
BET Protein Inhibitor JQ1 Modulates Mitochondrial Dysfunction and Oxidative Stress Induced by Chronic Kidney Disease

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Unilateral Ureteral Obstruction (UUO) Model
2.3. Cell Culture
2.4. Study of Proteins
2.5. Gene Expression
2.6. Mitochondrial Viability
2.7. Superoxide Anion Production from Mitochondria
2.8. Mitochondrial Membrane Potential
2.9. Statistical Analysis
3. Results
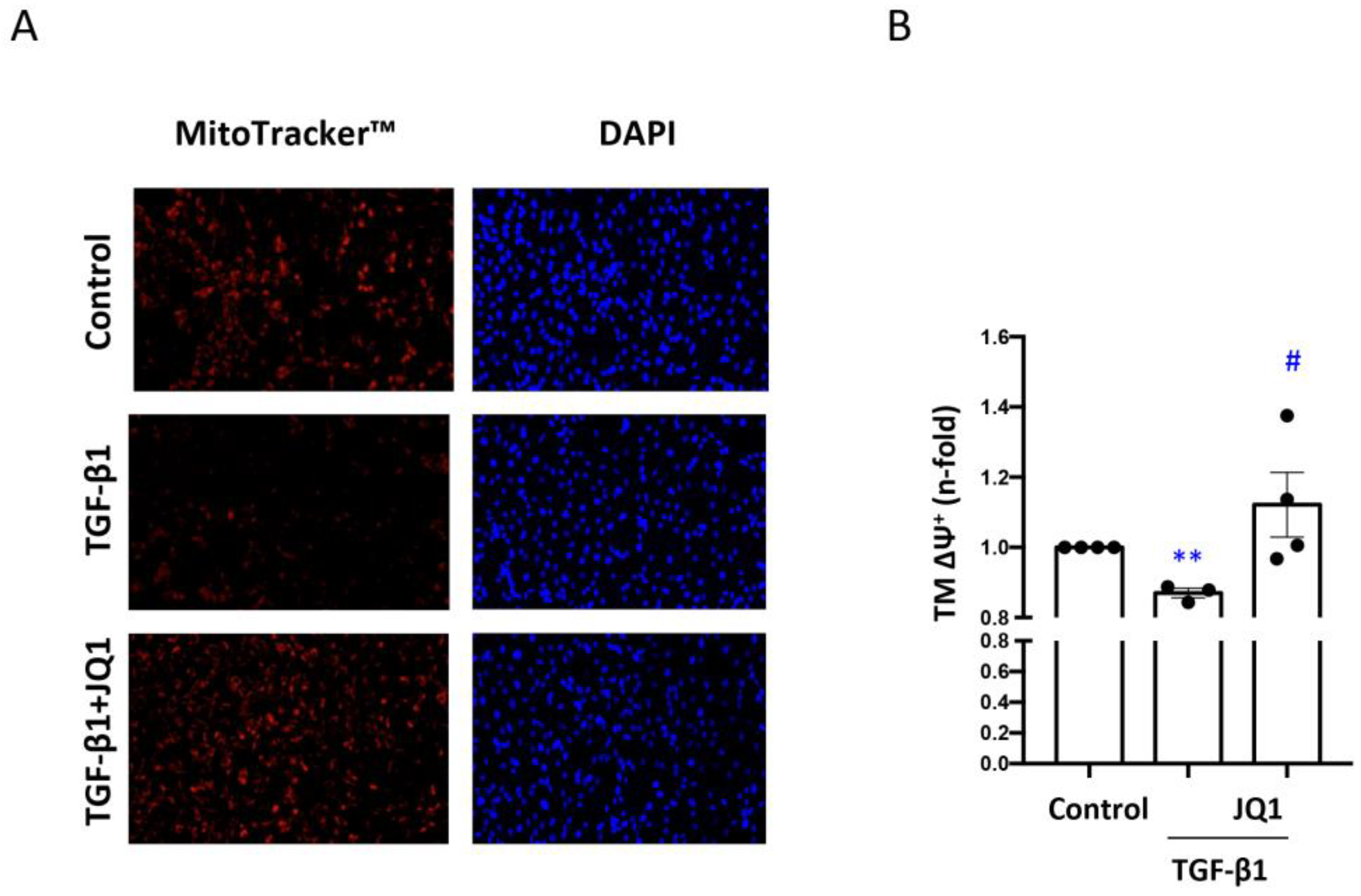
3.1. JQ1 Prevented the Loss of Mitochondrial Viability Induced by TGF-β1 in Human Tubular Cells
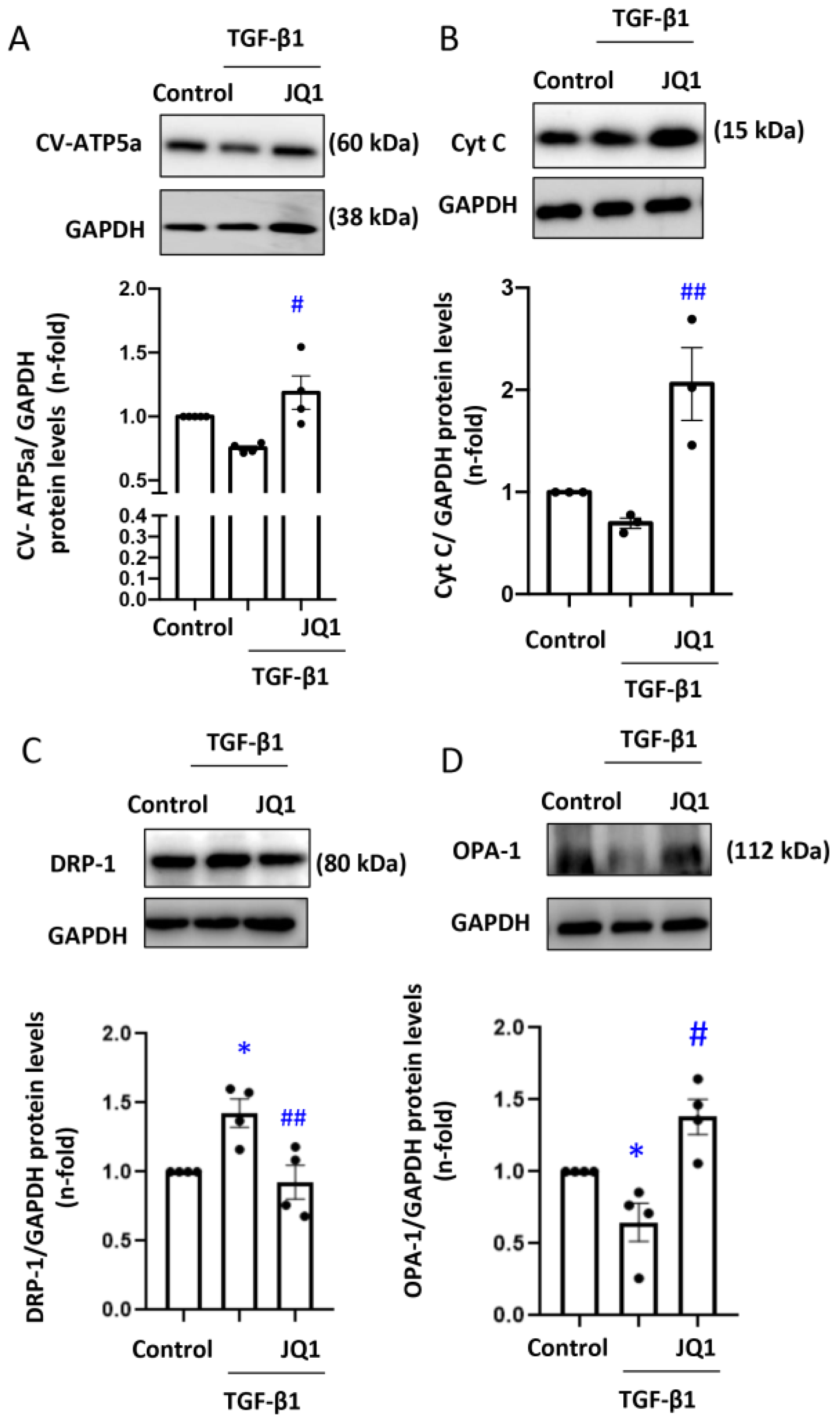
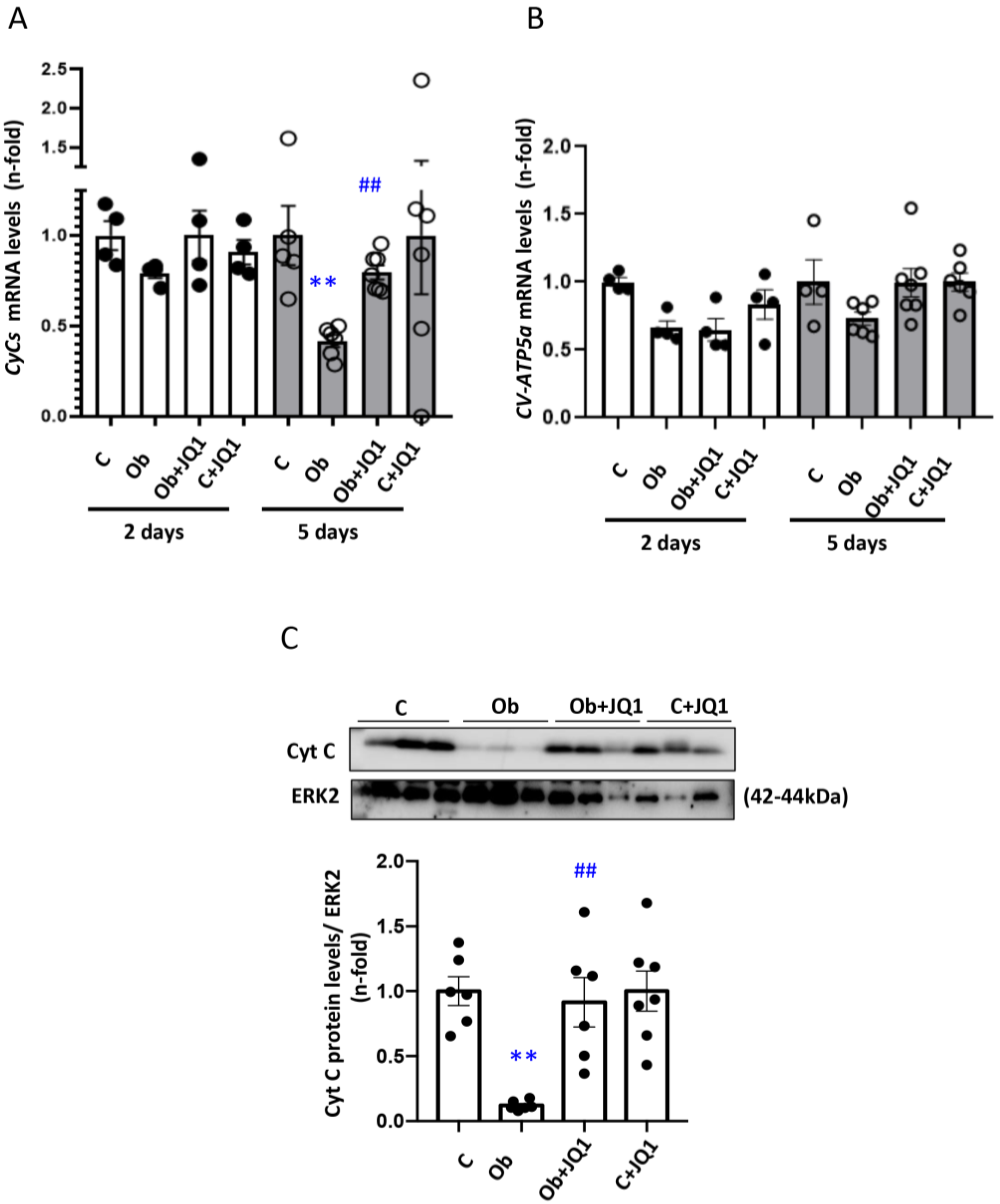
3.2. JQ1 Prevented the Loss of Mitochondrial Membrane Potential (∆Ψm) and Expression of Components of the Electron Transport Chain (ETC) Induced by TGF-β1 in Human Tubular Cells
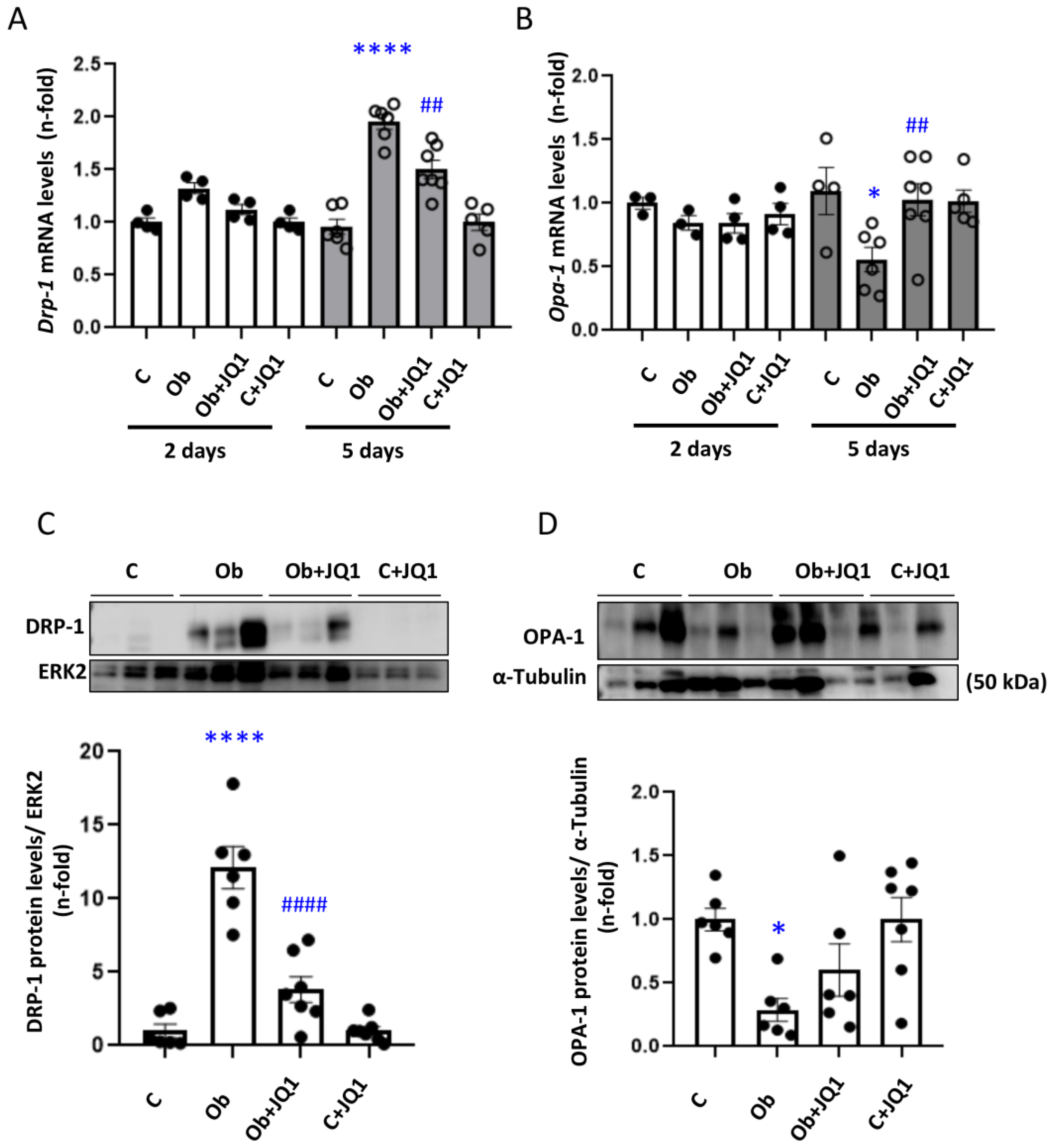
3.3. JQ1 Prevented Dysregulated Mitochondrial Dynamics Induced by TGF-β1 in Human Tubular Cells
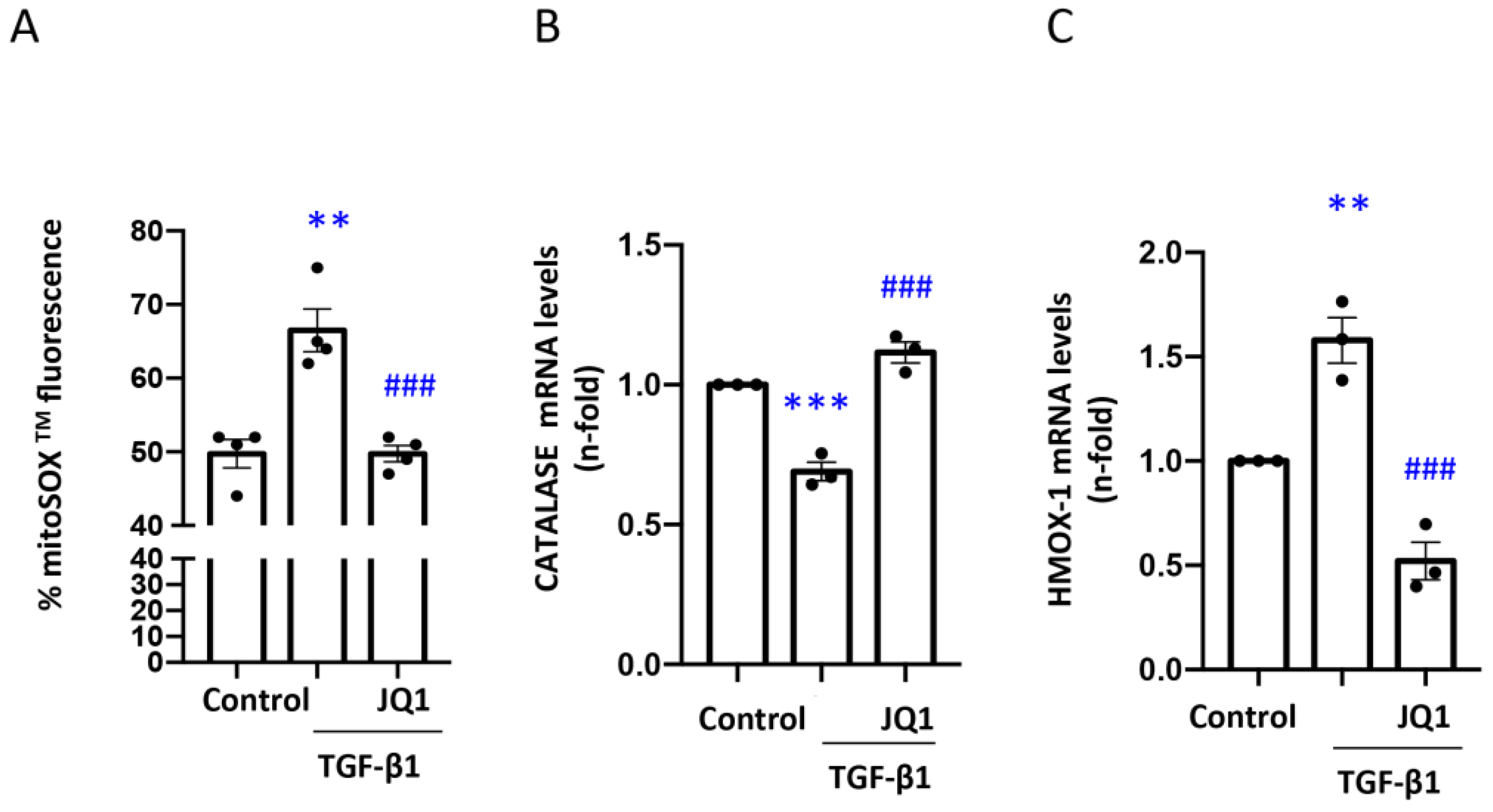
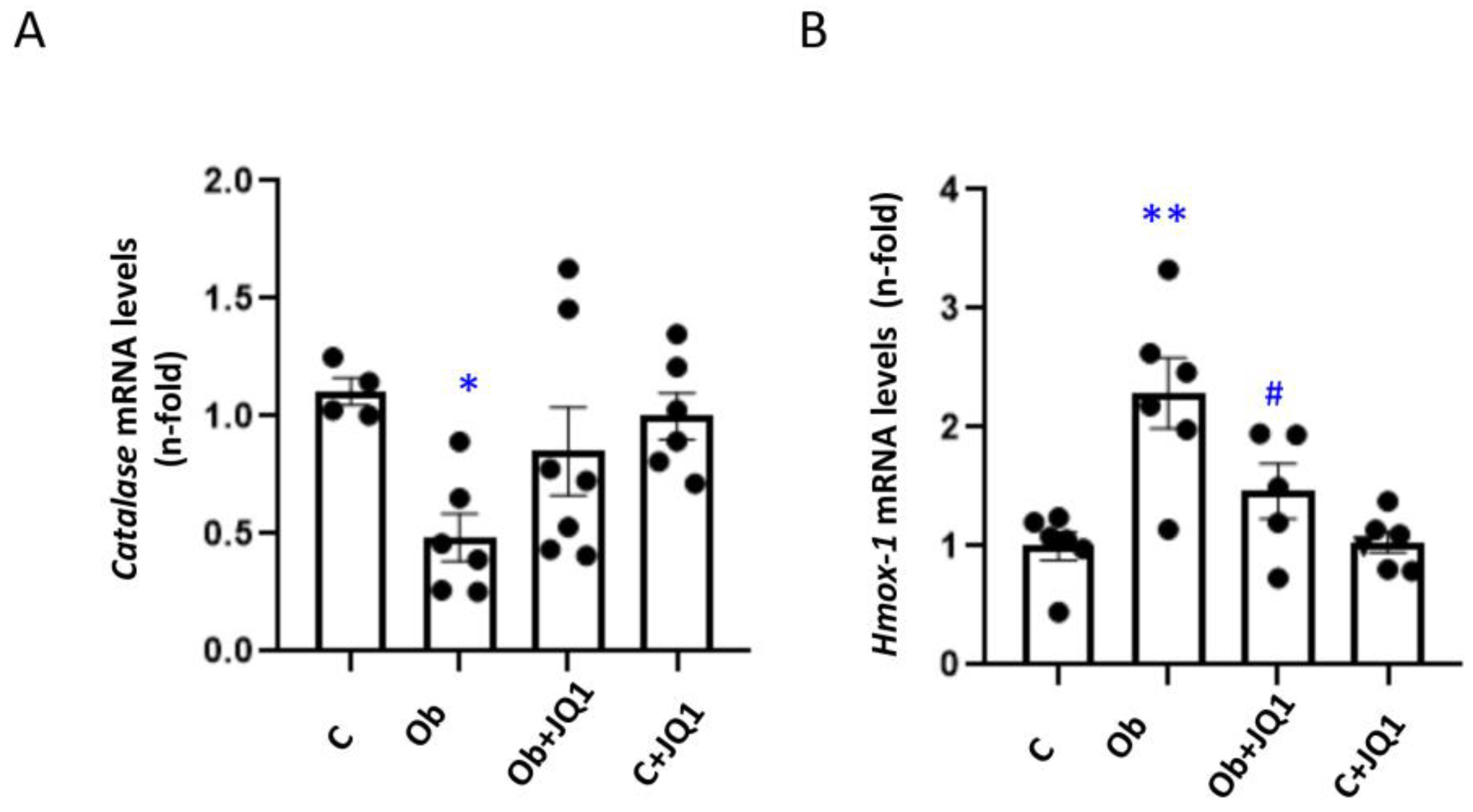
3.4. JQ1 Decreased Oxidative Stress, Including Mitochondrial Oxidative Stress, Induced by TGF-β1 in Human Tubular Cells
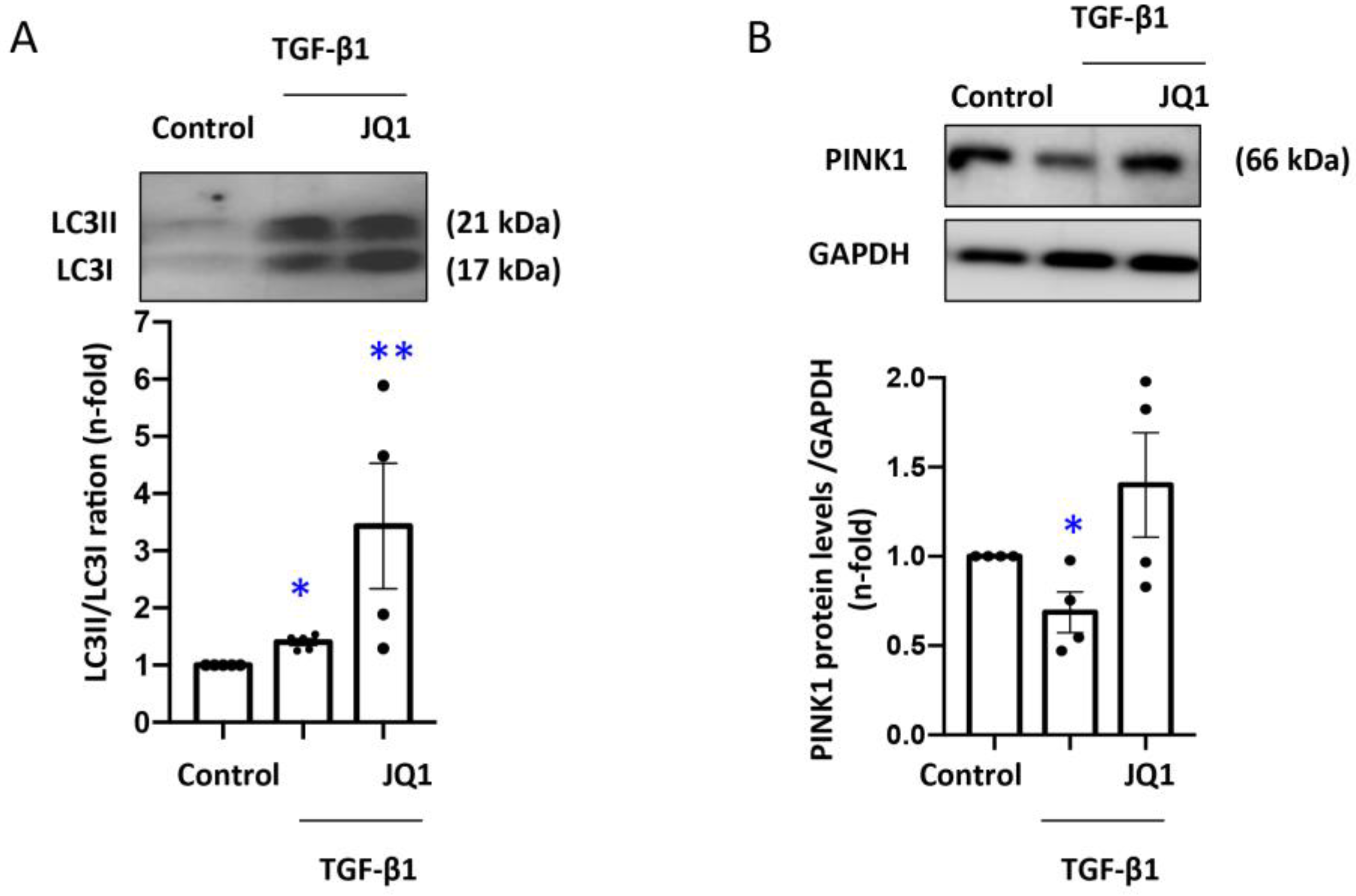
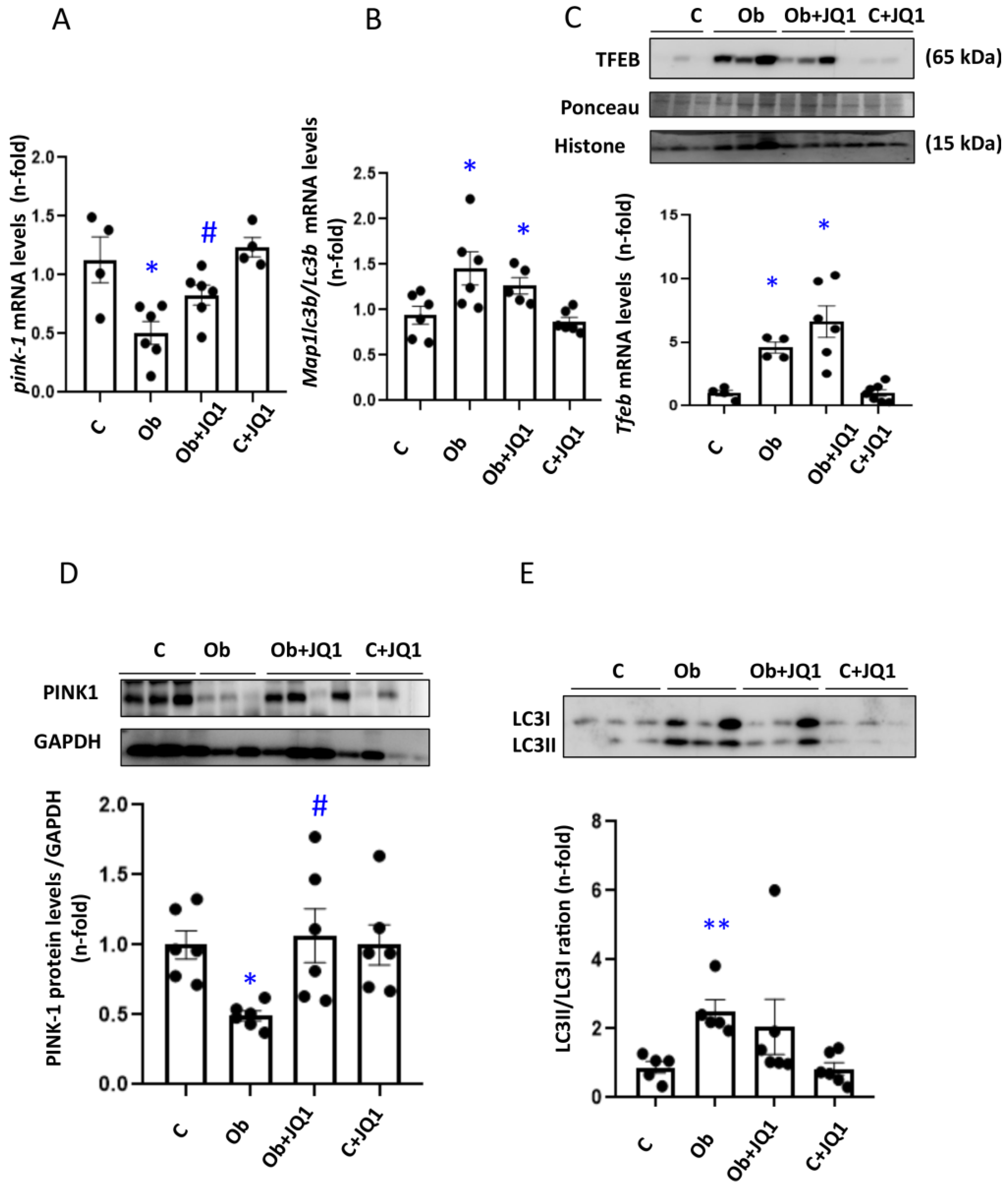
3.5. JQ1 Favored Mitophagy Induced by TGF-β1 in Human Tubular Cells
3.6. JQ1 Prevented the Kidney Downregulation of Electron Transport Chain (ETC) Components in Murine Unilateral Ureter Obstruction
3.7. JQ1 Prevented the Altered Kidney Mitochondrial Dynamics in Murine Unilateral Ureteral Obstruction
3.8. JQ1 Prevented the Kidney Dysregulation of Components of the Antioxidant Response in Murine Unilateral Ureteral Obstruction
