

Oxidants and Cardiorenal Vascular Remodeling—Insights from Rare Genetic Tubulopathies: Bartter’s and Gitelman’s Syndromes


 and
and
Abstract
:
1. Introduction
2. Gitelman’s and Bartter’s Syndromes
3. ROS, Oxidative Stress and Renin Angiotensin System Activity
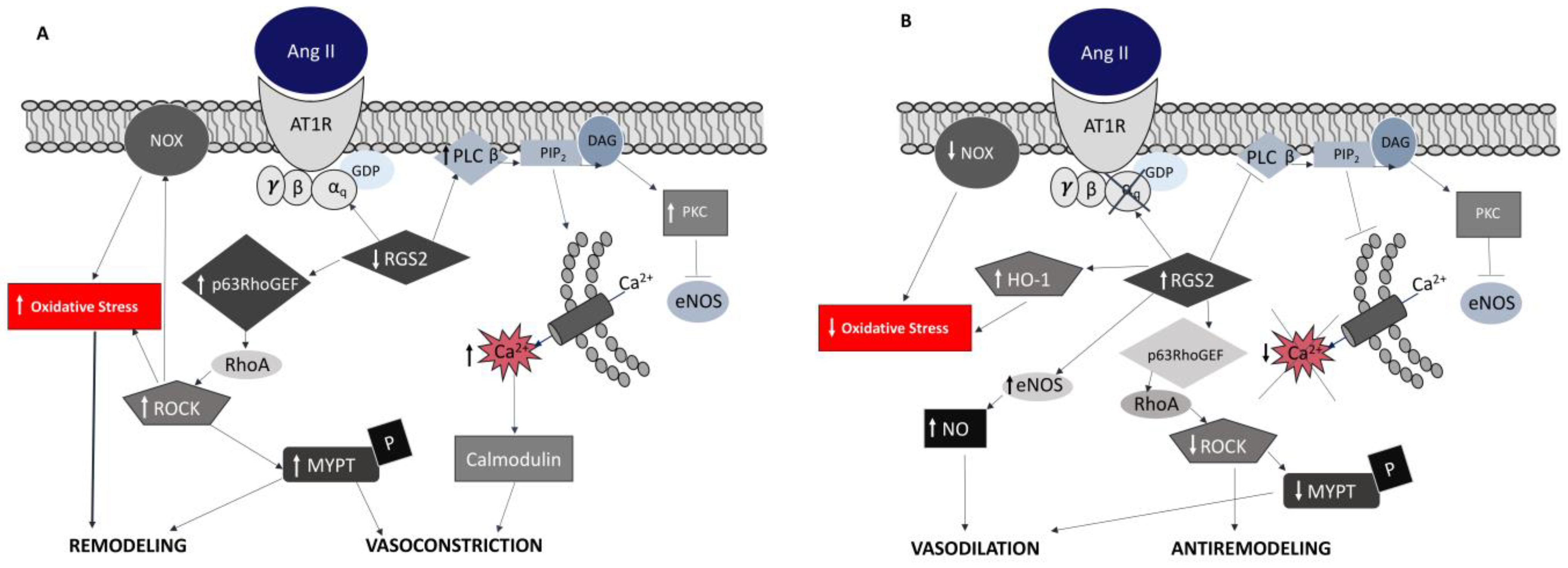
4. RAS and ROS in Gitelman’s and Bartter’s Syndrome Patients
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Konrad, M.; Nijenhuis, T.; Ariceta, G.; Bertholet-Thomas, A.; Calo, L.A.; Capasso, G.; Emma, F.; Schlingmann, K.P.; Singh, M.; Trepiccione, F.; et al. Diagnosis and management of Bartter syndrome: Executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int. 2021, 99, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.; Bockenhauer, D.; Bolignano, D.; Calò, L.A.; Cosyns, E.; Devuyst, O.; Ellison, D.H.; Karet Frankl, F.E.; Knoers, N.V.A.M.; Konrad, M.; et al. Gitelman syndrome: Consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calò, L.A.; Davis, P.A.; Rossi, G.P. Understanding the mechanisms of angiotensin II signaling involved in hypertension and its long-term sequelae. J. Hypertens 2014, 32, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Ravarotto, V.; Bertoldi, G.; Stefanelli, L.F.; Nalesso, F.; Calò, L.A. Gitelman’s and Bartter’s Syndromes: From Genetics to the Molecular Basis of Hypertension and More. Kidney Blood Press Res. 2022, 47, 556–564. [Google Scholar] [CrossRef]
- Gitelman, H.J.; Graham, J.B.; Welt, L.G. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans. Assoc. Am. Physicians 1966, 79, 221–235. [Google Scholar] [PubMed]
- Ji, W.; Foo, J.N.; O’Roak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Cheh, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008, 40, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Dong, C.; Xi, Y.G.; Su, X. Thiazide-sensitive Na+-Cl- cotransporter: Genetic polymorphisms and human diseases. Acta Biochim. Biophys Sin. 2015, 47, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative Stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative stress: Introductory remarks. In Oxidative Stress; Sies, H., Ed.; Academic Press: London, UK, 1985; pp. 1–7. [Google Scholar]
- Nistala, R.; Whaley-Connell, A.; Sowers, J.R. Redox control of renal function and hypertension. Antioxid Redox Signal 2008, 10, 2047–2089. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, P.M. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants 2020, 9, 1292. [Google Scholar] [CrossRef] [PubMed]
