

NAD+ Metabolism and Immune Regulation: New Approaches to Inflammatory Bowel Disease Therapies

Abstract
:1. Introduction
2. Immunometabolism and Inflammatory Bowel Disease
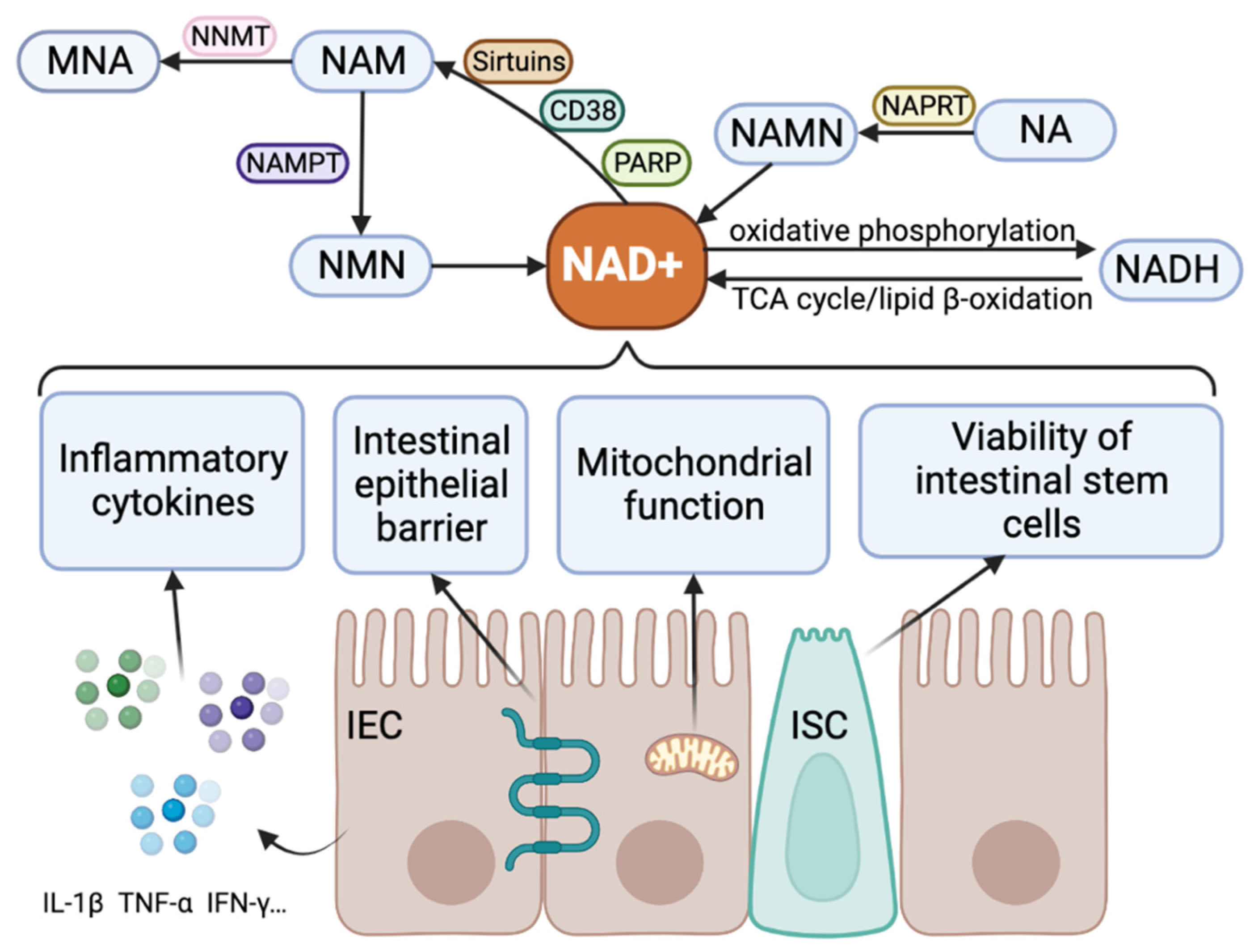
3. NAD+ Metabolism
4. The Role of NAD+ in Regulating IBD
4.1. NAD+ and IBD
4.2. NAD+ Metabolic Enzyme
4.2.1. Sirtuins
4.2.2. CD38
4.2.3. PARP
4.2.4. NAMPT and NAPRT
4.2.5. NNMT
4.3. IBD and NAD+ Regulation
4.3.1. NAD+ and Mitochondrial Dysfunction
4.3.2. Intestinal Epithelial Barrier
4.3.3. Intestinal Stem Cells
5. IBD NAD+ Regulation: Clinical Possibility
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Olén, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Colorectal Cancer in Ulcerative Colitis: A Scandinavian Population-Based Cohort Study. Lancet 2020, 395, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Wijnands, A.M.; de Jong, M.E.; Lutgens, M.W.M.D.; Hoentjen, F.; Elias, S.G.; Oldenburg, B. Dutch Initiative on Crohn and Colitis (ICC) Prognostic Factors for Advanced Colorectal Neoplasia in Inflammatory Bowel Disease: Systematic Review and Meta-Analysis. Gastroenterology 2021, 160, 1584–1598. [Google Scholar] [CrossRef] [PubMed]
- Zaiatz Bittencourt, V.; Jones, F.; Doherty, G.; Ryan, E.J. Targeting Immune Cell Metabolism in the Treatment of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1684–1693. [Google Scholar] [CrossRef]
- Caruso, R.; Lo, B.C.; Núñez, G. Host-Microbiota Interactions in Inflammatory Bowel Disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut Microbiota and IBD: Causation or Correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ Homeostasis in Health and Disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Conlon, N.; Ford, D. A Systems-Approach to NAD+ Restoration. Biochem. Pharmacol. 2022, 198, 114946. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in Cellular Functions and Cell Death: Regulation and Biological Consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef]
- Koch-Nolte, F.; Haag, F.; Guse, A.H.; Lund, F.; Ziegler, M. Emerging Roles of NAD+ and Its Metabolites in Cell Signaling. Sci. Signal. 2009, 2, mr1. [Google Scholar] [CrossRef]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD+-Dependent Activation of Sirt1 Corrects the Phenotype in a Mouse Model of Mitochondrial Disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wang, D.D.-H.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD Level Suppresses Inflammatory Activation of PBMCs in Heart Failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 Infection: The Perspectives on Immune Responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Deng, Y.; Pang, H.; Ma, T.; Ye, Q.; Chen, Q.; Chen, H.; Hu, Z.; Qin, C.-F.; Xu, Z. Treatment of SARS-CoV-2-Induced Pneumonia with NAD+ and NMN in Two Mouse Models. Cell Discov. 2022, 8, 38. [Google Scholar] [CrossRef]
- Altay, O.; Arif, M.; Li, X.; Yang, H.; Aydın, M.; Alkurt, G.; Kim, W.; Akyol, D.; Zhang, C.; Dinler-Doganay, G.; et al. Combined Metabolic Activators Accelerates Recovery in Mild-to-Moderate COVID-19. Adv. Sci. (Weinh) 2021, 8, 2101222. [Google Scholar] [CrossRef] [PubMed]
- Adriouch, S.; Hubert, S.; Pechberty, S.; Koch-Nolte, F.; Haag, F.; Seman, M. NAD+ Released during Inflammation Participates in T Cell Homeostasis by Inducing ART2-Mediated Death of Naive T Cells in Vivo. J. Immunol. 2007, 179, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.H.; Tucker, S.A.; Quevedo, S.F.; Inal, A.; Korzenik, J.R.; Haigis, M.C. Metabolic Analyses Reveal Dysregulated NAD+ Metabolism and Altered Mitochondrial State in Ulcerative Colitis. PLoS ONE 2022, 17, e0273080. [Google Scholar] [CrossRef]
- Gerner, R.R.; Klepsch, V.; Macheiner, S.; Arnhard, K.; Adolph, T.E.; Grander, C.; Wieser, V.; Pfister, A.; Moser, P.; Hermann-Kleiter, N.; et al. NAD Metabolism Fuels Human and Mouse Intestinal Inflammation. Gut 2018, 67, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Navarro, M.N.; Gómez de las Heras, M.M.; Mittelbrunn, M. Nicotinamide Adenine Dinucleotide Metabolism in the Immune Response, Autoimmunity and Inflammageing. Br. J. Pharmacol. 2022, 179, 1839–1856. [Google Scholar] [CrossRef]
- Smids, C.; Horjus Talabur Horje, C.S.; Drylewicz, J.; Roosenboom, B.; Groenen, M.J.M.; van Koolwijk, E.; van Lochem, E.G.; Wahab, P.J. Intestinal T Cell Profiling in Inflammatory Bowel Disease: Linking T Cell Subsets to Disease Activity and Disease Course. J. Crohn’s Colitis 2018, 12, 465–475. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gómez de las Heras, M.M.; Gabandé-Rodríguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A Key Player in the Inflammatory Response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diskin, C.; Ryan, T.A.J.; O’Neill, L.A.J. Modification of Proteins by Metabolites in Immunity. Immunity 2021, 54, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria Are the Powerhouses of Immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways In Immune Cell Activation And Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Assmann, N.; Finlay, D.K. Metabolic Regulation of Immune Responses: Therapeutic Opportunities. J. Clin. Investig. 2016, 126, 2031–2039. [Google Scholar] [CrossRef]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting Metabolism to Regulate Immune Responses in Autoimmunity and Cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum Zonulin in Patients with Inflammatory Bowel Disease: A Pilot Study. Minerva Med. 2019, 110, 95–100. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current State of the Art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host-Microbiota Maladaptation in Colorectal Cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef]
- Meserve, J.; Facciorusso, A.; Holmer, A.K.; Annese, V.; Sandborn, W.J.; Singh, S. Safety and Tolerability of Immune Checkpoint Inhibitors in Patients with Pre-Existing Inflammatory Bowel Diseases: A Systematic Review and Meta-Analysis. Aliment. Pharmacol. Ther. 2021, 53, 374–382. [Google Scholar]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New Insights into the Interplay between Autophagy, Gut Microbiota and Inflammatory Responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Michaudel, C.; Sokol, H. The Gut Microbiota at the Service of Immunometabolism. Cell Metab. 2020, 32, 514–523. [Google Scholar] [CrossRef]
- Harden, A.; Young, W.J.; Martin, C.J. The Alcoholic Ferment of Yeast-Juice. Proc. R. Soc. London. Ser. B Contain. Pap. A Biol. Character 1997, 77, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S. Emerging Therapeutic Roles for NAD(+) Metabolism in Mitochondrial and Age-Related Disorders. Clin. Transl. Med. 2016, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, H.R.; Raghava, G.P.S. Identification of NAD Interacting Residues in Proteins. BMC Bioinform. 2010, 11, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal. Transduct. Target Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. NAD+ as a Signaling Molecule Modulating Metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omran, H.M.; Almaliki, M.S. Influence of NAD+ as an Ageing-Related Immunomodulator on COVID 19 Infection: A Hypothesis. J. Infect. Public. Health 2020, 13, 1196–1201. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in Aging, Metabolism, and Neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Burgos, E.S. NAMPT in Regulated NAD Biosynthesis and Its Pivotal Role in Human Metabolism. Curr. Med. Chem. 2011, 18, 1947–1961. [Google Scholar] [CrossRef]
