
Chiisanoside Mediates the Parkin/ZNF746/PGC-1α Axis by Downregulating MiR-181a to Improve Mitochondrial Biogenesis in 6-OHDA-Caused Neurotoxicity Models In Vitro and In Vivo: Suggestions for Prevention of Parkinson’s Disease


Abstract
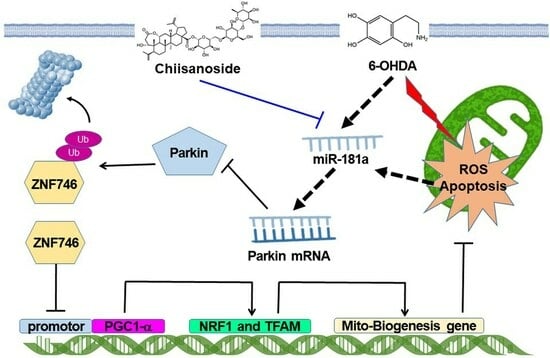
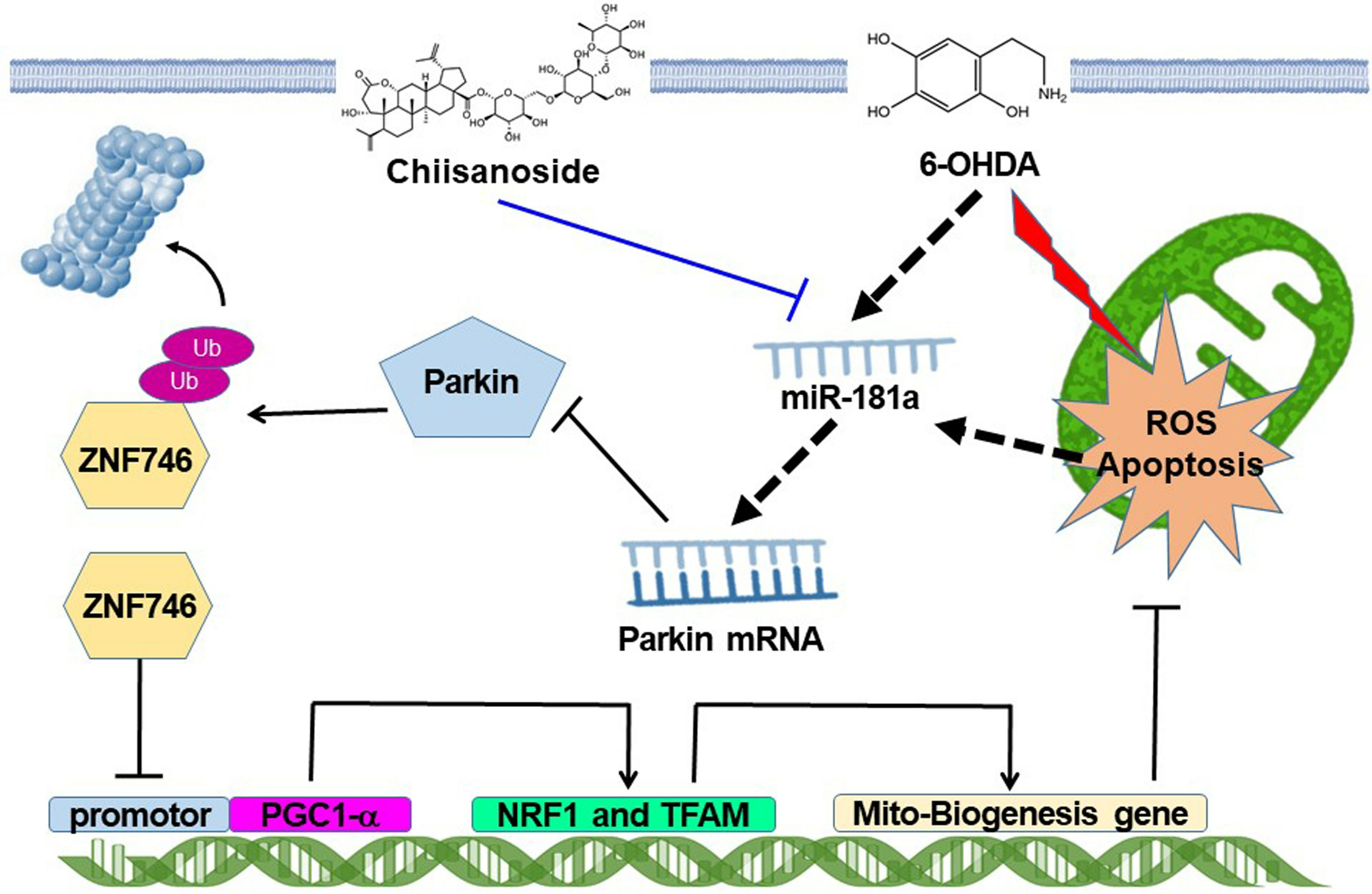
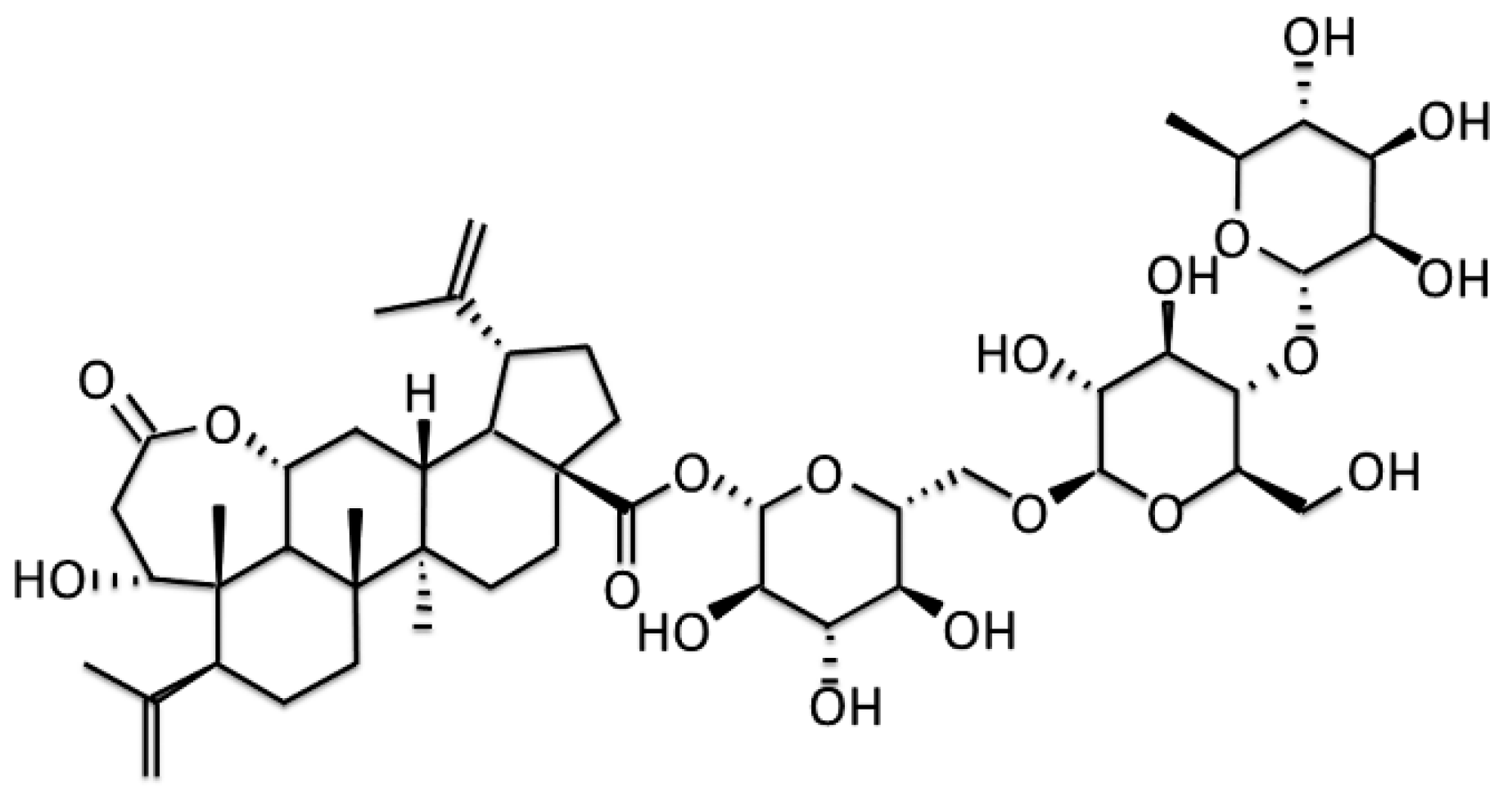
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Maintenance/Treatment of SH-SY5Y Cell Line
2.2. Determination of SH-SY5Y Cell Viability
2.3. Measurement of Mitochondrial Membrane Potential in SH-SY5Y Cells
2.4. TUNEL Assay of SH-SY5Y Cells
2.5. Flow Cytometry of FITC-Tagged Annexin-V and Propidium Iodide Staining on Apoptosis of SH-SY5Y Cells
2.6. Protein Expression Detection by Western Blotting in SH-SY5Y Cells
2.7. Quantification of Intracellular Reactive Oxygen Species in SH-SY5Y Cells
2.8. Mitochondrial Staining in SH-SY5Y Cells
2.9. Citrate Synthase Activity Assay of Mitochondrial in SH-SY5Y Cells
2.10. Immunoprecipitation (IP) Assay for SH-SY5Y Cells
2.11. Transient Transfection of Small Interfering RNAs in SH-SY5Y Cells
2.12. Reverse Transcriptase–Real-Time Polymerase Chain Reaction (RT-qPCR) for Parkin Gene in SH-SY5Y Cells
2.13. Measuring the Expression Level of MiR-181a by RT-qPCR in SH-SY5Y Cells
2.14. Treatment of Human MiR-181a Inhibitors and Mimics in SH-SY5Y Cells
2.15. Maintenance and Synchronization of Nematodes (C. elegans)
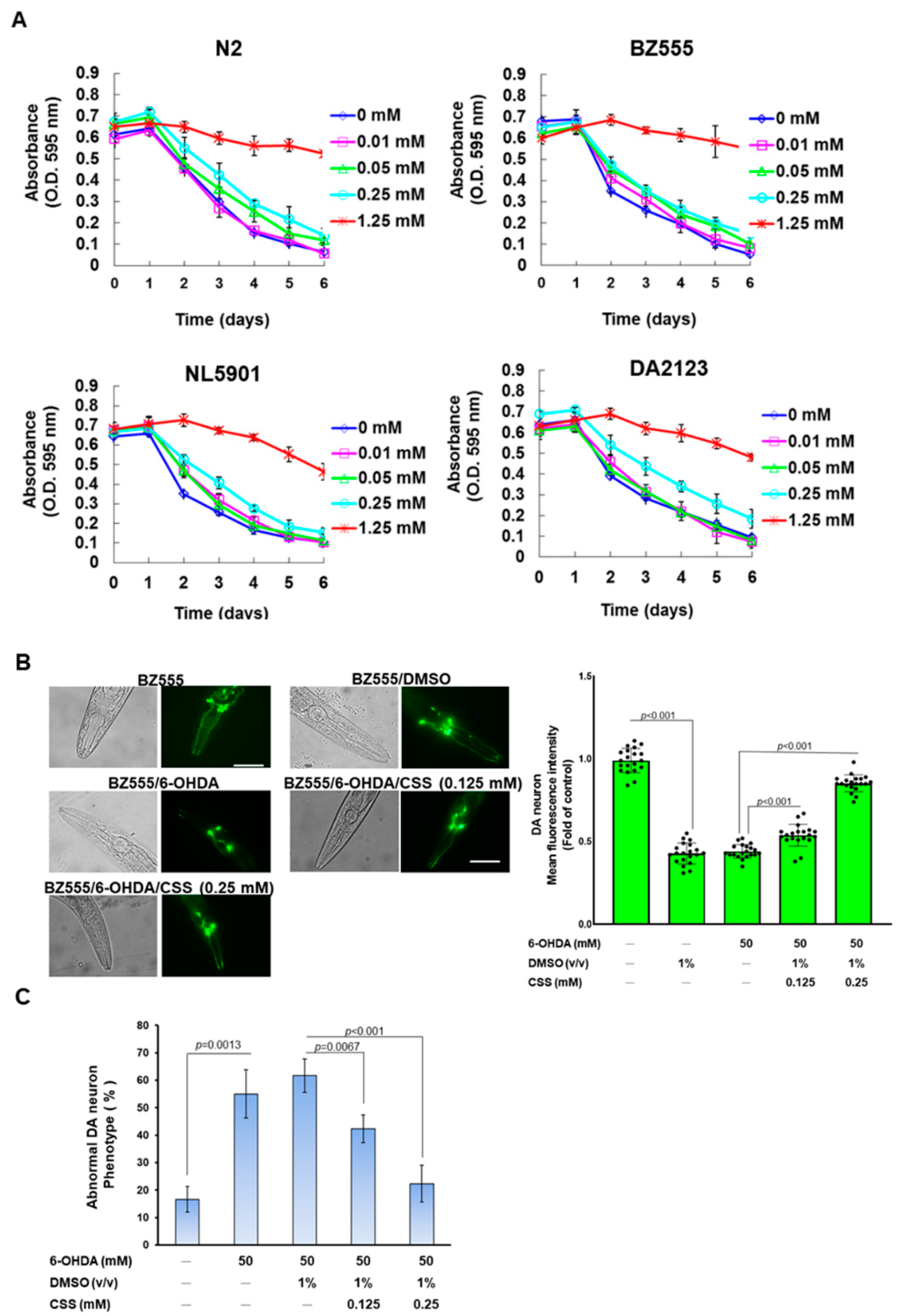
2.16. Food Clearance Test to Choice the Concentration of CSS for Nematode Treatment
2.17. CSS Pretreatment and 6-OHDA Exposure in Nematodes
2.18. The Analysis of DA Neuron Degeneration in Nematodes
2.19. Food-Sensitive Behavior Test in Nematodes
2.20. Lifespan Evaluation in Nematodes
2.21. Determination of Reactive Oxygen Species Level in Nematodes
2.22. Isolation of Total RNA and Determining of Gene Expression in Nematodes
2.23. Analysis of a-Synuclein Accumulation in Nematodes
2.24. Detection of Autophagy Activity in Nematodes
2.25. Statistical Methods in Experiments
3. Results
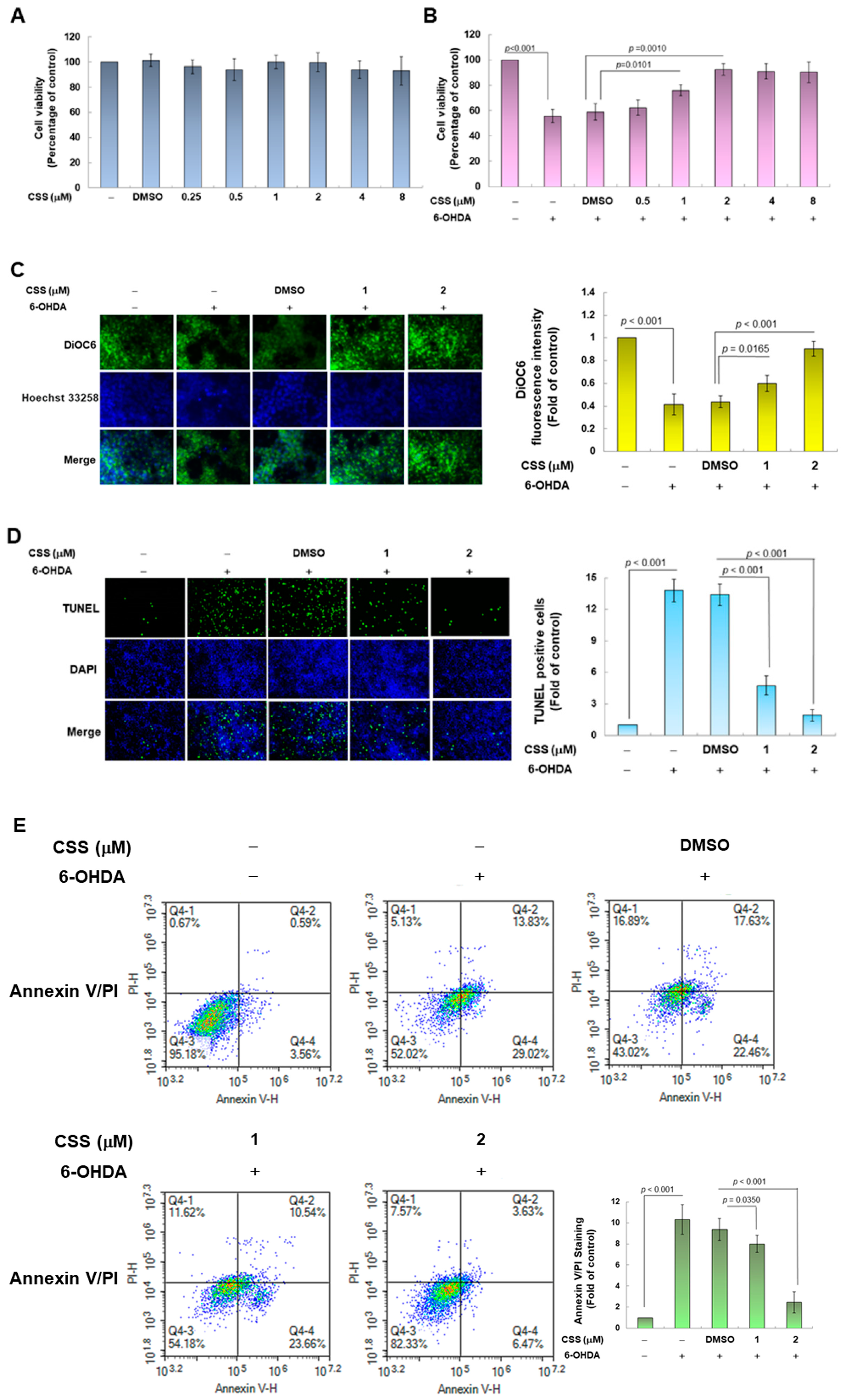
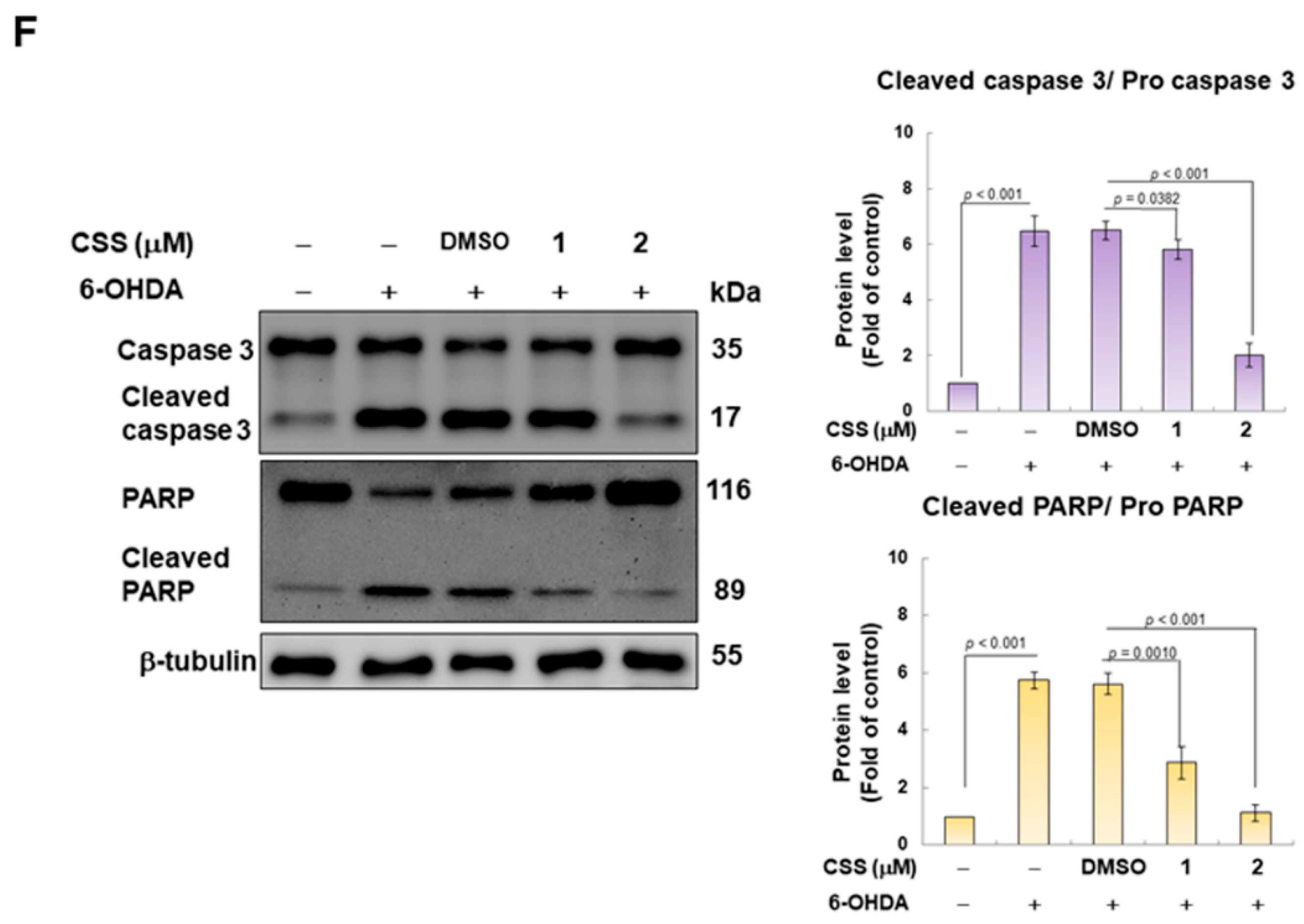
3.1. Chiisanoside (CSS) Inhibited Apoptosis in the SH-SY5Y Cell Model Exposed to 6-OHDA
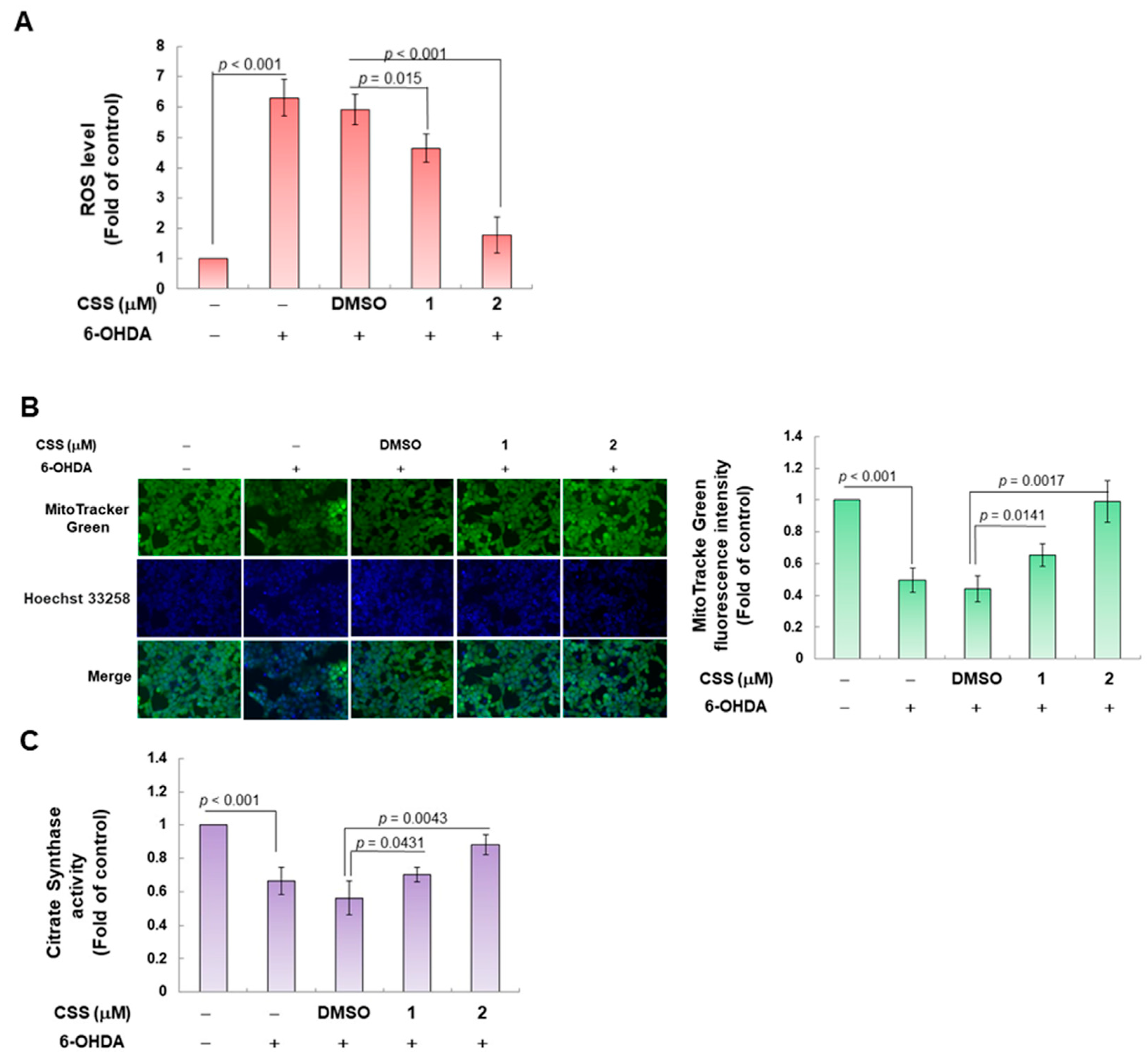
3.2. CSS Pretreatment Reduces the Generation of Cellular ROS Induced by 6-OHDA and Increases the Biogenesis of Mitochondria in the SH-SY5Y Cell Model
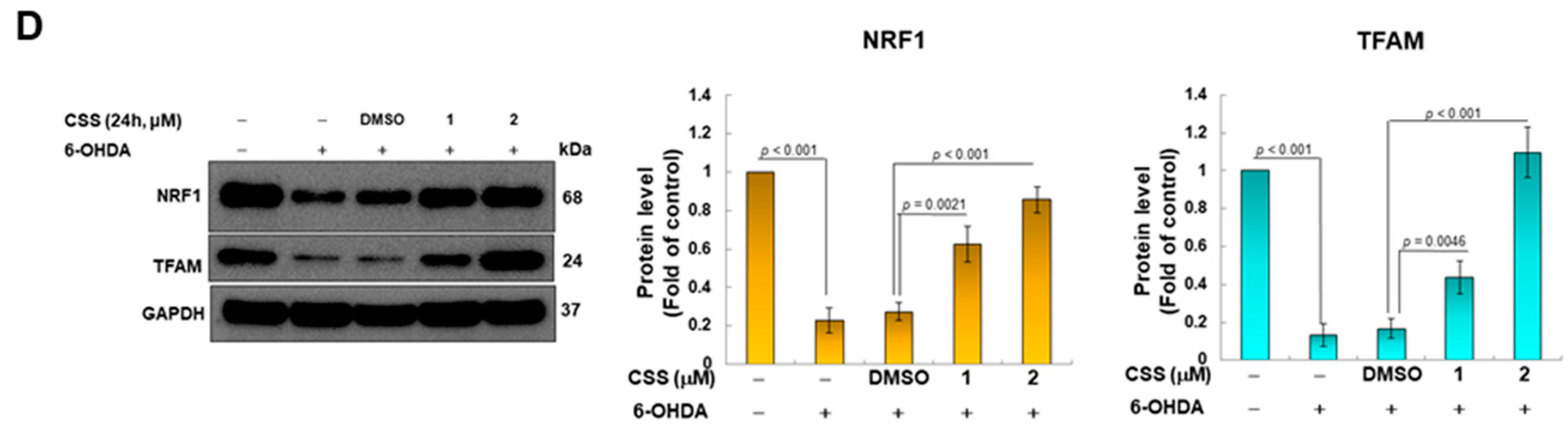
3.3. CSS Pretreatment Can Increase the Expression of Genes Related to Mitochondrial Biogenesis in SH-SY5Y Cells
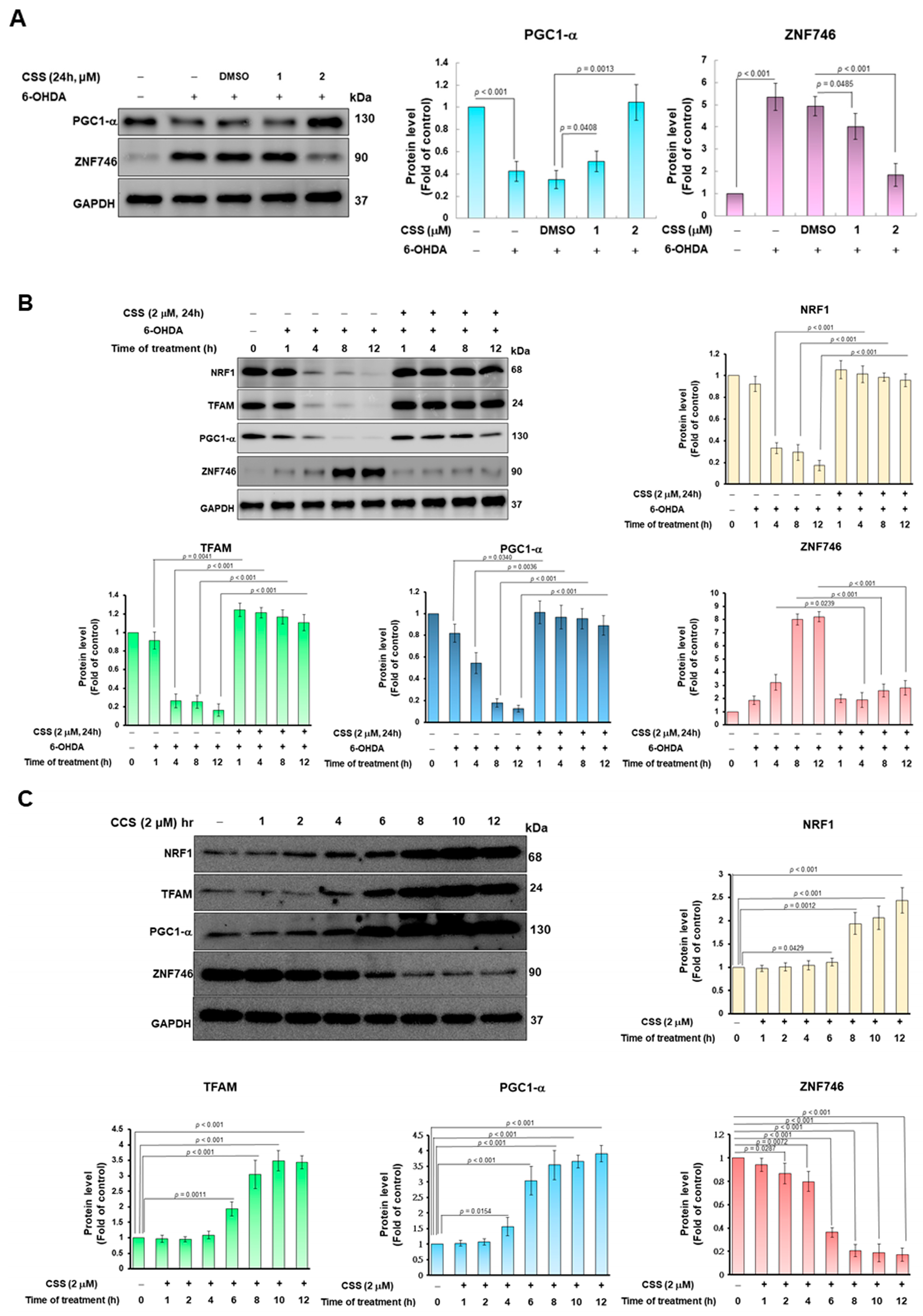
3.4. 6-OHDA Induces the Inhibition of ZNF746 on PGC-1α, Which Can Be Prevented by CSS Pretreatment
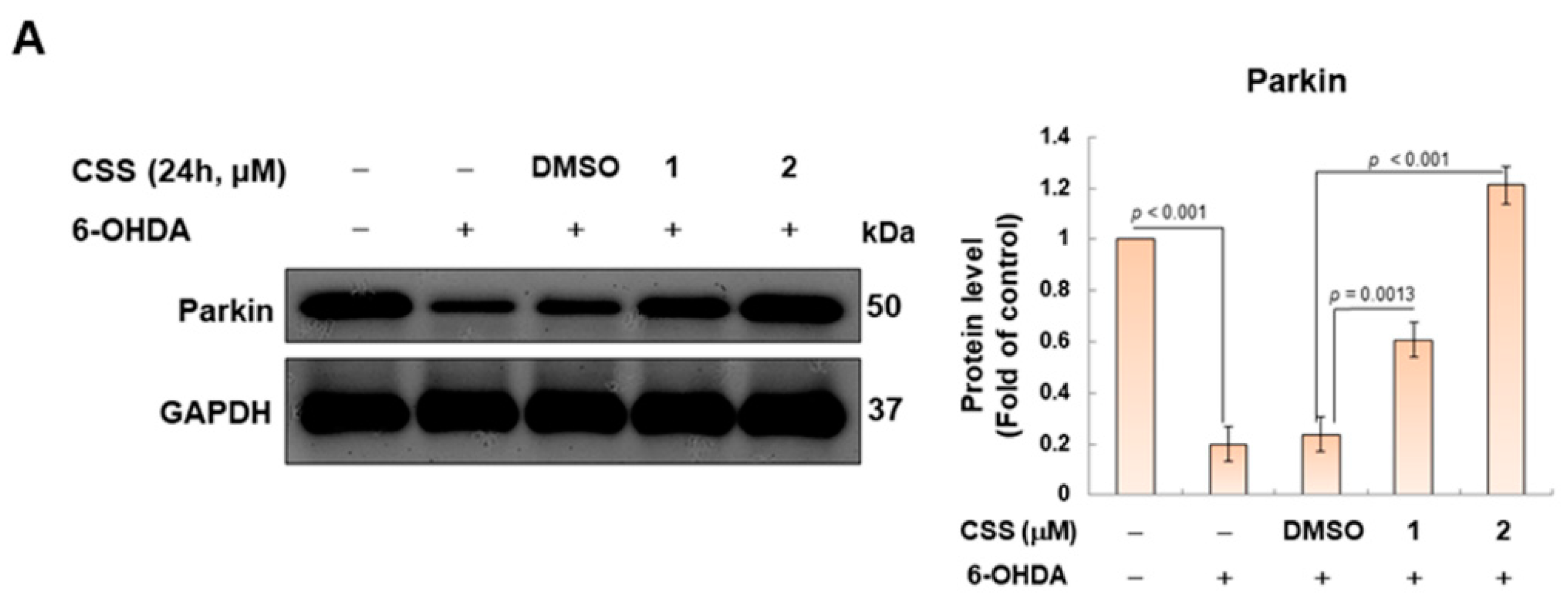
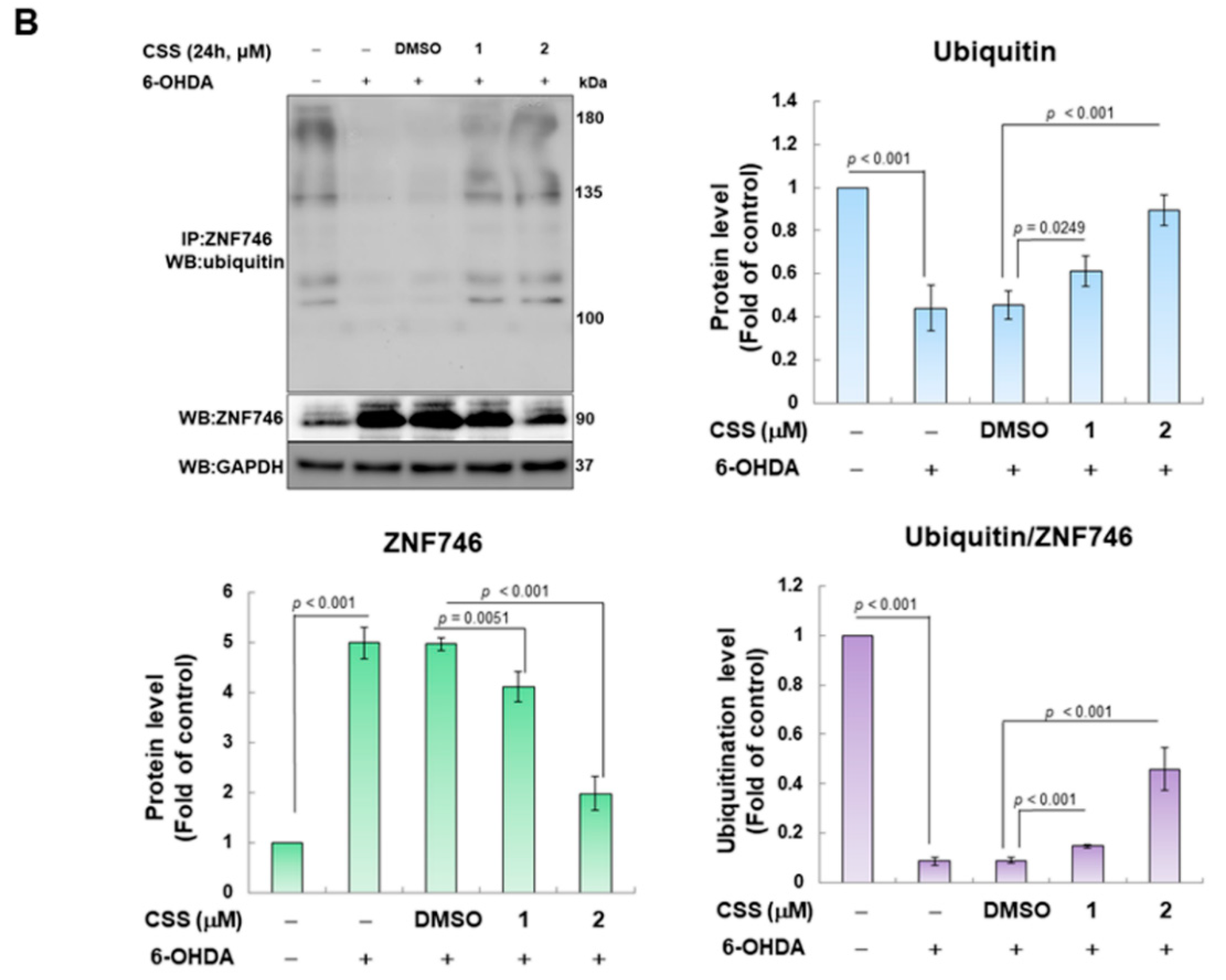
3.5. Parkin-Dependent Ubiquitination Degradation of ZNF746 Blocked by 6-OHDA Is Enhanced by CSS Pretreatment
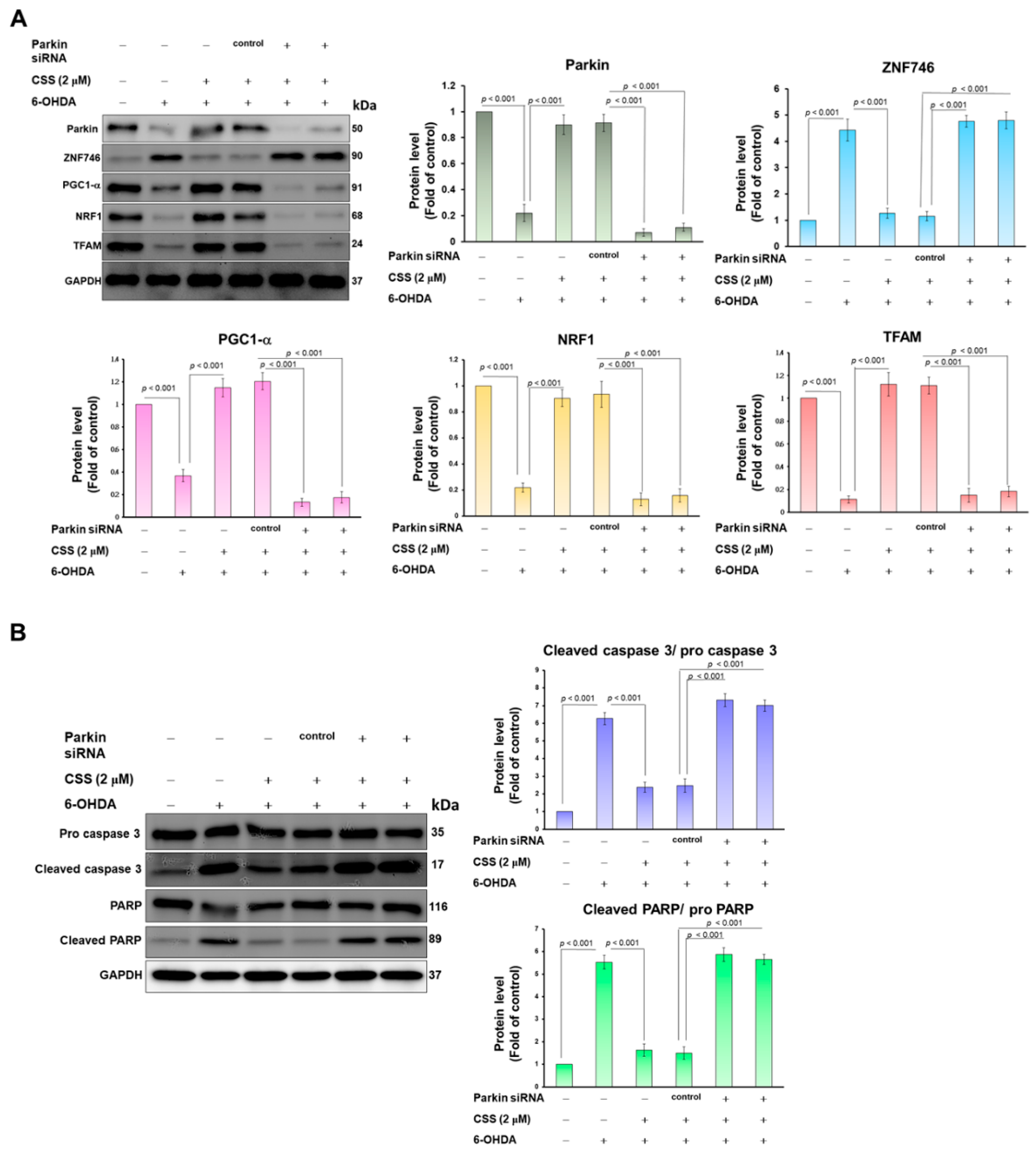