- Seccia, T.M.; Caroccia, B.; Calò, L.A. Hypertensive nephropathy. Moving from classic to emerging pathogenetic mechanisms. J. Hypertens 2017, 35, 205–212. [Google Scholar] [CrossRef]
- Ravarotto, V.; Bertoldi, G.; Rigato, M.; Pagnin, E.; Gobbi, L.; Davis, P.A.; Calò, L.A. Tracing Angiotensin II’s Yin—Yang effects on cardiovascular-renal pathophysiology. Yin and yang systems of Ang II. Minerva Med. 2021, 114, 56–67. [Google Scholar] [PubMed]
- Poznyak, A.V.; Bharadwaj, D.; Prasad, G.; Grechko, A.V.; Sazonova, M.A.; Orekhov, A.N. Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. Int. J. Mol. Sci. 2021, 22, 6702. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Hernandez, W.; Ansari, R.A.; Ferder, L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 2015, 6, 209–217. [Google Scholar] [CrossRef]
- Ravarotto, V.; Simioni, F.; Pagnin, E.; Davis, P.A.; Calò, L.A. Oxidative stress—chronic kidney disease—cardiovascular disease: A vicious circle. Life Sci. 2018, 210, 125–131. [Google Scholar] [CrossRef]
- Griendling, K.K.; Soresen, D.; Ushio-Fukai, M. NAD(P)H oxidase. Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta—Mol. Basis Dis. 2014, 1842, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.; Haskard, D.; Khamis, R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis—Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc. Med. 2019, 29, 22–26. [Google Scholar] [CrossRef]
- Jalife, J.; Kaur, K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 2015, 25, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sovari, A.A. Cellular and Molecular Mechanisms of Arrhythmia by Oxidative Stress. Cardiol. Res. Pract. 2016, 2016, 9656078. [Google Scholar] [CrossRef] [Green Version]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Seccia, T.M.; Caroccia, B.; Gioco, F.; Piazza, M.; Buccella, V.; Guidolin, D.; Guerzoni, E.; Montini, B.; Petrelli, L.; Pagnin, E.; et al. Endothelin-1 drives epithelial-mesenchymal transition in hypertensive nephroangiosclerosis. J. Am. Heart Assoc. 2016, 5, e003888. [Google Scholar] [CrossRef] [Green Version]
- Chitalia, N.; Ross, L.; Krishnamoorthy, M.; Kapustin, A.; Shanahan, C.M.; Kaski, J.C.; Roy-Chaudhury, P.; Chemla, E.; Banerjee, D. Neointimal Hyperplasia and Calcification in Medium Sized Arteries in Adult Patients with Chronic Kidney Disease. Semin. Dial. 2015, 28, E35–E40. [Google Scholar] [CrossRef]
- Ishii, Y.; Schuessler, R.B.; Gaynor, S.L.; Yamada, K.; Fu, A.S.; Boineau, J.P.; Damiano, R.J. Inflammation of Atrium After Cardiac Surgery Is Associated With Inhomogeneity of Atrial Conduction and Atrial Fibrillation. Circulation 2005, 111, 2881–2888. [Google Scholar] [CrossRef] [Green Version]