- Sano, A.; Endo, N.; Takitani, S. Fluorometric Assay of Rat Tissue N-Methyltransferases with Nicotinamide and Four Isomeric Methylnicotinamides. Chem. Pharm. Bull. (Tokyo) 1992, 40, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The in Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljungberg, M.C.; Ali, Y.O.; Zhu, J.; Wu, C.-S.; Oka, K.; Zhai, R.G.; Lu, H.-C. CREB-Activity and Nmnat2 Transcription Are down-Regulated Prior to Neurodegeneration, While NMNAT2 over-Expression Is Neuroprotective, in a Mouse Model of Human Tauopathy. Hum. Mol. Genet. 2012, 21, 251–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trammell, S.A.J.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Kim, E.Y.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-Enamorado, I.; Di Francesco, A.; et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018, 27, 667–676.e4. [Google Scholar] [CrossRef] [Green Version]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.; et al. Nicotinamide N-Methyltransferase Knockdown Protects against Diet-Induced Obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet Induced Obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Kannt, A.; Rajagopal, S.; Kadnur, S.V.; Suresh, J.; Bhamidipati, R.K.; Swaminathan, S.; Hallur, M.S.; Kristam, R.; Elvert, R.; Czech, J.; et al. A Small Molecule Inhibitor of Nicotinamide N-Methyltransferase for the Treatment of Metabolic Disorders. Sci. Rep. 2018, 8, 3660. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-P.; Oka, S.; Shao, D.; Hariharan, N.; Sadoshima, J. Nicotinamide Phosphoribosyltransferase Regulates Cell Survival through NAD+ Synthesis in Cardiac Myocytes. Circ. Res. 2009, 105, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mázala, D.A.G.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ Repletion Improves Muscle Function in Muscular Dystrophy and Counters Global PARylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De Novo NAD+ Biosynthetic Impairment in Acute Kidney Injury in Humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD Metabolome--a Key Determinant of Cancer Cell Biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Buonvicino, D.; Mazzola, F.; Zamporlini, F.; Resta, F.; Ranieri, G.; Camaioni, E.; Muzzi, M.; Zecchi, R.; Pieraccini, G.; Dölle, C.; et al. Identification of the Nicotinamide Salvage Pathway as a New Toxification Route for Antimetabolites. Cell. Chem. Biol. 2018, 25, 471–482.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vachharajani, V.; Liu, T.; McCall, C.E. Epigenetic Coordination of Acute Systemic Inflammation: Potential Therapeutic Targets. Expert Rev. Clin. Immunol. 2014, 10, 1141–1150. [Google Scholar] [CrossRef]
- Schilling, E.; Wehrhahn, J.; Klein, C.; Raulien, N.; Ceglarek, U.; Hauschildt, S. Inhibition of Nicotinamide Phosphoribosyltransferase Modifies LPS-Induced Inflammatory Responses of Human Monocytes. Innat. Immun. 2012, 18, 518–530. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Shan, G.; Sun, Z.; Zhang, F.; Xu, C.; Lou, X.; Li, S.; Du, H.; Chen, H.; Xu, G. Quantitative Proteomic Analysis Reveals the Deregulation of Nicotinamide Adenine Dinucleotide Metabolism and CD38 in Inflammatory Bowel Disease. Biomed. Res. Int. 2019, 2019, 3950628. [Google Scholar] [CrossRef] [Green Version]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Han, X.; Uchiyama, T.; Sappington, P.L.; Yaguchi, A.; Yang, R.; Fink, M.P.; Delude, R.L. NAD+ Ameliorates Inflammation-Induced Epithelial Barrier Dysfunction in Cultured Enterocytes and Mouse Ileal Mucosa. J. Pharmacol. Exp. Ther. 2003, 307, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Clemente, N.; Zito, A.; Bracci, C.; Colombo, F.S.; Sangaletti, S.; Jachetti, E.; Ribaldone, D.G.; Caviglia, G.P.; Pastorelli, L.; et al. Neutralization of Extracellular NAMPT (Nicotinamide Phosphoribosyltransferase) Ameliorates Experimental Murine Colitis. J. Mol. Med. (Berl.) 2020, 98, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Guarente, L. MTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, R.; Saito, Y.; Nogami, K.; Kajiyama, Y.; Suzuki, Y.; Kawase, Y.; Nakaoka, T.; Muramatsu, T.; Kimura, M.; Saito, H. Epigenetic Silencing of Lgr5 Induces Senescence of Intestinal Epithelial Organoids during the Process of Aging. NPJ Aging Mech. Dis. 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Wang, K.; Qiao, S.; Yang, L.; Xin, Y.; Dai, Y.; Wei, Z. Norisoboldine, a Natural AhR Agonist, Promotes Treg Differentiation and Attenuates Colitis via Targeting Glycolysis and Subsequent NAD+/SIRT1/SUV39H1/H3K9me3 Signaling Pathway. Cell Death Dis. 2018, 9, 258. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased Nuclear NAD Biosynthesis and SIRT1 Activation Prevent Axonal Degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Fulco, M.; Schiltz, R.L.; Iezzi, S.; King, M.T.; Zhao, P.; Kashiwaya, Y.; Hoffman, E.; Veech, R.L.; Sartorelli, V. Sir2 Regulates Skeletal Muscle Differentiation as a Potential Sensor of the Redox State. Mol. Cell. 2003, 12, 51–62. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of P53 by Sir2alpha Promotes Cell Survival under Stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A Role for the NAD-Dependent Deacetylase Sirt1 in the Regulation of Autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental Defects and P53 Hyperacetylation in Sir2 Homolog (SIRT1)-Deficient Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [Green Version]
- Li, X. SIRT1 and Energy Metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Arnal, L.; Katada, S.; Orozco-Solis, R.; Sassone-Corsi, P. NAD+-SIRT1 Control of H3K4 Trimethylation through Circadian Deacetylation of MLL1. Nat. Struct. Mol. Biol. 2015, 22, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellet, M.M.; Nakahata, Y.; Boudjelal, M.; Watts, E.; Mossakowska, D.E.; Edwards, K.A.; Cervantes, M.; Astarita, G.; Loh, C.; Ellis, J.L.; et al. Pharmacological Modulation of Circadian Rhythms by Synthetic Activators of the Deacetylase SIRT1. Proc. Natl. Acad. Sci. USA 2013, 110, 3333–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 Mediates Central Circadian Control in the SCN by a Mechanism That Decays with Aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-ΚB-Dependent Transcription and Cell Survival by the SIRT1 Deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 Functionally Interacts with the Metabolic Regulator and Transcriptional Coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Shin, J.; Bae, J.; Han, D.; Park, S.-R.; Shin, J.; Lee, S.K.; Park, H.-W. SIRT1 Alleviates LPS-Induced IL-1β Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells 2020, 9, 728. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. NAD+-Dependent SIRT1 Deacetylase Participates in Epigenetic Reprogramming during Endotoxin Tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, T.T.; Li, X. Surprising Sirtuin Crosstalk in the Heart. Aging (Albany N. Y.) 2010, 2, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 Activation as a Novel Mechanism Underlying the Prevention of Alzheimer Disease Amyloid Neuropathology by Calorie Restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.F.; Vachharajani, V.; Millet, P.; Bharadwaj, M.S.; Molina, A.J.; McCall, C.E. Sequential Actions of SIRT1-RELB-SIRT3 Coordinate Nuclear-Mitochondrial Communication during Immunometabolic Adaptation to Acute Inflammation and Sepsis. J. Biol. Chem. 2015, 290, 396–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Sasso, G.; Menzies, K.J.; Mottis, A.; Piersigilli, A.; Perino, A.; Yamamoto, H.; Schoonjans, K.; Auwerx, J. SIRT2 Deficiency Modulates Macrophage Polarization and Susceptibility to Experimental Colitis. PLoS ONE 2014, 9, e103573. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.-L.; Zhou, M.; Kang, C.; Lang, H.-D.; Chen, M.-T.; Hui, S.-C.; Wang, B.; Mi, M.-T. Crosstalk between Gut Microbiota and Sirtuin-3 in Colonic Inflammation and Tumorigenesis. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1β Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 2017, 19, 2331–2344. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Bu, H.-F.; Geng, H.; De Plaen, I.G.; Gao, C.; Wang, P.; Wang, X.; Kurowski, J.A.; Yang, H.; Qian, J.; et al. Sirtuin-6 Preserves R-Spondin-1 Expression and Increases Resistance of Intestinal Epithelium to Injury in Mice. Mol. Med. 2017, 23, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Caruso, R.; Marafini, I.; Franzè, E.; Stolfi, C.; Zorzi, F.; Monteleone, I.; Caprioli, F.; Colantoni, A.; Sarra, M.; Sedda, S.; et al. Defective Expression of SIRT1 Contributes to Sustain Inflammatory Pathways in the Gut. Mucosal. Immunol. 2014, 7, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Wellman, A.S.; Metukuri, M.R.; Kazgan, N.; Xu, X.; Xu, Q.; Ren, N.S.X.; Czopik, A.; Shanahan, M.T.; Kang, A.; Chen, W.; et al. Intestinal Epithelial Sirtuin 1 Regulates Intestinal Inflammation during Aging in Mice by Altering the Intestinal Microbiota. Gastroenterology 2017, 153, 772–786. [Google Scholar] [CrossRef]