3.9. JQ1 Maintains Kidney Mitophagy in Murine Unilateral Ureteral Obstruction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gosik, R.; Danel, K. The Rising Risk of Chronic Kidney Disease (CKD) and How it is Dealt with: A Review of Current and Potential Phosphate Binders (PB). Mini Rev. Med. Chem. 2021, 21, 3052–3061. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transpl. 2016, 31, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.Y.; Fokkens, M.R.; Reggiori, F.; Mari, M.; Verbeek, D.S. Parkinson’s disease-associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl. Neurodegener. 2021, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta. Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Garonzik-Wang, J.M.; Lonze, B.E.; Ruck, J.M.; Luo, X.; Massie, A.B.; Melancon, K.; Burdick, J.F.; Segev, D.L.; Sun, Z. Mitochondrial membrane potential and delayed graft function following kidney transplantation. Am. J. Transpl. 2019, 19, 585–590. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Tatsuta, T. Protein quality control in mitochondria. J. Biochem. 2009, 146, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lin, P.H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Mitochondrial bioenergetics, redox state, dynamics and turnover alterations in renal mass reduction models of chronic kidney diseases and their possible implications in the progression of this illness. Pharmacol. Res. 2018, 135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef]
- Legros, F.; Lombès, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.J.; McKay, G.J.; Maxwell, A.P.; McKnight, A.J. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics 2014, 9, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediat. Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. [Google Scholar] [CrossRef]
- Schwalm, M.P.; Knapp, S. BET bromodomain inhibitors. Curr. Opin. Chem. Biol. 2022, 68, 102148. [Google Scholar] [CrossRef]
- Suarez-Alvarez, B.; Morgado-Pascual, J.L.; Rayego-Mateos, S.; Rodriguez, R.M.; Rodrigues-Diez, R.; Cannata-Ortiz, P.; Sanz, A.B.; Egido, J.; Tharaux, P.L.; Ortiz, A.; et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. J. Am. Soc. Nephrol. 2017, 28, 504–519. [Google Scholar] [CrossRef]
- Schooling, C.M.; Zhao, J.V. How Might Bromodomain and Extra-Terminal (BET) Inhibitors Operate in Cardiovascular Disease? Am. J. Cardiovasc. Drugs 2019, 19, 107–111. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Suarez-Alvarez, B.; Marchant, V.; Basantes, P.; Tharaux, P.L.; Ortiz, A.; Lopez-Larrea, C.; Ruiz-Ortega, M.; Rayego-Mateos, S. Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis. Int. J. Mol. Sci. 2022, 24, 486. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Y.; Ren, D.; Xia, Y.; He, W.; Wu, Q.; Zhang, J.; Liu, M.; Du, Y.; Ren, C.; et al. JQ1, a bromodomain inhibitor, suppresses Th17 effectors by blocking p300-mediated acetylation of RORγt. Br. J. Pharmacol. 2020, 177, 2959–2973. [Google Scholar] [CrossRef]
- Mu, J.; Zhang, D.; Tian, Y.; Xie, Z.; Zou, M. hui BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell. Cardiol. 