Plasma ceramides target skeletal muscle in type 2 diabetes. Diabetes 2013, 62, 352–354. [Google Scholar] [CrossRef] [Green Version]
- Turpin–Nolan, S.M.; Brüning, J.C. The role of ceramides in metabolic disorders: When size and localization matters. Nat. Rev. Endocrinol. 2020, 16, 224–233. [Google Scholar] [CrossRef]
- Bismuth, J.; Lin, P.; Yao, Q.; Chen, C. Ceramide: A common pathway for atherosclerosis? Atherosclerosis 2008, 196, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Vorkas, P.A.; Shalhoub, J.; Isaac, G.; Want, E.J.; Nicholson, J.K.; Holmes, E.; Davies, A.H. Metabolic Phenotyping of Atherosclerotic Plaques Reveals Latent Associations between Free Cholesterol and Ceramide Metabolism in Atherogenesis. J. Proteome Res. 2015, 14, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Devlin, C.M.; Leventhal, A.R.; Kuriakose, G.; Schuchman, E.H.; Williams, K.J.; Tabas, I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1723–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yang, X.; Xing, S.; Bian, F.; Yao, W.; Bai, X.; Zheng, T.; Wu, G.; Jin, S. Endogenous ceramide contributes to the transcytosis of oxLDL across endothelial cells and promotes its subendothelial retention in vascular wall. Oxidative Med. Cell Longev. 2014, 2014, 823071. [Google Scholar] [CrossRef] [Green Version]
- Mah, M.; Febbraio, M.; Turpin–Nolan, S. Circulating Ceramides– Are Origins Important for Sphingolipid Biomarkers and Treatments? Front. Endocrinol. 2021, 12, 684448. [Google Scholar] [CrossRef]
- Rome, S.; Forterre, A.; Mizgier, M.L.; Bouzakri, K. Skeletal Muscle–Released Extracellular Vesicles: State of the Art. Front. Physiol. 2019, 10, 929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta–analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef]
- Jayaraman, A.; Pike, C.J. Alzheimer’s disease and type 2 diabetes: Multiple mechanisms contribute to interactions. Curr. Diabetes Rep. 2014, 14, 476. [Google Scholar] [CrossRef] [Green Version]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.; Cha, R.; Kokmen, E.; O’brien, P.; Palumbo, P. Risk of dementia among persons with diabetes mellitus: A population–based cohort study. Am. J. Epidemiol. 1997, 145, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-C.; Chung, C.-M.; Leu, H.-B.; Lin, L.-Y.; Chiu, C.-C.; Hsu, C.-Y.; Chiang, C.-H.; Huang, P.-H.; Chen, T.-J.; Lin, S.-J. Diabetes mellitus and the risk of Alzheimer’s disease: A nationwide population–based study. PLoS ONE 2014, 9, e87095. [Google Scholar] [CrossRef]
- Xia, W.; Wang, S.; Sun, Z.; Bai, F.; Zhou, Y.; Yang, Y.; Wang, P.; Huang, Y.; Yuan, Y. Altered baseline brain activity in type 2 diabetes: A resting–state fMRI study. Psychoneuroendocrinology 2013, 38, 2493–2501. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Z.; Liang, Y.; Tan, X.; Li, Y.; Qin, C.; Feng, Y.; Ma, X.; Mo, Z.; Xia, J.; et al. Classification of type 2 diabetes mellitus with or without cognitive impairment from healthy controls using high–order functional connectivity. Hum. Brain Mapp. 2021, 42, 4671–4684. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.; Phan, T.G.; Chen, J.; Blizzard, L.; Beare, R.; Venn, A.; Münch, G.; Wood, A.G.; Forbes, J.; Greenaway, T.M.; et al. Brain atrophy in type 2 diabetes: Regional distribution and influence on cognition. Diabetes Care 2013, 36, 4036–4042. [Google Scholar] [CrossRef] [Green Version]