- Lo Sasso, G.; Ryu, D.; Mouchiroud, L.; Fernando, S.C.; Anderson, C.L.; Katsyuba, E.; Piersigilli, A.; Hottiger, M.O.; Schoonjans, K.; Auwerx, J. Loss of Sirt1 Function Improves Intestinal Anti-Bacterial Defense and Protects from Colitis-Induced Colorectal Cancer. PLoS ONE 2014, 9, e102495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizaki, T.; Milne, J.C.; Imamura, T.; Schenk, S.; Sonoda, N.; Babendure, J.L.; Lu, J.-C.; Smith, J.J.; Jirousek, M.R.; Olefsky, J.M. SIRT1 Exerts Anti-Inflammatory Effects and Improves Insulin Sensitivity in Adipocytes. Mol. Cell. Biol. 2009, 29, 1363–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrosa, M.; Yañéz-Gascón, M.J.; Selma, M.V.; González-Sarrías, A.; Toti, S.; Cerón, J.J.; Tomás-Barberán, F.; Dolara, P.; Espín, J.C. Effect of a Low Dose of Dietary Resveratrol on Colon Microbiota, Inflammation and Tissue Damage in a DSS-Induced Colitis Rat Model. J. Agric. Food Chem. 2009, 57, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Montes, C.; Santos-Argumedo, L. CD38 Is Expressed Selectively during the Activation of a Subset of Mature T Cells with Reduced Proliferation but Improved Potential to Produce Cytokines. J. Leukoc. Biol. 2005, 77, 513–521. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-Faceted Ecto-Enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zocchi, E.; Franco, L.; Guida, L.; Benatti, U.; Bargellesi, A.; Malavasi, F.; Lee, H.C.; De Flora, A. A Single Protein Immunologically Identified as CD38 Displays NAD+ Glycohydrolase, ADP-Ribosyl Cyclase and Cyclic ADP-Ribose Hydrolase Activities at the Outer Surface of Human Erythrocytes. Biochem. Biophys. Res. Commun. 1993, 196, 1459–1465. [Google Scholar] [CrossRef]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid Apigenin Is an Inhibitor of the NAD+ Ase CD38: Implications for Cellular NAD+ Metabolism, Protein Acetylation, and Treatment of Metabolic Syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Kellenberger, E.; Kuhn, I.; Schuber, F.; Muller-Steffner, H. Flavonoids as Inhibitors of Human CD38. Bioorg. Med. Chem. Lett. 2011, 21, 3939–3942. [Google Scholar] [CrossRef]
- Glaría, E.; Valledor, A.F. Roles of CD38 in the Immune Response to Infection. Cells 2020, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.-L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-Ribose Gating of the Calcium-Permeable LTRPC2 Channel Revealed by Nudix Motif Homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef]
- Young, G.S.; Choleris, E.; Lund, F.E.; Kirkland, J.B. Decreased CADPR and Increased NAD+ in the Cd38−/− Mouse. Biochem. Biophys. Res. Commun. 2006, 346, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Schumacher, V.; Lischke, T.; Lücke, K.; Meyer-Schwesinger, C.; Velden, J.; Koch-Nolte, F.; Mittrücker, H.-W. CD38 Is Expressed on Inflammatory Cells of the Intestine and Promotes Intestinal Inflammation. PLoS ONE 2015, 10, e0126007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosse, M.E.; Menckeberg, C.L.; de Ruiter, L.F.; Raatgeep, H.R.C.; van Berkel, L.A.; Simons-Oosterhuis, Y.; Tindemans, I.; Muskens, A.F.M.; Hendriks, R.W.; Hoogenboezem, R.M.; et al. Frequencies of Circulating Regulatory TIGIT+CD38+ Effector T Cells Correlate with the Course of Inflammatory Bowel Disease. Mucosal. Immunol. 2019, 12, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Moschen, A.R.; Gerner, R.R.; Tilg, H. Pre-B Cell Colony Enhancing Factor/NAMPT/Visfatin in Inflammation and Obesity-Related Disorders. Curr. Pharm. Des. 2010, 16, 1913–1920. [Google Scholar] [CrossRef]
- Carbone, F.; Liberale, L.; Bonaventura, A.; Vecchiè, A.; Casula, M.; Cea, M.; Monacelli, F.; Caffa, I.; Bruzzone, S.; Montecucco, F.; et al. Regulation and Function of Extracellular Nicotinamide Phosphoribosyltransferase/Visfatin. Compr. Physiol. 2017, 7, 603–621. [Google Scholar] [CrossRef]
- Jia, S.H.; Li, Y.; Parodo, J.; Kapus, A.; Fan, L.; Rotstein, O.D.; Marshall, J.C. Pre–B Cell Colony–Enhancing Factor Inhibits Neutrophil Apoptosis in Experimental Inflammation and Clinical Sepsis. J. Clin. Investig. 2004, 113, 1318–1327. [Google Scholar] [CrossRef]
- Meier, F.M.P.; Frommer, K.W.; Peters, M.A.; Brentano, F.; Lefèvre, S.; Schröder, D.; Kyburz, D.; Steinmeyer, J.; Rehart, S.; Gay, S.; et al. Visfatin/Pre-B-Cell Colony-Enhancing Factor (PBEF), a Proinflammatory and Cell Motility-Changing Factor in Rheumatoid Arthritis. J. Biol. Chem. 2012, 287, 28378–28385. [Google Scholar] [CrossRef] [Green Version]
- El-Mesallamy, H.O.; Kassem, D.H.; El-Demerdash, E.; Amin, A.I. Vaspin and Visfatin/Nampt Are Interesting Interrelated Adipokines Playing a Role in the Pathogenesis of Type 2 Diabetes Mellitus. Metabolism 2011, 60, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Qi, L.; Li, X.; Wang, J.; Yu, J.; Zhou, B.; Guo, C.; Chen, J.; Zheng, S. Targeting the NAD+ Salvage Pathway Suppresses APC Mutation-Driven Colorectal Cancer Growth and Wnt/β-Catenin Signaling via Increasing Axin Level. Cell Commun. Signal. 2020, 18, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, A.M.; Castoldi, A.; Sanin, D.E.; Flachsmann, L.J.; Field, C.S.; Puleston, D.J.; Kyle, R.L.; Patterson, A.E.; Hässler, F.; Buescher, J.M.; et al. Inflammatory Macrophage Dependence on NAD+ Salvage Is a Consequence of Reactive Oxygen Species-Mediated DNA Damage. Nat. Immunol. 2019, 20, 420–432. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and Gene Regulation: Progress and Puzzles. Mol. Aspects. Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.M.; Lewis, P.J.; Davies, M.I.; Skidmore, C.J.; Shall, S. The Effect of Gamma Radiation and Neocarzinostatin of NAD and ATP Levels in Mouse Leukaemia Cells. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1978, 543, 576–582. [Google Scholar] [CrossRef]
- Skidmore, C.J.; Davies, M.I.; Goodwin, P.M.; Halldorsson, H.; Lewis, P.J.; Shall, S.; Zia’ee, A.A. The Involvement of Poly(ADP-Ribose) Polymerase in the Degradation of NAD Caused by Gamma-Radiation and N-Methyl-N-Nitrosourea. Eur. J. Biochem. 1979, 101, 135–142. [Google Scholar] [CrossRef]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.-S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting Poly ADP-Ribosylation Increases Fatty Acid Oxidation and Protects against Fatty Liver Disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Varga, Z.V.; Gariani, K.; Ryu, D.; Cao, Z.; Holovac, E.; Park, O.; Zhou, Z.; et al. PARP Inhibition Protects against Alcoholic and Non-Alcoholic Steatohepatitis. J. Hepatol. 2017, 66, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Mangerich, A.; Bürkle, A. Pleiotropic Cellular Functions of PARP1 in Longevity and Aging: Genome Maintenance Meets Inflammation. Oxid. Med. Cell. Longev. 2012, 2012, 321653. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, P.; Szabó, C. Poly(ADP-Ribose) Polymerase and the Therapeutic Effects of Its Inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Larmonier, C.B.; Shehab, K.W.; Laubitz, D.; Jamwal, D.R.; Ghishan, F.K.; Kiela, P.R. Transcriptional Reprogramming and Resistance to Colonic Mucosal Injury in Poly(ADP-Ribose) Polymerase 1 (PARP1)-Deficient Mice. J. Biol. Chem. 2016, 291, 8918–8930. [Google Scholar] [CrossRef] [Green Version]