2020, 149, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Zhang, X.; Schiattarella, G.G.; Altamirano, F.; Ramos, T.A.R.; French, K.M.; Jiang, N.; Szweda, P.A.; Evers, B.M.; May, H.I.; et al. Epigenetic Reader BRD4 (Bromodomain-Containing Protein 4) Governs Nucleus-Encoded Mitochondrial Transcriptome to Regulate Cardiac Function. Circulation 2020, 142, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Meng, Y.; Wu, X.; Li, J.; Sun, Y. Bromodomain-containing protein 4 inhibitor JQ1 promotes melanoma cell apoptosis by regulating mitochondrial dynamics. Cancer Sci. 2021, 112, 4013–4025. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Leelahavanichkul, A.; Yan, Q.; Hu, X.; Eisner, C.; Huang, Y.; Chen, R.; Mizel, D.; Zhou, H.; Wright, E.C.; Kopp, J.B.; et al. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010, 78, 1136–1153. [Google Scholar] [CrossRef]
- Bosch-Panadero, E.; Mas, S.; Civantos, E.; Abaigar, P.; Camarero, V.; Ruiz-Priego, A.; Ortiz, A.; Egido, J.; González-Parra, E. Bisphenol A is an exogenous toxin that promotes mitochondrial injury and death in tubular cells. Environ. Toxicol. 2018, 33, 325–332. [Google Scholar] [CrossRef]
- Ramamoorthy, H.; Abraham, P.; Isaac, B. Mitochondrial dysfunction and electron transport chain complex defect in a rat model of tenofovir disoproxil fumarate nephrotoxicity. J. Biochem. Mol. Toxicol. 2014, 28, 246–255. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Chang, C.C.; Chiu, P.F.; Wu, C.L.; Kuo, C.L.; Huang, C.S.; Liu, C.S.; Huang, C.H. Urinary cell-free mitochondrial and nuclear deoxyribonucleic acid correlates with the prognosis of chronic kidney diseases. BMC Nephrol. 2019, 20, 391. [Google Scholar] [CrossRef]