- Manschot, S.M.; Brands, A.M.; van der Grond, J.; Kessels, R.P.; Algra, A.; Kappelle, L.J.; Biessels, G.J. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 2006, 55, 1106–1113. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Suain Bon, R.; Ariaratnam, S.; Mat Saher, Z.; Mohamad, M.; Lee, F.S. Cognitive Impairment and Its Associated Risk Factors in the Elderly With Type 2 Diabetes Mellitus. Front. Psychiatry 2021, 12, 669725. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Dembitskaya, Y.; Piette, C.; Perez, S.; Berry, H.; Magistretti, P.J.; Venance, L. Lactate supply overtakes glucose when neural computational and cognitive loads scale up. Proc. Natl. Acad. Sci. USA 2022, 119, e2212004119. [Google Scholar] [CrossRef]
- Newington, J.T.; Harris, R.A.; Cumming, R.C. Reevaluating Metabolism in Alzheimer’s Disease from the Perspective of the Astrocyte–Neuron Lactate Shuttle Model. J. Neurodegener. Dis. 2013, 2013, 234572. [Google Scholar] [CrossRef] [Green Version]
- Kuehn, B.M. In Alzheimer Research, Glucose Metabolism Moves to Center Stage. JAMA 2020, 323, 297–299. [Google Scholar] [CrossRef]
- Hammond, T.C.; Lin, A.L. Glucose Metabolism is a Better Marker for Predicting Clinical Alzheimer’s Disease than Amyloid or Tau. J. Cell Immunol. 2022, 4, 15–18. [Google Scholar] [PubMed]
- Hoyer, S. Oxidative energy metabolism in Alzheimer brain: Studies in early–onset and late–onset cases. Mol. Chem. Neuropathol. 1992, 16, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I.; Horwitz, B.; Grady, C.L.; Haxby, J.V.; DeCarli, C.; Schapiro, M.B. Abnormal brain glucose metabolism in Alzheimer’s disease, as measured by positron emission tomography. In Fuel Homeostasis and the Nervous System, Advances in Experimental Medicine and Biology; Vranic, M., Efendic, S., Hollenberg, C.H., Eds.; Springer: Boston, MA, USA, 1991; Volume 291, pp. 231–248. [Google Scholar]
- Ou, Y.-N.; Xu, W.; Li, J.-Q.; Guo, Y.; Cui, M.; Chen, K.-L.; Huang, Y.-Y.; Dong, Q.; Tan, L.; Yu, J.-T.; et al. FDG–PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimer’s Res. Ther. 2019, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413–e422, quiz e423–e426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, N.L.; Heidebrink, J.L.; Clark, C.M.; Jagust, W.J.; Arnold, S.E.; Barbas, N.R.; DeCarli, C.S.; Scott Turner, R.; Koeppe, R.A.; Higdon, R.; et al. FDG–PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain 2007, 130, 2616–2635. [Google Scholar] [CrossRef]
- Khosravi, M.; Peter, J.; Wintering, N.A.; Serruya, M.; Shamchi, S.P.; Werner, T.J.; Alavi, A.; Newberg, A.B. 18F–FDG Is a Superior Indicator of Cognitive Performance Compared to 18F–Florbetapir in Alzheimer’s Disease and Mild Cognitive Impairment Evaluation: A Global Quantitative Analysis. J. Alzheimer’s Dis. 2019, 70, 1197–1207. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley–Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Potenza, M.A.; Sgarra, L.; Desantis, V.; Nacci, C.; Montagnani, M. Diabetes and Alzheimer’s Disease: Might Mitochondrial Dysfunction Help Deciphering the Common Path? Antioxidants 2021, 10, 1257. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, Defective Insulin Signaling, and Mitochondrial Dysfunction as Common Molecular Denominators Connecting Type 2 Diabetes to Alzheimer Disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Terzo, S.; Amato, A.; Mulè, F. From obesity to Alzheimer’s disease through insulin resistance. J. Diabetes Its Complicat. 2021, 35, 108026. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K. Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP–1 analogs. Neurodegener. Dis. Manag. 