- Popoff, I.; Jijon, H.; Monia, B.; Tavernini, M.; Ma, M.; McKay, R.; Madsen, K. Antisense Oligonucleotides to Poly(ADP-Ribose) Polymerase-2 Ameliorate Colitis in Interleukin-10-Deficient Mice. J. Pharmacol. Exp. Ther. 2002, 303, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Muñoz-Galvan, S.; Carnero, A. NAMPT Is a Potent Oncogene in Colon Cancer Progression That Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wnorowski, A.; Wnorowska, S.; Kurzepa, J.; Parada-Turska, J. Alterations in Kynurenine and NAD+ Salvage Pathways during the Successful Treatment of Inflammatory Bowel Disease Suggest HCAR3 and NNMT as Potential Drug Targets. Int. J. Mol. Sci. 2021, 22, 13497. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, J.; Wu, W.; Xie, S.; Yu, H.; Li, G.; Zhu, T.; Li, F.; Lu, J.; Wang, G.Y.; et al. Nicotinamide N-Methyltransferase Enhances Chemoresistance in Breast Cancer through SIRT1 Protein Stabilization. Breast Cancer Res. 2019, 21, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Yang, D.; Wang, W.; Zhang, L.; Liu, H.; Ma, S.; Guo, W.; Yao, M.; Zhang, K.; Li, W.; et al. Nicotinamide N-Methyltransferase Decreases 5-Fluorouracil Sensitivity in Human Esophageal Squamous Cell Carcinoma through Metabolic Reprogramming and Promoting the Warburg Effect. Mol. Carcinog. 2020, 59, 940–954. [Google Scholar] [CrossRef]
- Li, G.; Kong, B.; Tong, Q.; Li, Y.; Chen, L.; Zeng, J.; Yu, H.; Xie, X.; Zhang, J. Vanillin Downregulates NNMT and Attenuates NNMT-related Resistance to 5-fluorouracil via ROS-induced Cell Apoptosis in Colorectal Cancer Cells. Oncol. Rep. 2021, 45, 110. [Google Scholar] [CrossRef]
- Campagna, R.; Salvolini, E.; Pompei, V.; Pozzi, V.; Salvucci, A.; Molinelli, E.; Brisigotti, V.; Sartini, D.; Campanati, A.; Offidani, A.; et al. Nicotinamide N-Methyltransferase Gene Silencing Enhances Chemosensitivity of Melanoma Cell Lines. Pigment. Cell Melanoma. Res. 2021, 34, 1039–1048. [Google Scholar] [CrossRef]
- Hong, S.; Zhai, B.; Pissios, P. Nicotinamide N-Methyltransferase Interacts with Enzymes of the Methionine Cycle and Regulates Methyl Donor Metabolism. Biochemistry 2018, 57, 5775–5779. [Google Scholar] [CrossRef]
- Takahashi, R.; Kanda, T.; Komatsu, M.; Itoh, T.; Minakuchi, H.; Urai, H.; Kuroita, T.; Shigaki, S.; Tsukamoto, T.; Higuchi, N.; et al. The Significance of NAD + Metabolites and Nicotinamide N-Methyltransferase in Chronic Kidney Disease. Sci. Rep. 2022, 12, 6398. [Google Scholar] [CrossRef]
- Kanakkanthara, A.; Kurmi, K.; Ekstrom, T.L.; Hou, X.; Purfeerst, E.R.; Heinzen, E.P.; Correia, C.; Huntoon, C.J.; O’Brien, D.; Wahner Hendrickson, A.E.; et al. BRCA1 Deficiency Upregulates NNMT, Which Reprograms Metabolism and Sensitizes Ovarian Cancer Cells to Mitochondrial Metabolic Targeting Agents. Cancer Res. 2019, 79, 5920–5929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.C.; Mofarrahi, M.; Vassilakopoulos, T.; Maltais, F.; Sigala, I.; Debigare, R.; Bellenis, I.; Hussain, S.N.A. Expression and Functional Significance of Nicotinamide N-Methyl Transferase in Skeletal Muscles of Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 181, 797–805. [Google Scholar] [CrossRef]
- Savarimuthu Francis, S.M.; Larsen, J.E.; Pavey, S.J.; Duhig, E.E.; Clarke, B.E.; Bowman, R.V.; Hayward, N.K.; Fong, K.M.; Yang, I.A. Genes and Gene Ontologies Common to Airflow Obstruction and Emphysema in the Lungs of Patients with COPD. PLoS ONE 2011, 6, e17442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternak, M.; Khomich, T.I.; Jakubowski, A.; Szafarz, M.; Szczepański, W.; Białas, M.; Stojak, M.; Szymura-Oleksiak, J.; Chłopicki, S. Nicotinamide N-Methyltransferase (NNMT) and 1-Methylnicotinamide (MNA) in Experimental Hepatitis Induced by Concanavalin A in the Mouse. Pharmacol. Rep. 2010, 62, 483–493. [Google Scholar] [CrossRef]
- Fedorowicz, A.; Mateuszuk, Ł.; Kopec, G.; Skórka, T.; Kutryb-Zając, B.; Zakrzewska, A.; Walczak, M.; Jakubowski, A.; Łomnicka, M.; Słomińska, E.; et al. Activation of the Nicotinamide N-Methyltransferase (NNMT)-1-Methylnicotinamide (MNA) Pathway in Pulmonary Hypertension. Respir. Res. 2016, 17, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 and Other Sirtuins in Metabolism. Trends. Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Smith, J.S.; Brachmann, C.B.; Celic, I.; Kenna, M.A.; Muhammad, S.; Starai, V.J.; Avalos, J.L.; Escalante-Semerena, J.C.; Grubmeyer, C.; Wolberger, C.; et al. A Phylogenetically Conserved NAD+-Dependent Protein Deacetylase Activity in the Sir2 Protein Family. Proc. Natl. Acad. Sci. USA 2000, 97, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Tanny, J.C.; Dowd, G.J.; Huang, J.; Hilz, H.; Moazed, D. An Enzymatic Activity in the Yeast Sir2 Protein That Is Essential for Gene Silencing. Cell 1999, 99, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Byrnes, K.; Blessinger, S.; Bailey, N.T.; Scaife, R.; Liu, G.; Khambu, B. Therapeutic Regulation of Autophagy in Hepatic Metabolism. Acta. Pharm. Sin. B 2022, 12, 33–49. [Google Scholar] [CrossRef]
- Huang, Q.; Su, H.; Qi, B.; Wang, Y.; Yan, K.; Wang, X.; Li, X.; Zhao, D. A SIRT1 Activator, Ginsenoside Rc, Promotes Energy Metabolism in Cardiomyocytes and Neurons. J. Am. Chem. Soc. 2021, 143, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Salazar, G.; Patrushev, N.; Alexander, R.W. FoxO1 Mediates an Autofeedback Loop Regulating SIRT1 Expression. J. Biol. Chem. 2011, 286, 5289–5299. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. SIRT1 Longevity Factor Suppresses NF-KappaB -Driven Immune Responses: Regulation of Aging via NF-KappaB Acetylation? Bioessays 2008, 30, 939–942. [Google Scholar] [CrossRef]
- Serrano-Marco, L.; Chacón, M.R.; Maymó-Masip, E.; Barroso, E.; Salvadó, L.; Wabitsch, M.; Garrido-Sánchez, L.; Tinahones, F.J.; Palomer, X.; Vendrell, J.; et al. TNF-α Inhibits PPARβ/δ Activity and SIRT1 Expression through NF-ΚB in Human Adipocytes. Biochim. Biophys. Acta 2012, 1821, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and Sirtuins in Aging and Disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natoli, G. When Sirtuins and NF-KappaB Collide. Cell 2009, 136, 19–21. [Google Scholar] [CrossRef] [Green Version]
- Preyat, N.; Leo, O. Sirtuin Deacylases: A Molecular Link between Metabolism and Immunity. J. Leukoc. Biol. 2013, 93, 669–680. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E. The Many Faces of Sirtuins: Coupling of NAD Metabolism, Sirtuins and Lifespan. Nat. Med. 2014, 20, 25–27. [Google Scholar] [CrossRef]
- Chen, X.; Lu, Y.; Zhang, Z.; Wang, J.; Yang, H.; Liu, G. Intercellular Interplay between Sirt1 Signalling and Cell Metabolism in Immune Cell Biology. Immunology 2015, 145, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. NAD+-Dependent Sirtuin 1 and 6 Proteins Coordinate a Switch from Glucose to Fatty Acid Oxidation during the Acute Inflammatory Response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; de Oliveira, R.M.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 Promotes Fat Mobilization in White Adipocytes by Repressing PPAR-γ. Nature 2004, 429, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic Control through the PGC-1 Family of Transcription Coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NALP3/NLRP3 Inflammasome Instigates Obesity-Induced Autoinflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; Böni-Schnetzler, M.; Hubbard, B.P.; Bouzakri, K.; Brunner, A.; Cavelti-Weder, C.; Keller, C.; Meyer-Böni, M.; Meier, D.T.; Brorsson, C.; et al. Identification of a SIRT1 Mutation in a Family with Type 1 Diabetes. Cell Metab. 2013, 17, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Melhem, H.; Hansmannel, F.; Bressenot, A.; Battaglia-Hsu, S.-F.; Billioud, V.; Alberto, J.M.; Gueant, J.L.; Peyrin-Biroulet, L. Methyl-Deficient Diet Promotes Colitis and SIRT1-Mediated Endoplasmic Reticulum Stress. Gut 2016, 65, 595–606. [Google Scholar] [CrossRef]