3.6. Parkin RNAi Abrogates CSS Ability to Prevent 6-OHDA-Induced Downregulation of Mitochondrial Biogenesis Proteins and Activation of Apoptotic Proteins
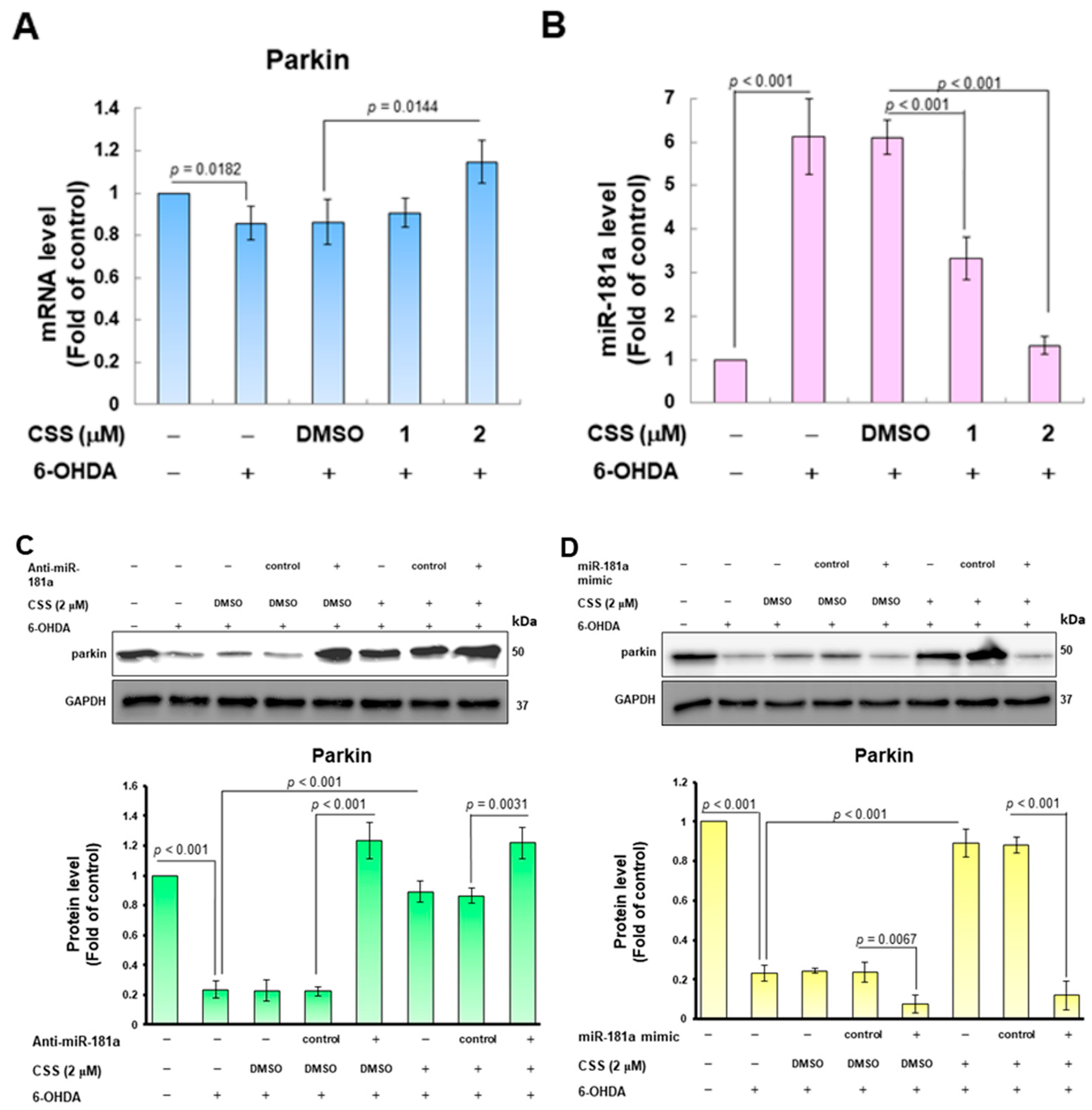
3.7. The Expression of Endogenous MiR-181a Induced by 6-OHDA Was Inhibited by CSS Pretreatment, Which Promoted the Increase in Parkin Level
3.8. Degeneration of DA Neurons in 6-OHDA-Exposed Caenorhabditis Elegans Animal Models Can Be Reduced by CSS Pretreatment
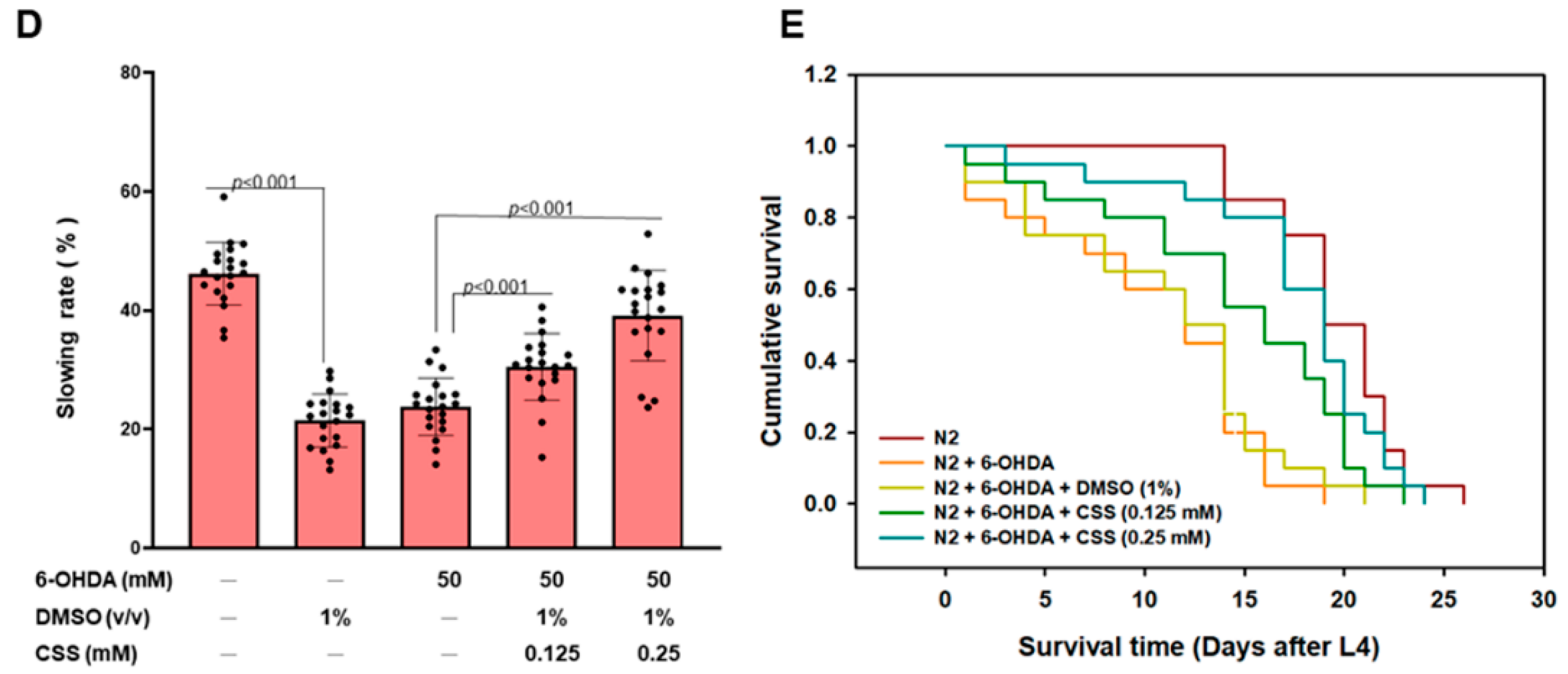
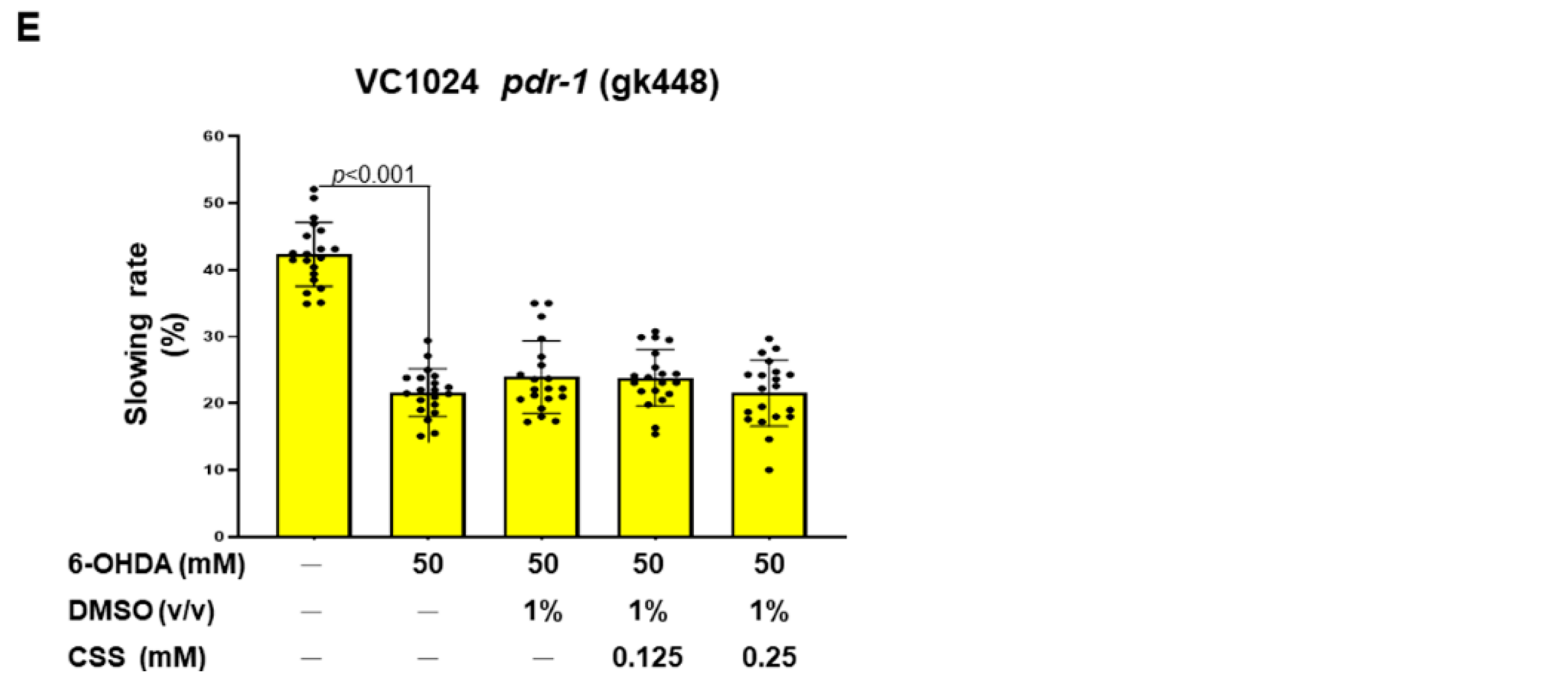
3.9. Dopamine-Mediated Deficits in Food-Sensitive Behaviors Induced by 6-OHDA Exposure in C. elegans Can Be Improved by CSS Pretreatment
3.10. The Lifespan of C. elegans Was Shortened by 6-OHDA Toxicity Can Be Extended by CSS Pretreatment
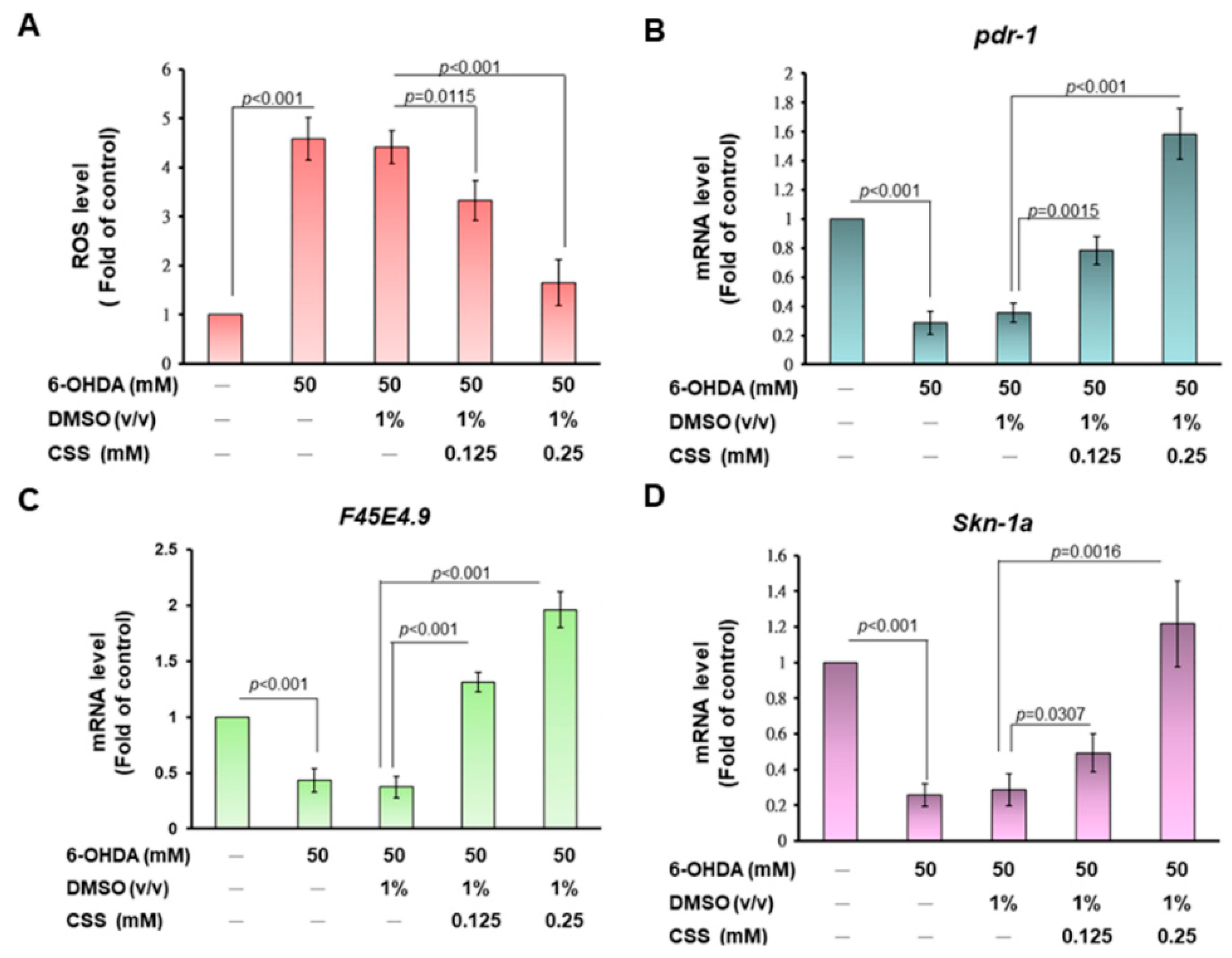
3.11. CSS Pretreatment Reduces the Level of Reactive Oxygen Species and Reverses the Downregulation of Pdr-1/Parkin, F45E4.9/TFAM, and SKN-1A/NRF1 in 6-OHDA-Exposed N2 Nematodes
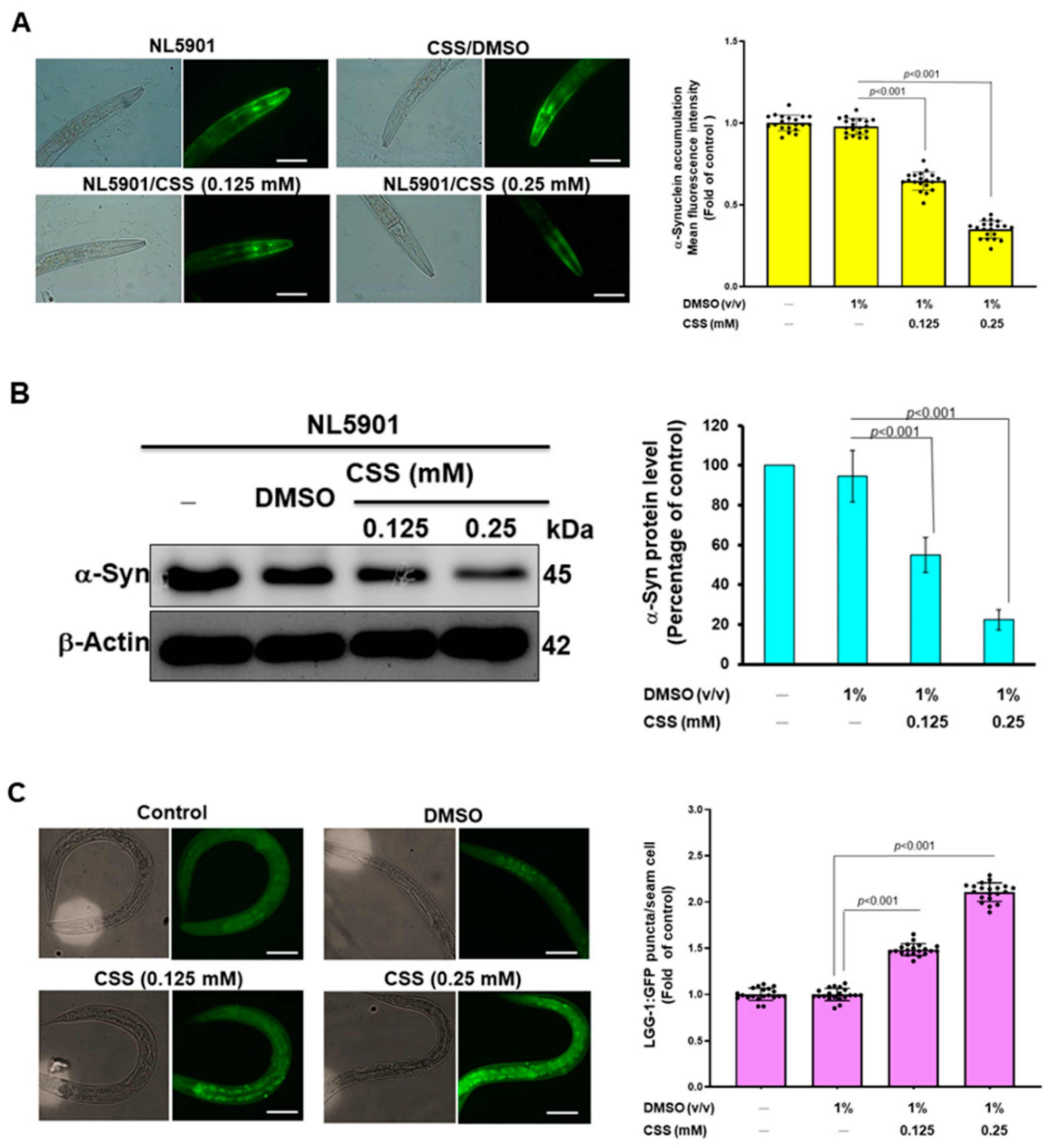
3.12. CSS Reduces α-Synuclein Accumulation by Promoting Autophagy Activity
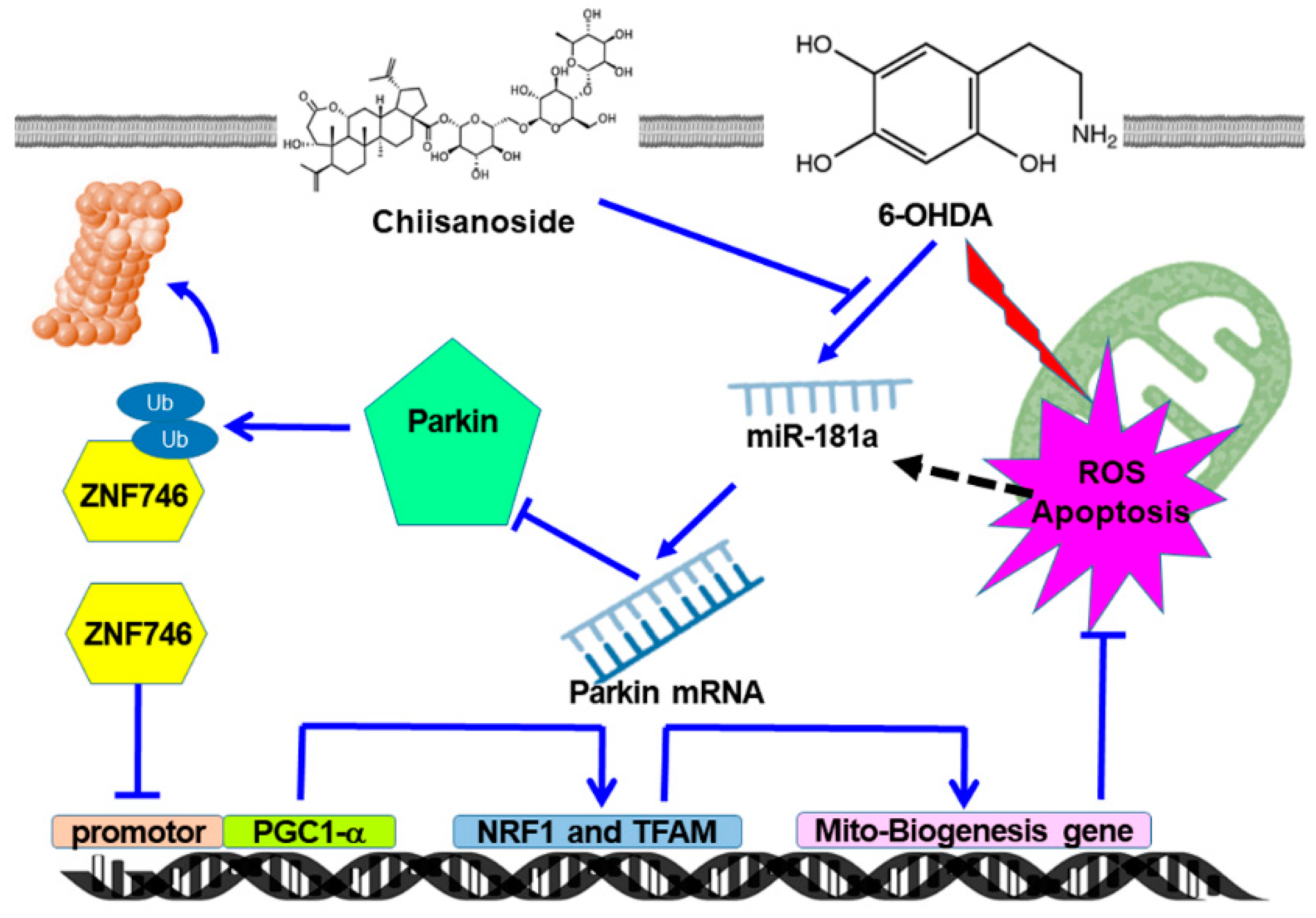
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef] [PubMed]
- Manini, A.; Abati, E.; Comi, G.P.; Corti, S.; Ronchi, D. Mitochondrial DNA homeostasis impairment and dopaminergic dysfunction: A trembling balance. Ageing Res. Rev. 2022, 76, 101578. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Zhou, B.; Ji, X. Mitochondrial quality control in acute ischemic stroke. J. Cereb. Blood Flow Metab. 2021, 41, 3157–3170. [Google Scholar] [CrossRef]
- Chodari, L.; Dilsiz Aytemir, M.; Vahedi, P.; Alipour, M.; Vahed, S.Z.; Khatibi, S.M.H.; Ahmadian, E.; Ardalan, M.; Eftekhari, A. Targeting Mitochondrial Biogenesis with Polyphenol Compounds. Oxid. Med. Cell. Longev. 2021, 2021, 4946711. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1alpha-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Front. Cell Dev. Biol. 2022, 10, 871357. [Google Scholar]
- Yao, K.; Zhang, W.W.; Yao, L.; Yang, S.; Nie, W.; Huang, F. Carvedilol promotes mitochondrial biogenesis by regulating the PGC-1/TFAM pathway in human umbilical vein endothelial cells (HUVECs). Biochem. Biophys. Res. Commun. 2016, 470, 961–966. [Google Scholar] [CrossRef]
- Panes, J.D.; Wendt, A.; Ramirez-Molina, O.; Castro, P.A.; Fuentealba, J. Deciphering the role of PGC-1alpha in neurological disorders: From mitochondrial dysfunction to synaptic failure. Neural Regen. Res. 2022, 17, 237–245. [Google Scholar]
- Koh, J.H.; Kim, Y.W.; Seo, D.Y.; Sohn, T.S. Mitochondrial TFAM as a Signaling Regulator between Cellular Organelles: A Perspective on Metabolic Diseases. Diabetes Metab. J. 2021, 45, 853–865. [Google Scholar] [CrossRef]
- Sekine, H.; Motohashi, H. Roles of CNC Transcription Factors NRF1 and NRF2 in Cancer. Cancers 2021, 13, 541. [Google Scholar] [CrossRef]
- Lee, Y.; Stevens, D.A.; Kang, S.U.; Jiang, H.; Lee, Y.I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef]