2014, 4, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.; Pernet, A.; Hallett, W.A.; Bingham, E.; Marsden, P.K.; Amiel, S.A. Lactate: A preferred fuel for human brain metabolism in vivo. J. Cereb. Blood Flow. Metab. 2003, 23, 658–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenn, T.C.; Martin, N.A.; Horning, M.A.; McArthur, D.L.; Hovda, D.A.; Vespa, P.; Brooks, G.A. Lactate: Brain fuel in human traumatic brain injury: A comparison with normal healthy control subjects. J. Neurotrauma 2015, 32, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte–neuron lactate transport is required for long–term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.A. Cell–cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Hashimoto, T.; Brooks, G.A. Mitochondrial lactate oxidation complex and an adaptive role for lactate production. Med. Sci. Sport. Exerc. 2008, 40, 486–494. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Pellerin, L. Astrocytes couple synaptic activity to glucose utilization in the brain. Physiology 1999, 14, 177–182. [Google Scholar] [CrossRef]
- Pellerin, L.; Pellegri, G.; Bittar, P.G.; Charnay, Y.; Bouras, C.; Martin, J.-L.; Stella, N.; Magistretti, P.J. Evidence supporting the existence of an activity–dependent astrocyte–neuron lactate shuttle. Dev. Neurosci. 1998, 20, 291–299. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes. Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF–1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- San–Millan, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez–Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al–Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. Elife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- Arismendi–Morillo, G.; Castellano–Ramirez, A.; Seyfried, T.N. Ultrastructural characterization of the Mitochondria–associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct. Pathol. 2017, 41, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.A.; Lundby, C. Mitochondria express enhanced quality as well as quantity in association with aerobic fitness across recreationally active individuals up to elite athletes. J. Appl. Physiol. 2013, 114, 344–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollnick, P.D.; Armstrong, R.B.; Saubert, C.W.t.; Piehl, K.; Saltin, B. Enzyme activity and fiber composition in skeletal muscle of untrained and trained men. J. Appl. Physiol. 1972, 33, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Daussin, F.N.; Zoll, J.; Ponsot, E.; Dufour, S.P.; Doutreleau, S.; Lonsdorfer, E.; Ventura–Clapier, R.; Mettauer, B.; Piquard, F.; Geny, B. Training at high exercise intensity promotes qualitative adaptations of mitochondrial function in human skeletal muscle. J. Appl. Physiol. 2008, 104, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, D.N.; Sinning, W.E.; Walro, J.; Sieck, G.C.; Lemon, P. Oxidative capacity of human muscle fiber types: Effects of age and training status. J. Appl. Physiol. 1995, 78, 2033–2038. [Google Scholar] [CrossRef]
- Turcotte, L.P.; Richter, E.A.; Kiens, B. Increased plasma FFA uptake and oxidation during prolonged exercise in trained vs. untrained humans. Am. J. Physiol. 1992, 262, E791–E799. [Google Scholar] [CrossRef]
- Kiens, B.; Essen–Gustavsson, B.; Christensen, N.J.; Saltin, B. Skeletal muscle substrate utilization during submaximal exercise in man: Effect of endurance training. J. Physiol. 1993, 469, 459–478. [Google Scholar] [CrossRef]
- Jansson, E.; Kaijser, L. Substrate utilization and enzymes in skeletal muscle of extremely endurance–trained men. J. Appl. Physiol. 1987, 62, 999–1005. [Google Scholar] [CrossRef]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [CrossRef]