- Talero, E.; Alcaide, A.; Ávila-Román, J.; García-Mauriño, S.; Vendramini-Costa, D.; Motilva, V. Expression Patterns of Sirtuin 1-AMPK-Autophagy Pathway in Chronic Colitis and Inflammation-Associated Colon Neoplasia in IL-10-Deficient Mice. Int. Immunopharmacol. 2016, 35, 248–256. [Google Scholar] [CrossRef]
- Ren, M.-T.; Gu, M.-L.; Zhou, X.-X.; Yu, M.-S.; Pan, H.-H.; Ji, F.; Ding, C.-Y. Sirtuin 1 Alleviates Endoplasmic Reticulum Stress-Mediated Apoptosis of Intestinal Epithelial Cells in Ulcerative Colitis. World J. Gastroenterol. 2019, 25, 5800–5813. [Google Scholar] [CrossRef]
- Xu, K.; Guo, Y.; Ping, L.; Qiu, Y.; Liu, Q.; Li, Z.; Wang, Z. Protective Effects of SIRT6 Overexpression against DSS-Induced Colitis in Mice. Cells 2020, 9, 1513. [Google Scholar] [CrossRef]
- Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Abedi, V.; Bassaganya-Riera, J. NLRX1 Modulates Immunometabolic Mechanisms Controlling the Host–Gut Microbiota Interactions during Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 363. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-X.; Yang, X.-Y.; Han, B.-S.; Hu, Y.-Y.; An, T.; Lv, B.-H.; Lian, J.; Wang, T.-Y.; Bao, X.-L.; Gao, L.; et al. Naringenin Regulates Gut Microbiota and SIRT1/PGC-1ɑ Signaling Pathway in Rats with Letrozole-Induced Polycystic Ovary Syndrome. Biomed. Pharmacother. 2022, 153, 113286. [Google Scholar] [CrossRef]
- Lee, H.C. Mechanisms of Calcium Signaling by Cyclic ADP-Ribose and NAADP. Physiol. Rev. 1997, 77, 1133–1164. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Physiological Functions of Cyclic ADP-Ribose and NAADP as Calcium Messengers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Menteyne, A.; Burdakov, A.; Charpentier, G.; Petersen, O.H.; Cancela, J.-M. Generation of Specific Ca(2+) Signals from Ca(2+) Stores and Endocytosis by Differential Coupling to Messengers. Curr. Biol. 2006, 16, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarragó, M.G.; Puranik, A.S.; et al. CD38 Ecto-Enzyme in Immune Cells Is Induced during Aging Regulating NAD+ and NMN Levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.T.P.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The Enzyme CD38 (a NAD Glycohydrolase, EC 3.2.2.5) Is Necessary for the Development of Diet-Induced Obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Airoldi, I.; Marimpietri, D.; Bracci, C.; Faini, A.C.; Gramignoli, R. CD38, a Receptor with Multifunctional Activities: From Modulatory Functions on Regulatory Cell Subsets and Extracellular Vesicles, to a Target for Therapeutic Strategies. Cells 2019, 8, 1527. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent Cells Promote Tissue NAD+ Decline during Ageing via the Activation of CD38+ Macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Deaglio, S.; Mallone, R.; Baj, G.; Donati, D.; Giraudo, G.; Corno, F.; Bruzzone, S.; Geuna, M.; Ausiello, C.; Malavasi, F. Human CD38 and Its Ligand CD31 Define a Unique Lamina Propria T Lymphocyte Signaling Pathway. FASEB J. 2001, 15, 580–582. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.C.J.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W.H.I. Monoclonal Antibodies Targeting CD38 in Hematological Malignancies and Beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P.; Canto, C.; Brunyánszki, A.; Huber, A.; Szántó, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 Regulates SIRT1 Expression and Whole-Body Energy Expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Pirinen, E.; Canto, C.; Jo, Y.-S.; Morato, L.; Zhang, H.; Menzies, K.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A High-Fat Diet and NAD+ Activate Sirt1 to Rescue Premature Aging in Cockayne Syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Oliver, F.J.; Ménissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to Endotoxic Shock as a Consequence of Defective NF-KappaB Activation in Poly (ADP-Ribose) Polymerase-1 Deficient Mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef]
- Boughton-Smith, N.K.; Evans, S.M.; Hawkey, C.J.; Cole, A.T.; Balsitis, M.; Whittle, B.J.; Moncada, S. Nitric Oxide Synthase Activity in Ulcerative Colitis and Crohn’s Disease. Lancet 1993, 342, 338–340. [Google Scholar] [CrossRef]
- Singer, I.I.; Kawka, D.W.; Scott, S.; Weidner, J.R.; Mumford, R.A.; Riehl, T.E.; Stenson, W.F. Expression of Inducible Nitric Oxide Synthase and Nitrotyrosine in Colonic Epithelium in Inflammatory Bowel Disease. Gastroenterology 1996, 111, 871–885. [Google Scholar] [CrossRef]
- Brunyanszki, A.; Olah, G.; Coletta, C.; Szczesny, B.; Szabo, C. Regulation of Mitochondrial Poly(ADP-Ribose) Polymerase Activation by the β-Adrenoceptor/CAMP/Protein Kinase A Axis during Oxidative Stress. Mol. Pharmacol. 2014, 86, 450–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory Networks Underlying Colorectal Cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef]
- Dörsam, B.; Seiwert, N.; Foersch, S.; Stroh, S.; Nagel, G.; Begaliew, D.; Diehl, E.; Kraus, A.; McKeague, M.; Minneker, V.; et al. PARP-1 Protects against Colorectal Tumor Induction, but Promotes Inflammation-Driven Colorectal Tumor Progression. Proc. Natl. Acad. Sci. USA 2018, 115, E4061–E4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannone, P.J.; Alcamo, A.A.; Schanbacher, B.L.; Nankervis, C.A.; Besner, G.E.; Bauer, J.A. Poly(ADP-Ribose) Polymerase-1: A Novel Therapeutic Target in Necrotizing Enterocolitis. Pediatr. Res. 2011, 70, 67–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an Adipocytokine with Proinflammatory and Immunomodulating Properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [Green Version]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and Characterization of the CDNA Encoding a Novel Human Pre-B-Cell Colony-Enhancing Factor. Mol. Cell. Biol. 1994, 14, 1431–1437. [Google Scholar]
- Li, Y.; Zhang, Y.; Dorweiler, B.; Cui, D.; Wang, T.; Woo, C.W.; Brunkan, C.S.; Wolberger, C.; Imai, S.; Tabas, I. Extracellular Nampt Promotes Macrophage Survival via a Nonenzymatic Interleukin-6/STAT3 Signaling Mechanism. J. Biol. Chem. 2008, 283, 34833–34843. [Google Scholar] [CrossRef] [Green Version]
- Van den Bergh, R.; Morin, S.; Sass, H.J.; Grzesiek, S.; Vekemans, M.; Florence, E.; Thanh Thi Tran, H.; Imiru, R.G.; Heyndrickx, L.; Vanham, G.; et al. Monocytes Contribute to Differential Immune Pressure on R5 versus X4 HIV through the Adipocytokine Visfatin/NAMPT. PLoS ONE 2012, 7, e35074. [Google Scholar] [CrossRef]
- Camp, S.M.; Ceco, E.; Evenoski, C.L.; Danilov, S.M.; Zhou, T.; Chiang, E.T.; Moreno-Vinasco, L.; Mapes, B.; Zhao, J.; Gursoy, G.; et al. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFκB Signaling and Inflammatory Lung Injury. Sci. Rep. 2015, 5, 13135. [Google Scholar] [CrossRef] [Green Version]
- Managò, A.; Audrito, V.; Mazzola, F.; Sorci, L.; Gaudino, F.; Gizzi, K.; Vitale, N.; Incarnato, D.; Minazzato, G.; Ianniello, A.; et al. Extracellular Nicotinate Phosphoribosyltransferase Binds Toll like Receptor 4 and Mediates Inflammation. Nat. Commun. 2019, 10, 4116. [Google Scholar] [CrossRef] [Green Version]
- Colombo, G.; Travelli, C.; Porta, C.; Genazzani, A.A. Extracellular Nicotinamide Phosphoribosyltransferase Boosts IFNγ-Induced Macrophage Polarization Independently of TLR4. Science 2022, 25, 104147. [Google Scholar] [CrossRef] [PubMed]