- Calò, L.A.; Ravarotto, V.; Bertoldi, G.; Pagnin, E.; Rossi, B.; Rigato, M.; Davis, P.A.; Proietti, R. Rho Kinase Activity, Connexin 40, and Atrial Fibrillation: Mechanistic Insights from End-Stage Renal Disease on Dialysis Patients. J. Clin. Med. 2020, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Ravarotto, V.; Bertoldi, G.; Stefanelli, L.F.; Nalesso, F.; Calò, L.A. Pathomechanism of oxidative stress in cardiovascularrenal remodeling and therapeutic strategies. Kidney Res. Clin. Pract. 2022, 41, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.; Ceolotto, G.; Milani, M.; Pagnin, E.; Van Den Heuvel, L.P.; Sartori, M.; Davis, P.A.; Costa, R.; Semplicini, A. Abnormalities of Gq-mediated cell signaling in Bartter and Gitelman syndromes1. Kidney Int. 2001, 60, 882–889. [Google Scholar] [CrossRef]
- Calò, L.; Davis, P.A.; Semplicini, A. Reduced content of α subunit of Gq protein content in monocytes of Bartter and Gitelman syndromes: Relationship with vascular hyporeactivity. Kidney Int. 2002, 61, 353–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calò, L.A.; Pagnin, E.; Davis, P.A.; Sartori, M.; Ceolotto, G.; Pessina, A.C.; Semplicini, A. Increased Expression of Regulator of G Protein Signaling-2 (RGS-2) in Bartter’s/Gitelman’s Syndrome. A Role in the Control of Vascular Tone and Implication for Hypertension. J. Clin. Endocrinol. Metab. 2004, 89, 4153–4157. [Google Scholar] [CrossRef]
- Gu, S.; Cifelli, C.; Wang, S.; Heximer, S.P. RGS proteins: Identifying new GAPs in the understanding of blood pressure regulation and cardiovascular function. Clin. Sci. 2009, 116, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semplicini, A.; Lenzini, L.; Sartori, M.; Papparella, I.; Calò, L.A.; Pagnin, E.; Strapazzon, G.; Benna, C.; Costa, R.; Avogaro, A.; et al. Reduced expression of regulator of G-protein signalling 2 (RGS2) in hypertensive patients increases calcium mobilization and ERK1/2 phosphorylation induced by angiotensin II. J. Hypertens 2006, 24, 1115–1124. [Google Scholar] [CrossRef]
- Calò, L.A.; Pagnin, E.; Ceolotto, G.; Davis, P.A.; Schiavo, S.; Papparella, I.; Semplicini, A.; Pessina, A.C. Silencing regulator of G protein signalling-2 (rgs-2) increases angiotensin Ii signalling: Insights into hypertension from findings in Bartter’s/gitelman’s syndromes. J. Hypertens 2008, 26, 938–945. [Google Scholar] [CrossRef]