- Gollnick, P.D.; King, D.W. Effect of exercise and training on mitochondria of rat skeletal muscle. Am. J. Physiol. 1969, 216, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, P.D.; Armstrong, R.B.; Saltin, B.; Saubert, C.W.t.; Sembrowich, W.L.; Shepherd, R.E. Effect of training on enzyme activity and fiber composition of human skeletal muscle. J. Appl. Physiol. 1973, 34, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.N.; Chen, C.C.; Hood, D.A. Mitochondria, muscle health, and exercise with advancing age. Physiology 2015, 30, 208–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubicic, V.; Joseph, A.M.; Adhihetty, P.J.; Huang, J.H.; Saleem, A.; Uguccioni, G.; Hood, D.A. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging 2009, 1, 818–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Rizza, R.A.; Coenen–Schimke, J.M.; Nair, K.S. Impact of Aerobic Exercise Training on Age–Related Changes in Insulin Sensitivity and Muscle Oxidative Capacity. Diabetes 2003, 52, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.L.; Thacker, E.M.; Kelley, G.A.; Musch, T.I.; Sinoway, L.I.; Foster, V.L.; Dickinson, A.L. Effect of training/detraining on submaximal exercise responses in humans. J. Appl. Physiol. 1987, 63, 1719–1724. [Google Scholar] [CrossRef]
- Klausen, K.; Andersen, L.B.; Pelle, I. Adaptive changes in work capacity, skeletal muscle capillarization and enzyme levels during training and detraining. Acta Physiol. Scand. 1981, 113, 9–16. [Google Scholar] [CrossRef]
- Wibom, R.; Hultman, E.; Johansson, M.; Matherei, K.; Constantin–Teodosiu, D.; Schantz, P.G. Adaptation of mitochondrial ATP production in human skeletal muscle to endurance training and detraining. J. Appl. Physiol. 1992, 73, 2004–2010. [Google Scholar] [CrossRef]
- Toledo, F.G.S.; Watkins, S.; Kelley, D.E. Changes Induced by Physical Activity and Weight Loss in the Morphology of Intermyofibrillar Mitochondria in Obese Men and Women. J. Clin. Endocrinol. Metab. 2006, 91, 3224–3227. [Google Scholar] [CrossRef]
- Toledo, F.G.; Menshikova, E.V.; Azuma, K.; Radikova, Z.; Kelley, C.A.; Ritov, V.B.; Kelley, D.E. Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes 2008, 57, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Toledo, F.G.S.; Menshikova, E.V.; Ritov, V.B.; Azuma, K.; Radikova, Z.; DeLany, J.; Kelley, D.E. Effects of Physical Activity and Weight Loss on Skeletal Muscle Mitochondria and Relationship With Glucose Control in Type 2 Diabetes. Diabetes 2007, 56, 2142–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Jiang, D.-M.; Yu, R.-R.; Zhang, L.-L.; Liu, Y.-Z.; Chen, J.-X.; Chen, H.-C.; Liu, Y.-P. The Effect of Aerobic Exercise on the Oxidative Capacity of Skeletal Muscle Mitochondria in Mice with Impaired Glucose Tolerance. J. Diabetes Res. 2022, 2022, 3780156. [Google Scholar] [CrossRef] [PubMed]
- Taivassalo, T.; Shoubridge, E.A.; Chen, J.; Kennaway, N.G.; DiMauro, S.; Arnold, D.L.; Haller, R.G. Aerobic conditioning in patients with mitochondrial myopathies: Physiological, biochemical, and genetic effects. Ann. Neurol. 2001, 50, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Taivassalo, T.; Gardner, J.L.; Taylor, R.W.; Schaefer, A.M.; Newman, J.; Barron, M.J.; Haller, R.G.; Turnbull, D.M. Endurance training and detraining in mitochondrial myopathies due to single large–scale mtDNA deletions. Brain 2006, 129, 3391–3401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, A.Y.; Chen, Y.-C.; Hsu, C.-C.; Fu, T.-C.; Wang, J.-S. The Effects of Exercise Training on Mitochondrial Function in Cardiovascular Diseases: A Systematic Review and Meta–Analysis. Int. J. Mol. Sci. 2022, 23, 12559. [Google Scholar] [CrossRef]