- Grolla, A.A.; Travelli, C.; Genazzani, A.A.; Sethi, J.K. Extracellular Nicotinamide Phosphoribosyltransferase, a New Cancer Metabokine. Br. J. Pharmacol. 2016, 173, 2182–2194. [Google Scholar] [CrossRef] [Green Version]
- Galassi, L.; Di Stefano, M.; Brunetti, L.; Orsomando, G.; Amici, A.; Ruggieri, S.; Magni, G. Characterization of Human Nicotinate Phosphoribosyltransferase: Kinetic Studies, Structure Prediction and Functional Analysis by Site-Directed Mutagenesis. Biochimie 2012, 94, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.B.; Chaykin, S. The Management of Nicotinamide and Nicotinic Acid in the Mouse. J. Biol. Chem. 1972, 247, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Tsuchiya, M. Nicotinamide Phosphoribosyltransferase/Visfatin Does Not Catalyze Nicotinamide Mononucleotide Formation in Blood Plasma. PLoS ONE 2011, 6, e22781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaut, Z.N.; Solomon, H.M. Inhibition of Nicotinate Phosphoribosyltransferase in Human Platelet Lysate by Nicotinic Acid Analogs. Biochem. Pharmacol. 1971, 20, 2903–2906. [Google Scholar] [CrossRef]
- Ruggieri, S.; Orsomando, G.; Sorci, L.; Raffaelli, N. Regulation of NAD Biosynthetic Enzymes Modulates NAD-Sensing Processes to Shape Mammalian Cell Physiology under Varying Biological Cues. Biochim. Biophys. Acta 2015, 1854, 1138–1149. [Google Scholar] [CrossRef]
- Smith, L.D.; Gholson, R.K. Allosteric Properties of Bovine Liver Nicotinate Phosphoribosyltransferase. J. Biol. Chem. 1969, 244, 68–71. [Google Scholar] [CrossRef]
- Neubauer, K.; Bednarz-Misa, I.; Walecka-Zacharska, E.; Wierzbicki, J.; Agrawal, A.; Gamian, A.; Krzystek-Korpacka, M. Oversecretion and Overexpression of Nicotinamide Phosphoribosyltransferase/Pre-B Colony-Enhancing Factor/Visfatin in Inflammatory Bowel Disease Reflects the Disease Activity, Severity of Inflammatory Response and Hypoxia. Int. J. Mol. Sci. 2019, 20, 166. [Google Scholar] [CrossRef] [Green Version]
- Colombo, G.; Caviglia, G.P.; Ravera, A.; Tribocco, E.; Frara, S.; Rosso, C.; Travelli, C.; Genazzani, A.A.; Ribaldone, D.G. NAMPT and NAPRT Serum Levels Predict Response to Anti-TNF Therapy in Inflammatory Bowel Disease. Front. Med. (Lausanne) 2023, 10, 1116862. [Google Scholar] [CrossRef]
- Piacente, F.; Caffa, I.; Ravera, S.; Sociali, G.; Passalacqua, M.; Vellone, V.G.; Becherini, P.; Reverberi, D.; Monacelli, F.; Ballestrero, A.; et al. Nicotinic Acid Phosphoribosyltransferase Regulates Cancer Cell Metabolism, Susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 2017, 77, 3857–3869. [Google Scholar] [CrossRef] [Green Version]
- Roberti, A.; Fernández, A.F.; Fraga, M.F. Nicotinamide N-Methyltransferase: At the Crossroads between Cellular Metabolism and Epigenetic Regulation. Mol. Metab. 2021, 45, 101165. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Sartini, D.; Pozzi, V.; Wilk, D.; Emanuelli, M.; Yee, V.C. Structural Basis of Substrate Recognition in Human Nicotinamide N-Methyltransferase. Biochemistry 2011, 50, 7800–7808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human Liver Nicotinamide N-Methyltransferase. CDNA Cloning, Expression, and Biochemical Characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Burnett, D.; Bennett, P.; Waring, R.H.; Brown, H.M.; Williams, A.C.; Ramsden, D.B. A Direct Correlation between Nicotinamide N-Methyltransferase Activity and Protein Levels in Human Liver Cytosol. Biochim. Biophys. Acta 1998, 1442, 238–244. [Google Scholar] [CrossRef]
- Seifert, R.; Hoshino, J.; Kröger, H. Nicotinamide Methylation. Tissue Distribution, Developmental and Neoplastic Changes. Biochim. Biophys. Acta 1984, 801, 259–264. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, Ł.; Wojnar-Lason, K.; Kaczara, P.; Tworzydło, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-Methyltransferase in Endothelium Protects against Oxidant Stress-Induced Endothelial Injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef]
- Riederer, M.; Erwa, W.; Zimmermann, R.; Frank, S.; Zechner, R. Adipose Tissue as a Source of Nicotinamide N-Methyltransferase and Homocysteine. Atherosclerosis 2009, 204, 412–417. [Google Scholar] [CrossRef]
- Xu, J.; Capezzone, M.; Xu, X.; Hershman, J.M. Activation of Nicotinamide N-Methyltransferase Gene Promoter by Hepatocyte Nuclear Factor-1beta in Human Papillary Thyroid Cancer Cells. Mol. Endocrinol. 2005, 19, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Katsyuba, E.; Auwerx, J. Modulating NAD + Metabolism, from Bench to Bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef]
- Xie, X.; Yu, H.; Wang, Y.; Zhou, Y.; Li, G.; Ruan, Z.; Li, F.; Wang, X.; Liu, H.; Zhang, J. Nicotinamide N-Methyltransferase Enhances the Capacity of Tumorigenesis Associated with the Promotion of Cell Cycle Progression in Human Colorectal Cancer Cells. Arch. Biochem. Biophys. 2014, 564, 52–66. [Google Scholar] [CrossRef]
- Lu, X.M.; Long, H. Nicotinamide N-Methyltransferase as a Potential Marker for Cancer. Neoplasma 2018, 65, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Tanaka, A.; Namba, K.; Shia, J.; Wang, J.Y.; Roehrl, M.H.A. Tumor Stromal Nicotinamide N-Methyltransferase Overexpression as a Prognostic Biomarker for Poor Clinical Outcome in Early-Stage Colorectal Cancer. Sci. Rep. 2022, 12, 2767. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, H.; Brightwell, C.R.; Graber, T.G.; Maroto, R.; Leo Wang, H.-Y.; McHardy, S.F.; Papaconstantinou, J.; Fry, C.S.; Watowich, S.J. Small Molecule Nicotinamide N-Methyltransferase Inhibitor Activates Senescent Muscle Stem Cells and Improves Regenerative Capacity of Aged Skeletal Muscle. Biochem. Pharmacol. 2019, 163, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Guo, M.; He, L.; Martínez, M.-A.; Martínez, M.; Lopez-Torres, B.; Martínez-Larrañaga, M.-R.; Wang, X.; Anadón, A.; Ares, I. Nicotinamide N-Methyltransferase Protects against Deoxynivalenol-Induced Growth Inhibition by Suppressing pro-Inflammatory Cytokine Expression. Food Chem. Toxicol. 2022, 163, 112969. [Google Scholar] [CrossRef]
- Jakubowski, A.; Sternak, M.; Jablonski, K.; Ciszek-Lenda, M.; Marcinkiewicz, J.; Chlopicki, S. 1-Methylnicotinamide Protects against Liver Injury Induced by Concanavalin A via a Prostacyclin-Dependent Mechanism: A Possible Involvement of IL-4 and TNF-α. Int. Immunopharmacol. 2016, 31, 98–104. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT Activation Can Contribute to the Development of Fatty Liver Disease by Modulating the NAD+ Metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef] [Green Version]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef]
- Vragović, J.; Vraţić, H. Inflammatory Bowel Disease. Prog. Drug Res. 2016, 71, 117–122. [Google Scholar]
- Fritze, C.E.; Verschueren, K.; Strich, R.; Easton Esposito, R. Direct Evidence for SIR2 Modulation of Chromatin Structure in Yeast RDNA. EMBO J. 1997, 16, 6495–6509. [Google Scholar] [CrossRef] [Green Version]
- Bryan, S.; Baregzay, B.; Spicer, D.; Singal, P.K.; Khaper, N. Redox-Inflammatory Synergy in the Metabolic Syndrome. Can. J. Physiol. Pharmacol. 2013, 91, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Baixauli, F.; Acín-Pérez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabandé-Rodriguez, E.; Dolores Ledesma, M.; Blázquez, A.; Martin, M.A.; Falcón-Pérez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de Novo NAD+ Synthesis Specifies Immune Function in Aging and Inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial Complex I Deficiency Increases Protein Acetylation and Accelerates Heart Failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T Cells with Dysfunctional Mitochondria Induce Multimorbidity and Premature Senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Almeida, L.; Dhillon-LaBrooy, A.; Castro, C.N.; Adossa, N.; Carriche, G.M.; Guderian, M.; Lippens, S.; Dennerlein, S.; Hesse, C.; Lambrecht, B.N.; et al. Ribosome-Targeting Antibiotics Impair T Cell Effector Function and Ameliorate Autoimmunity by Blocking Mitochondrial Protein Synthesis. Immunity 2021, 54, 68–83.e6. [Google Scholar] [CrossRef]