- Qiao, Y.-N.; He, W.-Q.; Chen, C.-P.; Zhang, C.-H.; Zhao, W.; Wang, P.; Zhang, L.; Wu, Y.-Z.; Yang, X.; Peng, Y.-J.; et al. Myosin Phosphatase Target Subunit 1 (MYPT1) Regulates the Contraction and Relaxation of Vascular Smooth Muscle and Maintains Blood Pressure. J. Biol. Chem. 2014, 289, 22512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calò, L.A.; Pessina, A.C. RhoA/Rho-kinase pathway: Much more than just a modulation of vascular tone. Evidence from studies in humans. J. Hypertens 2007, 25, 259–264. [Google Scholar] [CrossRef]
- Pagnin, E.; Semplicini, A.; Sartori, M.; Pessina, A.C.; Calò, L.A. Reduced mRNA and protein content of rho guanine nucleotide exchange factor (RhoGEF) in Bartter’s and Gitelman’s syndromes: Relevance for the pathophysiology of hypertension. Am. J. Hypertens 2005, 18, 1200–1205. [Google Scholar] [CrossRef] [Green Version]
- Calò, L.A.; Davis, P.A.; Pagnin, E.; Dal Maso, L.; Maiolino, G.; Seccia, T.M.; Pessina, A.C.; Rossi, G.P. Increased level of p63RhoGEF and RhoA/Rho kinase activity in hypertensive patients. J. Hypertens 2014, 32, 331–338. [Google Scholar] [CrossRef]
- Calò, L.A.; Pagnin, E.; Davis, P.A.; Sartori, M.; Semplicini, A. Oxidative stress-related factors in Bartter’s and Gitelman’s syndromes: Relevance for angiotensin II signalling. Nephrol. Dial. Transpl. 2003, 18, 1518–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.M.; Holt, A.G. Why antioxidant therapies have failed in clinical trials. J. Theor. Biol. 2018, 457, 1–5. [Google Scholar] [CrossRef]
- Lazzarin, T.; Rufino Garcia, L.; Martins, D.; Rios Queiroz, D.A.; Rodrigues Tonon, C.; da Silva Balin, P.; Furlan Polegato, B.; Rupp de Paiva, S.A.; Schmidt Azevedo, P.; Minicucci, M.; et al. Role of Nutrients and Foods in Attenuation of Cardiac Remodeling through Oxidative Stress Pathways. Antioxidants 2022, 11, 2064. [Google Scholar] [CrossRef] [PubMed]
- Ravarotto, V.; Pagnin, E.; Maiolino, G.; Fragasso, A.; Carraro, G.; Rossi, B.; Calò, L.A. The blocking of angiotensin II type 1 receptor and RhoA/Rho kinase activity in hypertensive patients: Effect of olmesartan medoxomil and implication with cardiovascular-renal remodeling. J. Renin.-Angiotensin.-Aldosterone Syst. 2015, 16, 1245–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calò, L.A.; Vertolli, U.; Pagnin, E.; Ravarotto, V.; Davis, P.A.; Lupia, M.; Naso, E.; Maiolino, G.; Naso, A. Increased rho kinase activity in mononuclear cells of dialysis and stage 3–4 chronic kidney disease patients with left ventricular hypertrophy: Cardiovascular risk implications. Life Sci. 2016, 148, 80–85. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| GS | BS Type 1 | BS Type 2 | BS Type 3 | BS Type 4 | BS Type 4b | BS Type 5 | |