- Gu, C.; Yan, J.; Zhao, L.; Wu, G.; Wang, Y.-L. Regulation of mitochondrial dynamics by aerobic exercise in cardiovascular diseases. Front. Cardiovasc. Med. 2022, 8, 2001. [Google Scholar] [CrossRef]
- Hambrecht, R.; Niebauer, J.; Fiehn, E.; Kälberer, B.; Offner, B.; Hauer, K.; Riede, U.; Schlierf, G.; Kübler, W.; Schuler, G. Physical training in patients with stable chronic heart failure: Effects on cardiorespiratory fitness and ultrastructural abnormalities of leg muscles. J. Am. Coll. Cardiol. 1995, 25, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Hambrecht, R.; Fiehn, E.; Yu, J.; Niebauer, J.; Weigl, C.; Hilbrich, L.; Adams, V.; Riede, U.; Schuler, G. Effects of endurance training on mitochondrial ultrastructure and fiber type distribution in skeletal muscle of patients with stable chronic heart failure. J. Am. Coll. Cardiol. 1997, 29, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-C.; Tsai, H.-H.; Fu, T.-C.; Wang, J.-S. Exercise Training Enhances Platelet Mitochondrial Bioenergetics in Stroke Patients: A Randomized Controlled Trial. J. Clin. Med. 2019, 8, 2186. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-L.; Fu, T.-C.; Hsu, C.-C.; Huang, S.-C.; Lin, Y.-T.; Wang, J.-S. Cycling exercise training enhances platelet mitochondrial bioenergetics in patients with peripheral arterial disease: A randomized controlled trial. Thromb. Haemost. 2021, 121, 900–912. [Google Scholar]
- Jiang, H.K.; Wang, Y.H.; Sun, L.; He, X.; Zhao, M.; Feng, Z.H.; Yu, X.J.; Zang, W.J. Aerobic interval training attenuates mitochondrial dysfunction in rats post–myocardial infarction: Roles of mitochondrial network dynamics. Int. J. Mol. Sci. 2014, 15, 5304–5322. [Google Scholar] [CrossRef] [Green Version]
- Campos, J.C.; Queliconi, B.B.; Bozi, L.H.; Bechara, L.R.; Dourado, P.M.; Andres, A.M.; Jannig, P.R.; Gomes, K.M.; Zambelli, V.O.; Rocha–Resende, C. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy 2017, 13, 1304–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandolesi, L.; Polverino, A.; Montuori, S.; Foti, F.; Ferraioli, G.; Sorrentino, P.; Sorrentino, G. Effects of Physical Exercise on Cognitive Functioning and Wellbeing: Biological and Psychological Benefits. Front. Psychol. 2018, 9, 509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- De la Rosa, A.; Olaso–Gonzalez, G.; Arc–Chagnaud, C.; Millan, F.; Salvador–Pascual, A.; García–Lucerga, C.; Blasco–Lafarga, C.; Garcia–Dominguez, E.; Carretero, A.; Correas, A.G. Physical exercise in the prevention and treatment of Alzheimer’s disease. J. Sport. Health Sci. 2020, 9, 394–404. [Google Scholar] [CrossRef]
- Hamer, M.; Chida, Y. Physical activity and risk of neurodegenerative disease: A systematic review of prospective evidence. Psychol. Med. 2009, 39, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Farina, N.; Rusted, J.; Tabet, N. The effect of exercise interventions on cognitive outcome in Alzheimer’s disease: A systematic review. Int. Psychogeriatr. 2014, 26, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, A.; Boyle, P.; Yu, L.; Shah, R.; Wilson, R.; Bennett, D. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology 2012, 78, 1323–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cass, S.P. Alzheimer’s disease and exercise: A literature review. Curr. Sport. Med. Rep. 2017, 16, 19–22. [Google Scholar] [CrossRef]