- Roediger, W.E. The Colonic Epithelium in Ulcerative Colitis: An Energy-Deficiency Disease? Lancet 1980, 2, 712–715. [Google Scholar] [CrossRef]
- Haberman, Y.; Karns, R.; Dexheimer, P.J.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative Colitis Mucosal Transcriptomes Reveal Mitochondriopathy and Personalized Mechanisms Underlying Disease Severity and Treatment Response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Ogawa, S.A.; Chau, L.; Whelan, K.A.; Hamilton, K.E.; Chen, J.; Tan, L.; Chen, E.Z.; Keilbaugh, S.; Fogt, F.; et al. Mitochondrial Dysfunction in Inflammatory Bowel Disease Alters Intestinal Epithelial Metabolism of Hepatic Acylcarnitines. J. Clin. Investig. 2020, 131, e133371. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.B.; Xavier, R.J. Pathway Paradigms Revealed from the Genetics of Inflammatory Bowel Disease. Nature 2020, 578, 527–539. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahiri, A.; Hedl, M.; Yan, J.; Abraham, C. Human LACC1 Increases Innate Receptor-Induced Responses and a LACC1 Disease-Risk Variant Modulates These Outcomes. Nat. Commun. 2017, 8, 15614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muise, A.M.; Xu, W.; Guo, C.-H.; Walters, T.D.; Wolters, V.M.; Fattouh, R.; Lam, G.Y.; Hu, P.; Murchie, R.; Sherlock, M.; et al. NADPH Oxidase Complex and IBD Candidate Gene Studies: Identification of a Rare Variant in NCF2 That Results in Reduced Binding to RAC2. Gut 2012, 61, 1028–1035. [Google Scholar] [CrossRef] [Green Version]
- Rivas, M.A.; Beaudoin, M.; Gardet, A.; Stevens, C.; Sharma, Y.; Zhang, C.K.; Boucher, G.; Ripke, S.; Ellinghaus, D.; Burtt, N.; et al. Deep Resequencing of GWAS Loci Identifies Independent Rare Variants Associated with Inflammatory Bowel Disease. Nat. Genet. 2011, 43, 1066–1073. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T. Regulation of Intestinal Epithelial Permeability by Tight Junctions. Cell. Mol. Life Sci. 2013, 70, 631–659. [Google Scholar] [CrossRef]
- Garcia-Hernandez, V.; Quiros, M.; Nusrat, A. Intestinal Epithelial Claudins: Expression and Regulation in Homeostasis and Inflammation. Ann. N. Y. Acad. Sci. 2017, 1397, 66–79. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional Strands in Tight Junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Tight Junctions and the Molecular Basis for Regulation of Paracellular Permeability. Am. J. Physiol. 1995, 269, G467–G475. [Google Scholar] [CrossRef] [PubMed]
- Unno, N.; Fink, M.P. Intestinal Epithelial Hyperpermeability. Mechanisms and Relevance to Disease. Gastroenterol. Clin. N. Am. 1998, 27, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Delude, R.L.; Han, Y.Y.; Sappington, P.L.; Han, X.; Carcillo, J.A.; Fink, M.P. Liposomal NAD(+) Prevents Diminished O(2) Consumption by Immunostimulated Caco-2 Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1082–L1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, M.; Lu, C.; An, L.; Gao, Q.; Xie, W.; Miao, F.; Chen, X.; Pan, Y.; Wang, Q. SIRT1 Relieves Necrotizing Enterocolitis through Inactivation of Hypoxia-Inducible Factor (HIF)-1a. Cell Cycle 2020, 19, 2018–2027. [Google Scholar] [CrossRef]
- Berger, F.; Ramírez-Hernández, M.H.; Ziegler, M. The New Life of a Centenarian: Signalling Functions of NAD(P). Trends Biochem. Sci. 2004, 29, 111–118. [Google Scholar] [CrossRef]
- Pollak, N.; Dölle, C.; Ziegler, M. The Power to Reduce: Pyridine Nucleotides–Small Molecules with a Multitude of Functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Regulation of Genes Encoding NAD(P)H:Quinone Oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.T.; Hwang, J.H.; Kim, D.H.; Park, M.J.; Lee, I.H.; Nam, H.J.; Kang, J.K.; Kim, S.K.; Hwang, J.S.; Chung, H.K.; et al. Role of NADH: Quinone Oxidoreductase-1 in the Tight Junctions of Colonic Epithelial Cells. BMB Rep. 2014, 47, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.L.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The Role of Mitochondria in Stem Cell Fate and Aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 Reverses Aging-Associated Degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. STEM CELL AGING. A Mitochondrial UPR-Mediated Metabolic Checkpoint Regulates Hematopoietic Stem Cell Aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ Repletion Improves Mitochondrial and Stem Cell Function and Enhances Life Span in Mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biteau, B.; Hochmuth, C.E.; Jasper, H. Maintaining Tissue Homeostasis: Dynamic Control of Somatic Stem Cell Activity. Cell Stem Cell 2011, 9, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Tan, S.; Clevers, H. Lgr Proteins in Epithelial Stem Cell Biology. Development 2013, 140, 2484–2494. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of Stem Cells in Small Intestine and Colon by Marker Gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Cheng, C.-W.; Cao, A.Q.; Tripathi, S.; Mana, M.D.; Bauer-Rowe, K.E.; Abu-Remaileh, M.; Clavain, L.; Erdemir, A.; Lewis, C.A.; et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 2018, 22, 769–778.e4. [Google Scholar] [CrossRef] [Green Version]
- Nalapareddy, K.; Nattamai, K.J.; Kumar, R.S.; Karns, R.; Wikenheiser-Brokamp, K.A.; Sampson, L.L.; Mahe, M.M.; Sundaram, N.; Yacyshyn, M.-B.; Yacyshyn, B.; et al. Canonical Wnt Signaling Ameliorates Aging of Intestinal Stem Cells. Cell Rep. 2017, 18, 2608–2621. [Google Scholar] [CrossRef]
- Annunziata, F.; Rasa, S.M.M.; Krepelova, A.; Lu, J.; Minetti, A.; Omrani, O.; Nunna, S.; Adam, L.; Käppel, S.; Neri, F. Paneth Cells Drive Intestinal Stem Cell Competition and Clonality in Aging and Calorie Restriction. Eur. J. Cell Biol. 2022, 101, 151282. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ Metabolism, Stemness, the Immune Response, and Cancer. Signal. Transduct. Target Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Hong, S.M.; Lee, A.-Y.; Hwang, S.-M.; Ha, Y.-J.; Kim, M.-J.; Min, S.; Hwang, W.; Yoon, G.; Kwon, S.M.; Woo, H.G.; et al. NAMPT Mitigates Colitis Severity by Supporting Redox-Sensitive Activation of Phagocytosis in Inflammatory Macrophages. Redox Biol. 2022, 50, 102237. [Google Scholar] [CrossRef] [PubMed]
- Peritore, A.F.; D’Amico, R.; Cordaro, M.; Siracusa, R.; Fusco, R.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Di Paola, R.; Cuzzocrea, S.; et al. PEA/Polydatin: Anti-Inflammatory and Antioxidant Approach to Counteract DNBS-Induced Colitis. Antioxidants 2021, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Shi, L.; Wang, L.; Zhou, Z.; Wang, C.; Lin, Y.; Luo, D.; Qiu, J.; Chen, D. Activation of Sirtuin 1 by Catalpol-Induced down-Regulation of MicroRNA-132 Attenuates Endoplasmic Reticulum Stress in Colitis. Pharmacol. Res. 2017, 123, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Akimova, T.; Xiao, H.; Liu, Y.; Bhatti, T.R.; Jiao, J.; Eruslanov, E.; Singhal, S.; Wang, L.; Han, R.; Zacharia, K.; et al. Targeting Sirtuin-1 Alleviates Experimental Autoimmune Colitis by Induction of Foxp3+ T-Regulatory Cells. Mucosal. Immunol. 2014, 7, 1209–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabley, J.G.; Jagtap, P.; Perretti, M.; Getting, S.J.; Salzman, A.L.; Virág, L.; Szabó, E.; Soriano, F.G.; Liaudet, L.; Abdelkarim, G.E.; et al. Anti-Inflammatory Effects of a Novel, Potent Inhibitor of Poly (ADP-Ribose) Polymerase. Inflamm. Res. 2001, 50, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B.; O’Connor, M.; Hake, P.W. Inhibitors of Poly (ADP-Ribose) Polymerase Modulate Signal Transduction Pathways in Colitis. Eur. J. Pharmacol. 2003, 469, 183–194. [Google Scholar] [CrossRef]