|---|---|---|---|---|---|---|---|
| Gene | SCL12A3 | SCL12A1 | KCNJ1 | CLCNKb | BSND | CLCNKa+CLCNKb | MAGE-D2 |
| Protein | NCC | NKCC2 | ROMK | CIC-Kb | Barttin | CIC-Ka+CIC Kb | MAGE-D2 |
| Region of the Tubule | DCT | TAL | TAL+DCT | TAL+DCT | TAL+DCT | TAL+DCT | TAL+DCT |
| Onset | Adulthood | Prenatally | Prenatally | 0–5 years | Prenatally | Prenatally | prenatally |
| Features | HypoMg, HypoCa, HypoK, Mild dehydration | Polyuria HypoCl, HypoK, Alkalosis HyperCa nephrocalcinosis | Polyuria hypoCl, alkalosis, Transient HyperK, HyperCa | Failure to thrive HypoCl Alkalosis HypoK, HyperCa HypoMg | Polyuria hypoCl alkalosis HypoK, HyperCa deafness | Polyuria hypoCl alkalosis HypoK, HyperCa deafness | Polyuria, hypoCl, alkalosis, HypoK, HyperCa |
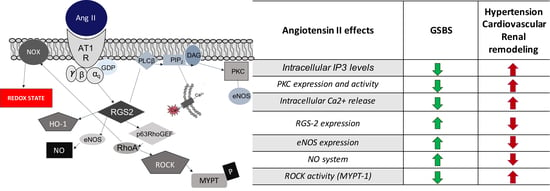
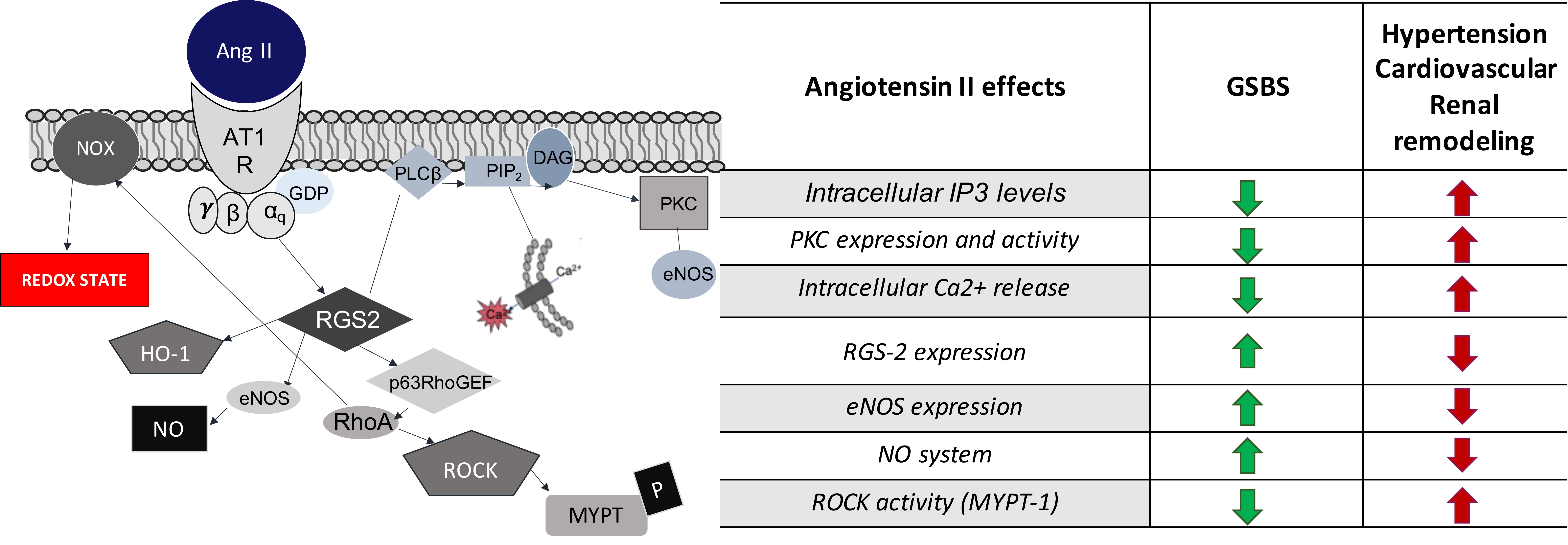
| Angiotensin II Effects | GSBS | Hypertension |
|---|---|---|
| PKC expression and activity | Decreased | Increased |
| Intracellular IP3 levels | Decreased | Increased |
| Intracellular Ca2+ release | Decreased | Increased |
| NO system | Increased | Decreased |
| eNOS expression | Increased | Decreased |
| ROCK activity (MYPT-1) | Decreased | Increased |
| RGS-2 expression | Increased | Decreased |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgarabotto, L.; Ravarotto, V.; Stefanelli, L.F.; Cacciapuoti, M.; Davis, P.A.; Nalesso, F.; Calò, L.A. Oxidants and Cardiorenal Vascular Remodeling—Insights from Rare Genetic Tubulopathies: Bartter’s and Gitelman’s Syndromes. Antioxidants 2023, 12, 811. https://doi.org/10.3390/antiox12040811
Sgarabotto L, Ravarotto V, Stefanelli LF, Cacciapuoti M, Davis PA, Nalesso F, Calò LA. Oxidants and Cardiorenal Vascular Remodeling—Insights from Rare Genetic Tubulopathies: Bartter’s and Gitelman’s Syndromes. Antioxidants. 2023; 12(4):811. https://doi.org/10.3390/antiox12040811
Chicago/Turabian StyleSgarabotto, Luca, Verdiana Ravarotto, Lucia Federica Stefanelli, Martina Cacciapuoti, Paul A. Davis, Federico Nalesso, and Lorenzo A. Calò. 2023. "Oxidants and Cardiorenal Vascular Remodeling—Insights from Rare Genetic Tubulopathies: Bartter’s and Gitelman’s Syndromes" Antioxidants 12, no. 4: 811. https://doi.org/10.3390/antiox12040811
APA StyleSgarabotto, L., Ravarotto, V., Stefanelli, L. F., Cacciapuoti, M., Davis, P. A., Nalesso, F., & Calò, L. A. (2023). Oxidants and Cardiorenal Vascular Remodeling—Insights from Rare Genetic Tubulopathies: Bartter’s and Gitelman’s Syndromes. Antioxidants, 12(4), 811. https://doi.org/10.3390/antiox12040811