- Fontecha-barriuso, M.; Martin-sanchez, D.; Martinez-moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-niño, M.D.; Ruiz-ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, N. Epigenetic Modification Drives Acute Kidney Injury-to-Chronic Kidney Disease Progression. Nephron 2021, 145, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Barrow, J.J.; Balsa, E.; Verdeguer, F.; Tavares, C.D.J.; Soustek, M.S.; Hollingsworth, L.R.; Jedrychowski, M.; Vogel, R.; Paulo, J.A.; Smeitink, J.; et al. Bromodomain Inhibitors Correct Bioenergetic Deficiency Caused by Mitochondrial Disease Complex I Mutations. Mol. Cell 2016, 64, 163–175. [Google Scholar] [CrossRef] [PubMed]
- García-Sánchez, O.; López-Hernández, F.J.; López-Novoa, J.M. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int. 2010, 77, 950–955. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Szeto, H.H. Pharmacologic Approaches to Improve Mitochondrial Function in AKI and CKD. J. Am. Soc. Nephrol. 2017, 28, 2856–2865. [Google Scholar] [CrossRef]
- O’Toole, J.F.; Patel, H.V.; Naples, C.J.; Fujioka, H.; Hoppel, C.L. Decreased cytochrome c mediates an age-related decline of oxidative phosphorylation in rat kidney mitochondria. Biochem. J. 2010, 427, 105–112. [Google Scholar] [CrossRef]
- Prieto-Carrasco, R.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; Rojas-Morales, P.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Tapia, E.; Pedraza-Chaverri, J. Progressive Reduction in Mitochondrial Mass Is Triggered by Alterations in Mitochondrial Biogenesis and Dynamics in Chronic Kidney Disease Induced by 5/6 Nephrectomy. Biology 2021, 10, 349. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.J.