- Kim, H.; Kang, H.; Lee, Y.; Park, C.H.; Jo, A.; Khang, R.; Shin, J.H. Identification of transketolase as a target of PARIS in substantia nigra. Biochem. Biophys. Res. Commun. 2017, 493, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, S.K.; Yuan, C.; Khan, M.R.; Karuppagounder, S.S.; Wang, L.; Xiong, Y.; Kang, S.U.; Lee, Y.; Dawson, V.L.; Dawson, T.M. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol. Neurodegener. 2020, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.; Medvedev, A. Atypical Ubiquitination and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 3705. [Google Scholar] [CrossRef] [PubMed]
- Ning, P.; Jiang, X.; Yang, J.; Zhang, J.; Yang, F.; Cao, H. Mitophagy: A potential therapeutic target for insulin resistance. Front. Physiol. 2022, 13, 957968. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Brouwer, J.M.; Tan, I.K.; Sandow, J.J.; Huang, S.; Stafford, C.A.; Bankovacki, A.; Riffkin, C.D.; Wardak, A.Z.; Czabotar, P.E.; et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. 2019, 38, e99916. [Google Scholar] [CrossRef]
- Silvian, L.F. PINK1/Parkin Pathway Activation for Mitochondrial Quality Control—Which Is the Best Molecular Target for Therapy? Front. Aging Neurosci. 2022, 14, 890823. [Google Scholar] [CrossRef]
- Leggio, L.; Vivarelli, S.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Marchetti, B.; Iraci, N. microRNAs in Parkinson’s Disease: From Pathogenesis to Novel Diagnostic and Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 2698. [Google Scholar] [CrossRef]
- Li, S.; Bi, G.; Han, S.; Huang, R. MicroRNAs Play a Role in Parkinson’s Disease by Regulating Microglia Function: From Pathogenetic Involvement to Therapeutic Potential. Front. Mol. Neurosci. 2021, 14, 744942. [Google Scholar] [CrossRef]
- Nguyen, T.P.N.; Kumar, M.; Fedele, E.; Bonanno, G.; Bonifacino, T. MicroRNA Alteration, Application as Biomarkers, and Therapeutic Approaches in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 4718. [Google Scholar] [CrossRef]
- Bian, X.; Liu, X.; Liu, J.; Zhao, Y.; Li, H.; Zhang, L.; Li, P.; Gao, Y. Hepatoprotective effect of chiisanoside from Acanthopanax sessiliflorus against LPS/D-GalN-induced acute liver injury by inhibiting NF-kappaB and activating Nrf2/HO-1 signaling pathways. J. Sci. Food Agric. 2019, 99, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, C.; Liu, R.; Wang, X.; Zhao, Y.; Yan, Z.; Cai, E.; Zhu, H. Potential myocardial protection of 3,4-seco-lupane triterpenoids from Acanthopanax sessiliflorus Leaves. Chem. Biodivers. 2021, 18, e2000830. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liu, X.; Liu, J.; Zhao, Y.; Li, H.; Cai, E.; Li, P.; Gao, Y. Study on antidepressant activity of chiisanoside in mice. Int. Immunopharmacol. 2018, 57, 33–42. [Google Scholar] [CrossRef]
- Yoshizumi, K.; Murota, K.; Watanabe, S.; Tomi, H.; Tsuji, T.; Terao, J. Chiisanoside is not absorbed but inhibits oil absorption in the small intestine of rodents. Biosci. Biotechnol. Biochem. 2008, 72, 1126–1129. [Google Scholar] [CrossRef]