- Intlekofer, K.A.; Cotman, C.W. Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol. Dis. 2013, 57, 47–55. [Google Scholar] [CrossRef]
- Dubé, J.J.; Broskey, N.T.; Despines, A.A.; Stefanovic–Racic, M.; Toledo, F.G.; Goodpaster, B.H.; Amati, F. Muscle Characteristics and Substrate Energetics in Lifelong Endurance Athletes. Med. Sci. Sport. Exerc. 2016, 48, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzen, A.M.; Andersen, S.P.; Qadri, K.A.N.; Thøgersen, F.D.; Krag, T.; Ørngreen, M.C.; Vissing, J.; Jeppesen, T.D. Effect of Aerobic Exercise Training and Deconditioning on Oxidative Capacity and Muscle Mitochondrial Enzyme Machinery in Young and Elderly Individuals. J. Clin. Med. 2020, 9, 3113. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.J.; Granata, C.; Eynon, N. Can we optimise the exercise training prescription to maximise improvements in mitochondria function and content? Biochim. Et. Biophys. Acta (BBA)—Gen. Subj. 2014, 1840, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Long, Q.; Huang, L.; Huang, K.; Yang, Q. Assessing Mitochondrial Bioenergetics in Isolated Mitochondria from Mouse Heart Tissues Using Oroboros 2k–Oxygraph. Methods Mol. Biol. 2019, 1966, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Horan, M.P.; Pichaud, N.; Ballard, J.W.O. Review: Quantifying Mitochondrial Dysfunction in Complex Diseases of Aging. J. Gerontol. Ser. A 2012, 67, 1022–1035. [Google Scholar] [CrossRef] [Green Version]
- Pesta, D.; Gnaiger, E. High–Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. In Mitochondrial Bioenergetics: Methods and Protocols; Palmeira, C.M., Moreno, A.J., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 25–58. [Google Scholar] [CrossRef]
- Boutagy, N.E.; Rogers, G.W.; Pyne, E.S.; Ali, M.M.; Hulver, M.W.; Frisard, M.I. Using Isolated Mitochondria from Minimal Quantities of Mouse Skeletal Muscle for High throughput Microplate Respiratory Measurements. J. Vis. Exp. 2015, 104, e53216. [Google Scholar] [CrossRef] [Green Version]
- Amara, C.E.; Marcinek, D.J.; Shankland, E.G.; Schenkman, K.A.; Arakaki, L.S.L.; Conley, K.E. Mitochondrial function in vivo: Spectroscopy provides window on cellular energetics. Methods 2008, 46, 312–318. [Google Scholar] [CrossRef]
- Kemp, G.J.; Ahmad, R.E.; Nicolay, K.; Prompers, J.J. Quantification of skeletal muscle mitochondrial function by 31P magnetic resonance spectroscopy techniques: A quantitative review. Acta Physiol. 2015, 213, 107–144. [Google Scholar] [CrossRef]
- Campbell, M.D.; Marcinek, D.J. Evaluation of in vivo mitochondrial bioenergetics in skeletal muscle using NMR and optical methods. Biochim. Et. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 716–724. [Google Scholar] [CrossRef]
- Jucker, B.M.; Dufour, S.; Ren, J.; Cao, X.; Previs, S.F.; Underhill, B.; Cadman, K.S.; Shulman, G.I. Assessment of mitochondrial energy coupling in vivo 13C/31P NMR. Proc. Natl. Acad. Sci. USA 2000, 97, 6880–6884. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
San-Millán, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12, 782. https://doi.org/10.3390/antiox12040782
San-Millán I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants. 2023; 12(4):782. https://doi.org/10.3390/antiox12040782
Chicago/Turabian StyleSan-Millán, Iñigo. 2023. "The Key Role of Mitochondrial Function in Health and Disease" Antioxidants 12, no. 4: 782. https://doi.org/10.3390/antiox12040782
APA StyleSan-Millán, I. (2023). The Key Role of Mitochondrial Function in Health and Disease. Antioxidants, 12(4), 782. https://doi.org/10.3390/antiox12040782