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Civenni, G.; Bosotti, R.; Timpanaro, A.; Vàzquez, R.; Merulla, J.; Pandit, S.; Rossi, S.; Albino, D.; Allegrini, S.; Mitra, A.; et al. Epigenetic Control of Mitochondrial Fission Enables Self-Renewal of Stem-like Tumor Cells in Human Prostate Cancer. Cell Metab. 2019, 30, 303–318.e6. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Go, G.; Yoon, S.; Lim, J.H.; Lee, G.; Lee, J.H.; Lee, S.H. Melatonin Treatment Improves Renal Fibrosis via miR-4516/SIAH3/PINK1 Axis. Cells 2021, 10, 1682. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Yin, X.M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Chiu, C.H.; Chen, J.B.; Chen, C.H.; Chang, H.W. Mitochondrial Fission Increases Apoptosis and Decreases Autophagy in Renal Proximal Tubular Epithelial Cells Treated with High Glucose. DNA Cell Biol. 2016, 35, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Zhao, F.; Tang, C.Y.; Luo, M.; Yang, S.K.; Cheng, W.; Li, X.W.; Duan, S. Bin Mitophagy Plays a Protective Role in Iodinated Contrast-Induced Acute Renal Tubular Epithelial Cells Injury. Cell. Physiol. Biochem. 2018, 46, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yi, H.; Shi, Y.; Cao, Q.; Shi, Y.; Cheng, D.; Braet, F.; Chen, X.M.; Pollock, C.A. KCa3.1 Mediates Dysregulation of Mitochondrial Quality Control in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2021, 9, 573814. [Google Scholar] [CrossRef]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genom. 2009, 10, 388. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.D.