- Jin, J.L.; Lee, S.; Lee, Y.Y.; Kim, J.M.; Heo, J.E.; Yun-Choi, H.S. Platelet anti-aggregating triterpenoids from the leaves of Acanthopanax senticosus and the fruits of A. sessiliflorus. Planta Med. 2004, 70, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, R.M. Neonatal 6-hydroxydopamine lesioning of rats and dopaminergic neurotoxicity: Proposed animal model of Parkinson’s disease. J. Neural. Transm. 2022, 129, 445–461. [Google Scholar] [CrossRef]
- Thirugnanam, T.; Santhakumar, K. Chemically induced models of Parkinson’s disease. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2022, 252, 109213. [Google Scholar] [CrossRef]
- Lopez-Suarez, L.; Awabdh, S.A.; Coumoul, X.; Chauvet, C. The SH-SY5Y human neuroblastoma cell line, a relevant in vitro cell model for investigating neurotoxicology in human: Focus on organic pollutants. Neurotoxicology 2022, 92, 131–155. [Google Scholar] [CrossRef]
- Surguchov, A. Invertebrate Models Untangle the Mechanism of Neurodegeneration in Parkinson’s Disease. Cells 2021, 10, 407. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liu, S.P.; Lin, H.L.; Chan, M.C.; Chen, Y.C.; Huang, Y.L.; Tsai, M.C.; Fu, R.H. Irisflorentin improves alpha-synuclein accumulation and attenuates 6-OHDA-induced dopaminergic neuron degeneration, implication for Parkinson’s disease therapy. Biomedicine 2015, 5, 4. [Google Scholar] [CrossRef]
- Xie, W.; Li, M.; Xu, N.; Lv, Q.; Huang, N.; He, J.; Zhang, Y. MiR-181a regulates inflammation responses in monocytes and macrophages. PLoS ONE 2013, 8, e58639. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, L.A.; Hamamichi, S.; Knight, A.L.; Harrington, A.J.; Caldwell, G.A.; Caldwell, K.A. Application of a C. elegans dopamine neuron degeneration assay for the validation of potential Parkinson’s disease genes. J. Vis. Exp. 2008, 17, 835. [Google Scholar]
- Fu, R.H.; Tsai, C.W.; Liu, S.P.; Chiu, S.C.; Chen, Y.C.; Chiang, Y.T.; Kuo, Y.H.; Shyu, W.C.; Lin, S.Z. Neuroprotective Capability of Narcissoside in 6-OHDA-Exposed Parkinson’s Disease Models through Enhancing the MiR200a/Nrf-2/GSH Axis and Mediating MAPK/Akt Associated Signaling Pathway. Antioxidants 2022, 11, 2089. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhao, J.; Zhang, R.; Zhang, L.; Zhang, Q.; Yang, H.; An, J. Neuroprotective effects of natural compounds on neurotoxin-induced oxidative stress and cell apoptosis. Nutr. Neurosci. 2022, 25, 1078–1099. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Gutierrez, M.T.; Serrano-Garcia, N.; Orozco-Ibarra, M. Rotenone-Induced Model of Parkinson’s Disease: Beyond Mitochondrial Complex I Inhibition. Mol. Neurobiol. 2023, 60, 1929–1948. [Google Scholar] [CrossRef]