- Sánchez-Fidalgo, S.; Villegas, I.; Martín, A.; Sánchez-Hidalgo, M.; Alarcón de la Lastra, C. PARP Inhibition Reduces Acute Colonic Inflammation in Rats. Eur. J. Pharmacol. 2007, 563, 216–223. [Google Scholar] [CrossRef]
- Lu, H.; Lin, J.; Xu, C.; Sun, M.; Zuo, K.; Zhang, X.; Li, M.; Huang, H.; Li, Z.; Wu, W.; et al. Cyclosporine Modulates Neutrophil Functions via the SIRT6-HIF-1α-Glycolysis Axis to Alleviate Severe Ulcerative Colitis. Clin. Transl. Med. 2021, 11, e334. [Google Scholar] [CrossRef]
- Huang, P.; Wang, X.; Wang, S.; Wu, Z.; Zhou, Z.; Shao, G.; Ren, C.; Kuang, M.; Zhou, Y.; Jiang, A.; et al. Treatment of Inflammatory Bowel Disease: Potential Effect of NMN on Intestinal Barrier and Gut Microbiota. Curr. Res. Food Sci. 2022, 5, 1403–1411. [Google Scholar] [CrossRef]
- Malhi, G.; Rumman, A.; Thanabalan, R.; Croitoru, K.; Silverberg, M.S.; Hillary Steinhart, A.; Nguyen, G.C. Vaccination in Inflammatory Bowel Disease Patients: Attitudes, Knowledge, and Uptake. J. Crohn’s Colitis 2015, 9, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Murray, M.F.; Nghiem, M.; Srinivasan, A. HIV Infection Decreases Intracellular Nicotinamide Adenine Dinucleotide [NAD]. Biochem. Biophys. Res. Commun. 1995, 212, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Vanham, G.; Toossi, Z.; Hirsch, C.S.; Wallis, R.S.; Schwander, S.K.; Rich, E.A.; Ellner, J.J. Examining a Paradox in the Pathogenesis of Human Pulmonary Tuberculosis: Immune Activation and Suppression/Anergy. Tuber. Lung Dis. 1997, 78, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Rozwarski, D.A.; Grant, G.A.; Barton, D.H.; Jacobs, W.R.; Sacchettini, J.C. Modification of the NADH of the Isoniazid Target (InhA) from Mycobacterium Tuberculosis. Science 1998, 279, 98–102. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| NAD+ Metabolic Enzyme | Intracellular or Extracellular | Regulation of NAD+ | Cellular Processes and Pathways | Illnesses | IBD-Related Studies | Mechanisms Associated with IBD |
|---|---|---|---|---|---|---|
| Sirtuins | Intracellular | Sirtuins are NAD+ substrates and cofactors in deacetylated ADP-ribosylation reactions. In chronic inflammatory diseases, NAD+ and SIRT are downregulated in specific tissues [66]. | Energy shifts [67], cell differentiation [68], apoptosis [69], autophagy [70], development [71], and metabolism [72]; regulates circadian clock proteins CLOCK/Per [73,74,75,76,77], NF-κB/p65 [78], PPAR-γ/pgc1α [79], NLRP3 inflammation [80], TLR4 [81]. | Fat deposits in obesity and inflammation, atherosclerosis [82], Alzheimer’s [83], and sepsis [84]. | Some sirtuins (SIRT2, 3, 5, 6) protect against IBD [85,86,87,88,89,90] and some sirtuins (SIRT1) might be pathogenic [91]. | Activate macrophages [85,92] and remodel the gut microbiota [90,93,94]. |
| CD38 | Intracellular and extracellular | Type II glycosylated membrane protein CD38 converts NAD+ to ADPr and cADPr and hydrolyzes cADPr to ADPr [95,96]. Low-level CD38 inhibition increases cellular NAD+ levels [97,98]. | Regulates cell adhesion, differentiation, and proliferation [99]; mobilizes intracellular Ca2+ [100,101]; improves glucose metabolism and maintains lipid homeostasis [97,102]; repels neutrophils [99]. | Aging [103]. | Potential pathogenicity in IBD [58,104,105,106] | Activation and proliferation of T cells [105]. |
| NAMPT | Intracellular and extracellular | NAMPT upregulation increases NAD+, and the precursor NMN that converts NAM to NAD+ is essential for cellular NAD+ supply and rate-limiting steps in the NAD+ remediation pathway [40]. | Regulates energy metabolism, circadian rhythm, and immunity production [107]; proliferation, anti-apoptotic, pro-inflammatory, pro-angiogenic, and metastatic properties [108]. | Sepsis, rheumatoid arthritis, diabetes [109,110,111]. | NAMPT inhibition prevents experimental colitis in humans and mice [18,62] and colon tumor pathogenesis in mice [18,112]. | Reduces NF-κB activation and inflammatory cell infiltration [18]; decreases cell ATP levels; inhibits IL-1β, IL-6, and TNF-α secretion in vitro [113]. |
| PARP | Intracellular | NAD+ decomposing enzyme, cleavage of the nicotinamide-glycosidic bond of NAD+ to generate ADPr polymer [114]; enhances PARP activity and has significant harmful effects on NAD+ pools [115,116]. | Pro-inflammatory, pro-proliferative, oxidative stress [117,118]; NF-κB pathway [119]. | Peritonitis, septic shock, ovarian cancer [120], and alcoholic fatty liver [117,118]. | PARP1 and PARP2 promote colitis in mice [121,122]. | Downregulates SIRT1 and causes mucosal atrophy [123]. |
| NNMT | Intracellular | NAD+-dependent pro-inflammatory signals are maintained by methylation and excretion of NAM, which reduces precursors for NAD+ synthesis [41,124]. | Modulates tumor resistance and chemotherapy sensitivity [125,126,127,128], glucose metabolism [129]; NF-κB pathway [130]. | Colorectal cancer, breast cancer, esophageal squamous cell carcinoma, colorectal cancer, melanoma [125,126,127,128], type 2 diabetes [47], obesity [131], COPD [132,133], liver injury [134,135]. | Infliximab-treated IBD patients show normalized NNMT expression [124]. | No studies were found. |
| Drugs | Effects on NAD+ Metabolic Pathways | Research Models | Mechanism | References |
|---|---|---|---|---|
| NMN | NAD+ precursor, activate the NAMPT-dependent NAD+ biosynthetic pathway, increase NAD+ content | DSS-induced colitis mice | Improves inflammatory intestine morphology, colon length, intestinal epithelial barrier, blood serum pro-inflammatory factors, and gut microbiota composition. | [261,269] |
| Resveratrol | Increases SIRT1 expression | TNBS-induced colitis mice | Inhibits NF-κB signaling and activates NRF2 antioxidant program. | [262] |
| Cay10591 | Activates SIRT1 | TNBS or oxazolidinone-induced colitis in mice | Block NF-κB signaling and cytokine production. | [89] |
| Catalpol | Activates SIRT1 | TNBS-induced colitis mice | Improves endoplasmic reticulum stress | [263] |
| EX-527 | Inhibits SIRT1 | DSS-induced colitis mice | Promotes the formation of Treg | [264] |
| Norisoboldine | Reduce NAD+ levels and inhibit SIRT1 | DSS-induced colitis mice | Enhances the differentiation of Treg, regulates the AhR/glycolysis axis | [65] |
| PJ34 | Inhibits PARP1 | DSS-induced colitis mice | [265] | |
| 1,5-Dihydroxyisoquinoline | Inhibits PARP1 | TNBS-induced colitis rats | Inhibits NF-κB pathway and AP-1 | [266,267] |
| FK866 | NAMPT inhibitors | Isolated lamina propria mononuclear cells (LPMCs) from IBD patients and DSS-induced colitis mice | Inhibits intestinal mucosa immunity and cytokines. | [18] |
| Cyclosporine A | Inhibits SIRT6 | Neutrophils in peripheral blood from patients with acute UC | Inhibits peripheral blood neutrophil function and migration in acute UC | [268] |
| NNMT | Restores NAD+ | Intestinal mucosa from IBD patients | Tryptophan metabolism | [124] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Yan, W.; Tao, M.; Fu, Y. NAD+ Metabolism and Immune Regulation: New Approaches to Inflammatory Bowel Disease Therapies. Antioxidants 2023, 12, 1230. https://doi.org/10.3390/antiox12061230
Chen C, Yan W, Tao M, Fu Y. NAD+ Metabolism and Immune Regulation: New Approaches to Inflammatory Bowel Disease Therapies. Antioxidants. 2023; 12(6):1230. https://doi.org/10.3390/antiox12061230
Chicago/Turabian StyleChen, Chaoyue, Wei Yan, Meihui Tao, and Yu Fu. 2023. "NAD+ Metabolism and Immune Regulation: New Approaches to Inflammatory Bowel Disease Therapies" Antioxidants 12, no. 6: 1230. https://doi.org/10.3390/antiox12061230
APA StyleChen, C., Yan, W., Tao, M., & Fu, Y. (2023). NAD+ Metabolism and Immune Regulation: New Approaches to Inflammatory Bowel Disease Therapies. Antioxidants, 12(6), 1230. https://doi.org/10.3390/antiox12061230