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011, 278, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Hussong, M.; Börno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sültmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The bromodomain protein BRD4 regulates the KEAP1/NRF2-dependent oxidative stress response. Cell Death Dis. 2014, 5, e1195. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Mercado, N.; Clarke, C.; Bhavsar, P.K.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Bromodomain and extraterminal proteins suppress NF-E2-related factor 2-mediated antioxidant gene expression. J. Immunol. 2014, 192, 4913–4920. [Google Scholar] [CrossRef]
- An, Q.-D.; Li, Y.-Y.; Zhang, H.-X.; Lu, J.; Yu, X.-D.; Lin, Q.; Zhang, Y.-G. Inhibition of bromodomain-containing protein 4 ameliorates oxidative stress-mediated apoptosis and cartilage matrix degeneration through activation of NF-E2-related factor 2-heme oxygenase-1 signaling in rat chondrocytes. J. Cell. Biochem. 2018, 119, 7719–7728. [Google Scholar] [CrossRef]
- Huang, J.; Yao, X.; Weng, G.; Qi, H.; Ye, X. Protective effect of curcumin against cyclosporine A-induced rat nephrotoxicity. Mol. Med. Rep. 2018, 17, 6038–6044. [Google Scholar] [CrossRef]
- Sun, L.; Liu, J.; Yuan, Y.; Zhang, I.; Dong, Z. Protective effect of the BET protein inhibitor JQ1 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2018, 315, F469–F478. [Google Scholar] [CrossRef]
- Zhou, B.; Mu, J.; Gong, Y.; Lu, C.; Zhao, Y.; He, T.; Qin, Z. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-β-mediated Nox4 expression. Redox Biol. 2017, 11, 390–402. [Google Scholar] [CrossRef]