- Hees, J.T.; Harbauer, A.B. Metabolic Regulation of Mitochondrial Protein Biogenesis from a Neuronal Perspective. Biomolecules 2022, 12, 1595. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.Y.; Park, S.J.; Kwag, E.; Kim, J.; Shin, J.H. S-nitrosylated PARIS Leads to the Sequestration of PGC-1alpha into Insoluble Deposits in Parkinson’s Disease Model. Cells 2022, 11, 3682. [Google Scholar] [CrossRef]
- Jo, A.; Lee, Y.; Kam, T.I.; Kang, S.U.; Neifert, S.; Karuppagounder, S.S.; Khang, R.; Kang, H.; Park, H.; Chou, S.C.; et al. PARIS farnesylation prevents neurodegeneration in models of Parkinson’s disease. Sci. Transl. Med. 2021, 13, eaax8891. [Google Scholar] [CrossRef]
- Goljanek-Whysall, K.; Soriano-Arroquia, A.; McCormick, R.; Chinda, C.; McDonagh, B. miR-181a regulates p62/SQSTM1, parkin, and protein DJ-1 promoting mitochondrial dynamics in skeletal muscle aging. Aging Cell 2020, 19, e13140. [Google Scholar] [CrossRef]
- Hoffmann-Conaway, S.; Brockmann, M.M.; Schneider, K.; Annamneedi, A.; Rahman, K.A.; Bruns, C.; Textoris-Taube, K.; Trimbuch, T.; Smalla, K.H.; Rosenmund, C.; et al. Parkin contributes to synaptic vesicle autophagy in Bassoon-deficient mice. eLife 2020, 9, e56590. [Google Scholar] [CrossRef]
- Mittal, P.; Dhankhar, S.; Chauhan, S.; Garg, N.; Bhattacharya, T.; Ali, M.; Chaudhary, A.A.; Rudayni, H.A.; Al-Zharani, M.; Ahmad, W.; et al. A Review on Natural Antioxidants for Their Role in the Treatment of Parkinson’s Disease. Pharmaceuticals 2023, 16, 908. [Google Scholar] [CrossRef]
- Tassone, A.; Meringolo, M.; Ponterio, G.; Bonsi, P.; Schirinzi, T.; Martella, G. Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson. Int. J. Mol. Sci. 2023, 24, 7221. [Google Scholar] [CrossRef]
- Jain, R.; Begum, N.; Tryphena, K.P.; Singh, S.B.; Srivastava, S.; Rai, S.N.; Vamanu, E.; Khatri, D.K. Inter and intracellular mitochondrial transfer: Future of mitochondrial transplant therapy in Parkinson’s disease. Biomed. Pharmacother. 2023, 159, 114268. [Google Scholar] [CrossRef]
- Lang, M.; Pramstaller, P.P.; Pichler, I. Crosstalk of organelles in Parkinson’s disease—MiT family transcription factors as central players in signaling pathways connecting mitochondria and lysosomes. Mol. Neurodegener. 2022, 17, 50. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef]
- Yang, J.J.; Wang, J.; Yang, Y.; Sun, F.; Yang, Y.; Li, J.; Tao, H.; Lu, C. Epigenetic control of mitochondrial fission enables hepatic stellate cells activation in liver fibrosis via PGC-1alpha-Drp1 pathway. Mitochondrion 2022, 66, 38–50. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Wang, J.; Zhang, D.; Wu, H.; Li, W.; Wei, H.; Ta, N.; Fan, Y.; Liu, Y.; et al. Mitophagy receptor FUNDC1 is regulated by PGC-1alpha/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021, 22, e50629. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, Q.; Tuccitto, A.; Chan, D.; Alqawlaq, S.; Won, G.J.; Sivak, J.M. The AMPK-PGC-1alpha signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury. Neurobiol. Dis. 2018, 113, 59–69. [Google Scholar] [CrossRef]