- Ou, Y.C.; Li, J.R.; Wu, C.C.; Yu, T.M.; Chen, W.Y.; Liao, S.L.; Kuan, Y.H.; Chen, Y.F.; Chen, C.J. Cadmium induces the expression of Interleukin-6 through Heme Oxygenase-1 in HK-2 cells and Sprague-Dawley rats. Food Chem. Toxicol. 2022, 161, 112846. [Google Scholar] [CrossRef]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; De Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayego-Mateos, S.; Basantes, P.; Morgado-Pascual, J.L.; Brazal Prieto, B.; Suarez-Alvarez, B.; Ortiz, A.; Lopez-Larrea, C.; Ruiz-Ortega, M. BET Protein Inhibitor JQ1 Modulates Mitochondrial Dysfunction and Oxidative Stress Induced by Chronic Kidney Disease. Antioxidants 2023, 12, 1130. https://doi.org/10.3390/antiox12051130
Rayego-Mateos S, Basantes P, Morgado-Pascual JL, Brazal Prieto B, Suarez-Alvarez B, Ortiz A, Lopez-Larrea C, Ruiz-Ortega M. BET Protein Inhibitor JQ1 Modulates Mitochondrial Dysfunction and Oxidative Stress Induced by Chronic Kidney Disease. Antioxidants. 2023; 12(5):1130. https://doi.org/10.3390/antiox12051130
Chicago/Turabian StyleRayego-Mateos, Sandra, Pamela Basantes, José Luis Morgado-Pascual, Beatriz Brazal Prieto, Beatriz Suarez-Alvarez, Alberto Ortiz, Carlos Lopez-Larrea, and Marta Ruiz-Ortega. 2023. "BET Protein Inhibitor JQ1 Modulates Mitochondrial Dysfunction and Oxidative Stress Induced by Chronic Kidney Disease" Antioxidants 12, no. 5: 1130. https://doi.org/10.3390/antiox12051130
APA StyleRayego-Mateos, S., Basantes, P., Morgado-Pascual, J. L., Brazal Prieto, B., Suarez-Alvarez, B., Ortiz, A., Lopez-Larrea, C., & Ruiz-Ortega, M. (2023). BET Protein Inhibitor JQ1 Modulates Mitochondrial Dysfunction and Oxidative Stress Induced by Chronic Kidney Disease. Antioxidants, 12(5), 1130. https://doi.org/10.3390/antiox12051130