- Zhou, H.; Shao, M.; Yang, X.; Li, C.; Cui, G.; Gao, C.; Di, L.; Zhong, H.; Wang, Y.; Zhang, Z.; et al. Tetramethylpyrazine Analogue T-006 Exerts Neuroprotective Effects against 6-Hydroxydopamine-Induced Parkinson’s Disease In Vitro and In Vivo. Oxid. Med. Cell. Longev. 2019, 2019, 8169125. [Google Scholar] [CrossRef]
- Tang, H.; Zheng, Z.; Wang, H.; Wang, L.; Zhao, G.; Wang, P. Vitamin K2 Modulates Mitochondrial Dysfunction Induced by 6-Hydroxydopamine in SH-SY5Y Cells via Mitochondrial Quality-Control Loop. Nutrients 2022, 14, 1504. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, J.; Qiu, J.; Wang, L.; Zhuo, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A protects dopaminergic neurons in experimental models of Parkinson’s disease by promoting mitochondrial biogenesis through the SIRT1/PGC-1alpha signaling pathway. Food Funct. 2022, 13, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Huang, Y.N.; Fu, R.H.; Liao, Y.H.; Kuo, T.Y.; Tsai, C.W. Promotion of mitochondrial biogenesis via the regulation of PARIS and PGC-1alpha by parkin as a mechanism of neuroprotection by carnosic acid. Phytomedicine 2021, 80, 153369. [Google Scholar] [CrossRef] [PubMed]
- Saihara, K.; Kamikubo, R.; Ikemoto, K.; Uchida, K.; Akagawa, M. Pyrroloquinoline Quinone, a Redox-Active o-Quinone, Stimulates Mitochondrial Biogenesis by Activating the SIRT1/PGC-1alpha Signaling Pathway. Biochemistry 2017, 56, 6615–6625. [Google Scholar] [CrossRef]
- Zhang, C.; He, X.; Sheng, Y.; Xu, J.; Yang, C.; Zheng, S.; Liu, J.; Li, H.; Ge, J.; Yang, M.; et al. Allicin Regulates Energy Homeostasis through Brown Adipose Tissue. iScience 2020, 23, 101113. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.F.F.; Binda, K.H.; Singulani, M.P.; Pereira, C.P.M.; Ferrari, G.D.; Alberici, L.C.; Real, C.C.; Britto, L.R. Physical exercise protects against mitochondria alterations in the 6-hidroxydopamine rat model of Parkinson’s disease. Behav. Brain Res. 2020, 387, 112607. [Google Scholar] [CrossRef]
- Hsu, H.T.; Yang, Y.L.; Chang, W.H.; Fang, W.Y.; Huang, S.H.; Chou, S.H.; Lo, Y.C. Hyperbaric Oxygen Therapy Improves Parkinson’s Disease by Promoting Mitochondrial Biogenesis via the SIRT-1/PGC-1alpha Pathway. Biomolecules 2022, 12, 661. [Google Scholar] [CrossRef]
- Yazar, V.; Kang, S.U.; Ha, S.; Dawson, V.L.; Dawson, T.M. Integrative genome-wide analysis of dopaminergic neuron-specific PARIS expression in Drosophila dissects recognition of multiple PPAR-gamma associated gene regulation. Sci. Rep. 2021, 11, 21500. [Google Scholar] [CrossRef]
- Li, C.Y.; Ou, R.W.; Chen, Y.P.; Gu, X.J.; Wei, Q.Q.; Cao, B.; Zhang, L.Y.; Hou, Y.B.; Liu, K.C.; Chen, X.P.; et al. Genetic Analysis of ZNF Protein Family Members for Early-Onset Parkinson’s Disease in Chinese Population. Mol. Neurobiol. 2021, 58, 3435–3442. [Google Scholar] [CrossRef]
- Bae, J.H.; Jeong, H.J.; Kim, H.; Leem, Y.E.; Ryu, D.; Park, S.C.; Lee, Y.I.; Cho, S.C.; Kang, J.S. ZNF746/PARIS overexpression induces cellular senescence through FoxO1/p21 axis activation in myoblasts. Cell Death Dis. 2020, 11, 359. [Google Scholar] [CrossRef]
- Kang, H.; Jo, A.; Kim, H.; Khang, R.; Lee, J.Y.; Kim, H.; Park, C.H.; Choi, J.Y.; Lee, Y.; Shin, J.H. PARIS reprograms glucose metabolism by HIF-1alpha induction in dopaminergic neurodegeneration. Biochem. Biophys. Res. Commun. 2018, 495, 2498–2504. [Google Scholar] [CrossRef]
- Nishida, T.; Yamada, Y. RNF4-mediated SUMO-targeted ubiquitination relieves PARIS/ZNF746-mediated transcriptional repression. Biochem. Biophys. Res. Commun. 2020, 526, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, S.; Lee, S.; Kim, S.; Yuan, C.; Karuppagounder, S.S.; Ge, P.; Shi, R.; Kim, E.J.; Liu, A.; Kim, D.; et al. Parkin interacting substrate zinc finger protein 746 is a pathological mediator in Parkinson’s disease. Brain 2019, 142, 2380–2401. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.L.; Grossmann, D.; Delcambre, S.; Hermann, A.; Grunewald, A. Novel insights into Parkin-mediated mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Curr. Opin. Neurobiol. 2023, 80, 102720. [Google Scholar] [CrossRef]
- Trempe, J.F.; Gehring, K. Structural Mechanisms of Mitochondrial Quality Control Mediated by PINK1 and Parkin. J. Mol. Biol. 2023, 435, 168090. [Google Scholar] [CrossRef]
- Patel, J.; Panicker, N.; Dawson, V.L.; Dawson, T.M. Cell Biology of Parkin: Clues to the Development of New Therapeutics for Parkinson’s Disease. CNS Drugs 2022, 36, 1249–1267. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Kam, T.I.; Wang, H.; Neifert, S.; Chou, S.C.; Kumar, M.; Brahmachari, S.; Jhaldiyal, A.; Hinkle, J.T.; Akkentli, F.; et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 2022, 110, 2422–2437.e9. [Google Scholar] [CrossRef]
- Indrieri, A.; Carrella, S.; Romano, A.; Spaziano, A.; Marrocco, E.; Fernandez-Vizarra, E.; Barbato, S.; Pizzo, M.; Ezhova, Y.; Golia, F.M.; et al. miR-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol. Med. 2019, 11, e8734. [Google Scholar] [CrossRef]
- Cheng, M.; Liu, L.; Lao, Y.; Liao, W.; Liao, M.; Luo, X.; Wu, J.; Xie, W.; Zhang, Y.; Xu, N. MicroRNA-181a suppresses parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis. Oncotarget 2016, 7, 42274–42287. [Google Scholar] [CrossRef]
- Stein, C.S.; McLendon, J.M.; Witmer, N.H.; Boudreau, R.L. Modulation of miR-181 influences dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Mol. Ther. Nucleic Acids 2022, 28, 1–15. [Google Scholar] [CrossRef]
- Barbato, A.; Iuliano, A.; Volpe, M.; D’Alterio, R.; Brillante, S.; Massa, F.; De Cegli, R.; Carrella, S.; Salati, M.; Russo, A.; et al. Integrated Genomics Identifies miR-181/TFAM Pathway as a Critical Driver of Drug Resistance in Melanoma. Int. J. Mol. Sci. 2021, 22, 1801. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.W.; Yang, P.; Zhao, L.L. Chlorpyrifos activates cell pyroptosis and increases susceptibility on oxidative stress-induced toxicity by miR-181/SIRT1/PGC-1alpha/Nrf2 signaling pathway in human neuroblastoma SH-SY5Y cells: Implication for association between chlorpyrifos and Parkinson’s disease. Environ. Toxicol. 2019, 34, 699–707. [Google Scholar] [PubMed]
- Liang, X.; Xu, W. miR-181a-5p regulates the proliferation and apoptosis of glomerular mesangial cells by targeting KLF6. Exp. Ther. Med. 2020, 20, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; An, X.H.; Zhang, X.Q.; Liu, J.H.; Wang, J.W.; Yang, Z.Y. Morphine induces the apoptosis of mouse hippocampal neurons HT-22 through upregulating miR-181-5p. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7114–7121. [Google Scholar] [PubMed]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Jung, H.J.; Nam, J.H.; Choi, J.; Lee, K.T.; Park, H.J. Antiinflammatory effects of chiisanoside and chiisanogenin obtained from the leaves of Acanthopanax chiisanensis in the carrageenan- and Freund’s complete adjuvant-induced rats. J. Ethnopharmacol. 2005, 97, 359–367. [Google Scholar] [CrossRef]
- Bian, X.; Wang, S.; Liu, J.; Zhao, Y.; Li, H.; Zhang, L.; Li, P. Hepatoprotective effect of chiisanoside against acetaminophen-induced acute liver injury in mice. Nat. Prod. Res. 2018, 33, 2704–2707. [Google Scholar] [CrossRef]
- Aune, D.; Schlesinger, S.; Mahamat-Saleh, Y.; Zheng, B.; Udeh-Momoh, C.T.; Middleton, L.T. Diabetes mellitus, prediabetes and the risk of Parkinson’s disease: A systematic review and meta-analysis of 15 cohort studies with 29.9 million participants and 86,345 cases. Eur. J. Epidemiol. 2023, 38, 591–604. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, Y.-L.; Chen, H.-J.; Gao, J.-X.; Yang, M.-Y.; Fu, R.-H. Chiisanoside Mediates the Parkin/ZNF746/PGC-1α Axis by Downregulating MiR-181a to Improve Mitochondrial Biogenesis in 6-OHDA-Caused Neurotoxicity Models In Vitro and In Vivo: Suggestions for Prevention of Parkinson’s Disease. Antioxidants 2023, 12, 1782. https://doi.org/10.3390/antiox12091782
Hsu Y-L, Chen H-J, Gao J-X, Yang M-Y, Fu R-H. Chiisanoside Mediates the Parkin/ZNF746/PGC-1α Axis by Downregulating MiR-181a to Improve Mitochondrial Biogenesis in 6-OHDA-Caused Neurotoxicity Models In Vitro and In Vivo: Suggestions for Prevention of Parkinson’s Disease. Antioxidants. 2023; 12(9):1782. https://doi.org/10.3390/antiox12091782
Chicago/Turabian StyleHsu, Yu-Ling, Hui-Jye Chen, Jia-Xin Gao, Ming-Yang Yang, and Ru-Huei Fu. 2023. "Chiisanoside Mediates the Parkin/ZNF746/PGC-1α Axis by Downregulating MiR-181a to Improve Mitochondrial Biogenesis in 6-OHDA-Caused Neurotoxicity Models In Vitro and In Vivo: Suggestions for Prevention of Parkinson’s Disease" Antioxidants 12, no. 9: 1782. https://doi.org/10.3390/antiox12091782
APA StyleHsu, Y. -L., Chen, H. -J., Gao, J. -X., Yang, M. -Y., & Fu, R. -H. (2023). Chiisanoside Mediates the Parkin/ZNF746/PGC-1α Axis by Downregulating MiR-181a to Improve Mitochondrial Biogenesis in 6-OHDA-Caused Neurotoxicity Models In Vitro and In Vivo: Suggestions for Prevention of Parkinson’s Disease. Antioxidants, 12(9), 1782. https://doi.org/10.3390/antiox12091782